
Parameters for Irreversible Inactivation of Monoamine Oxidase

 ,
,  , and
, and
Abstract
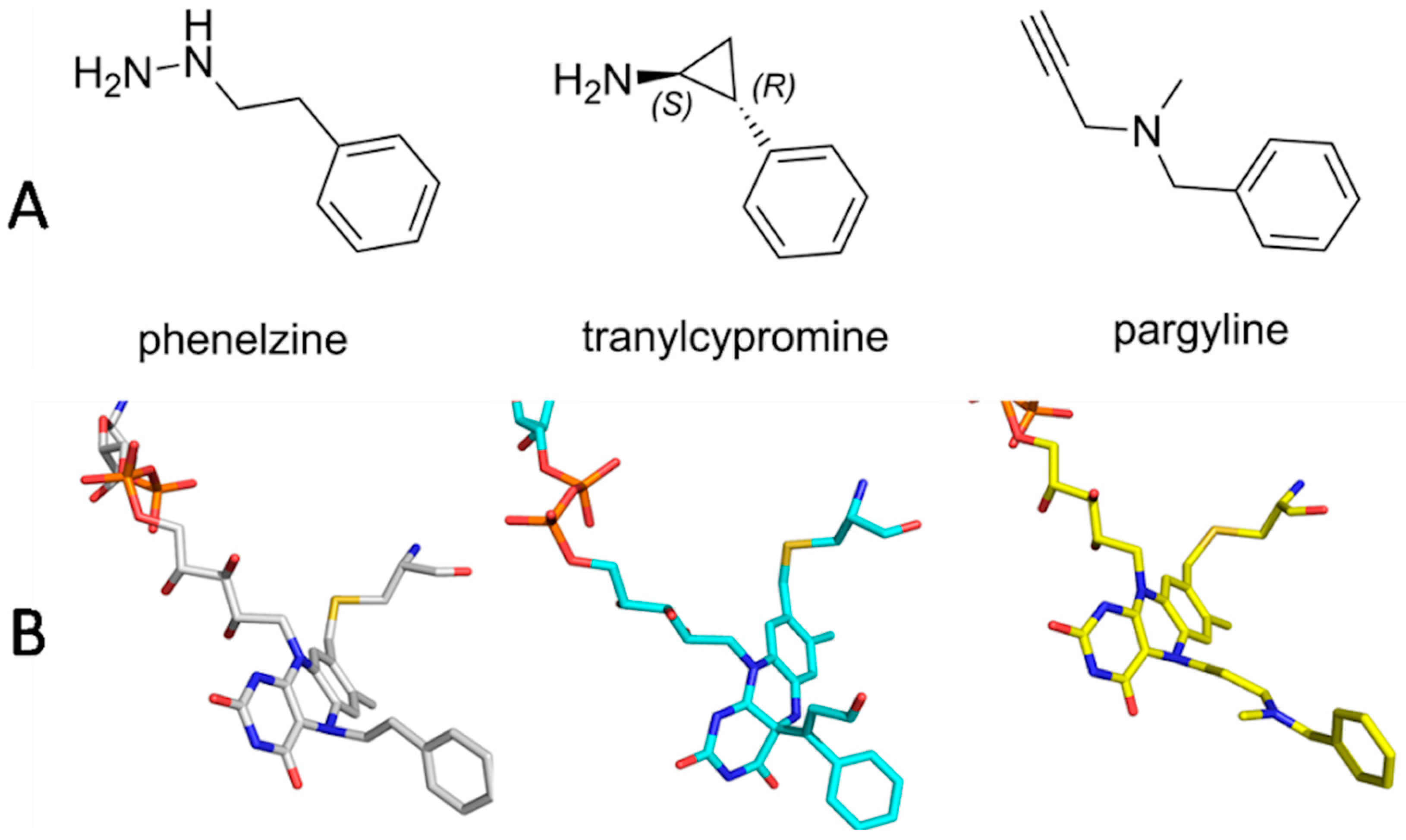
:1. Introduction
2. Results and Discussion
2.1. Kinetics for Assessment of Mechanism-Based Inhibitors
2.1.1. Reversible Binding of MAO Inactivators
2.1.2. IC50 after Preincubation with MAO
2.1.3. Oxidation of Clorgyline Is Required for Adduct Formation
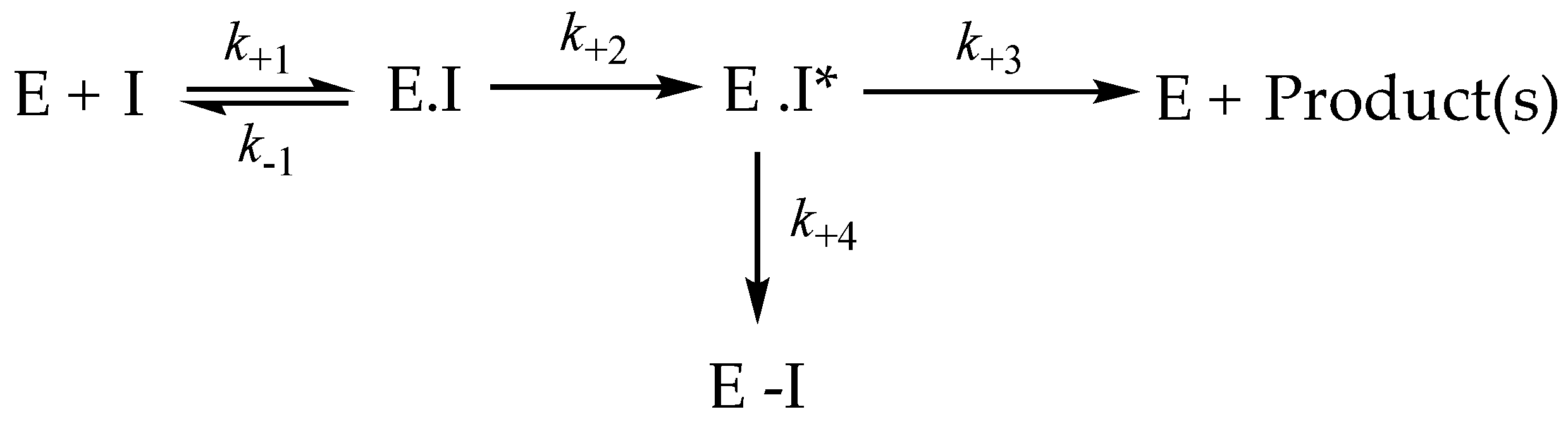
2.1.4. Kinetic Constants for Time-Dependent Inactivation
2.1.5. Partition Ratios
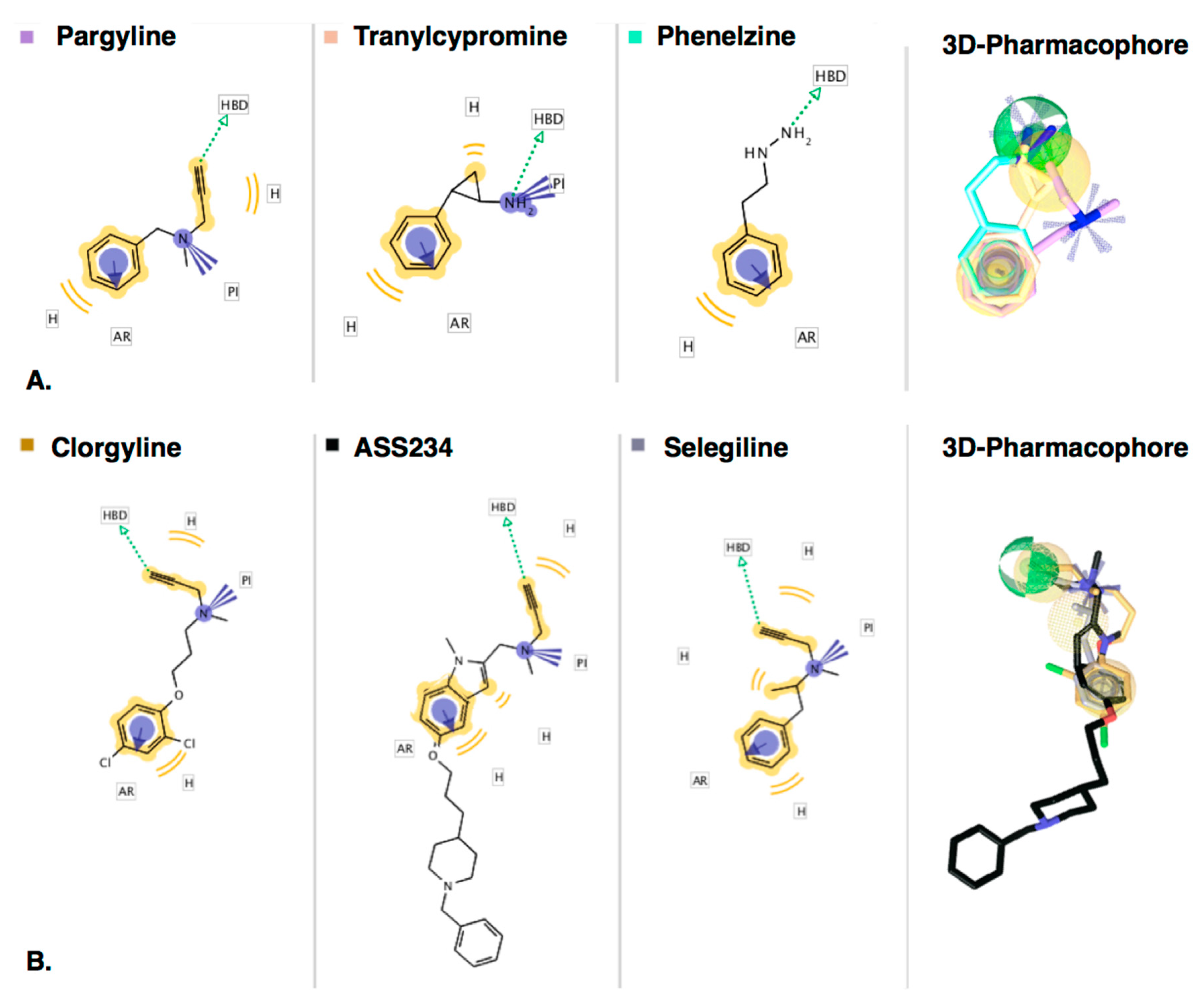
2.2. D-Chemical Feature-Based-Pharmacophore Interactions
2.2.1. Ligand-Based Modeling
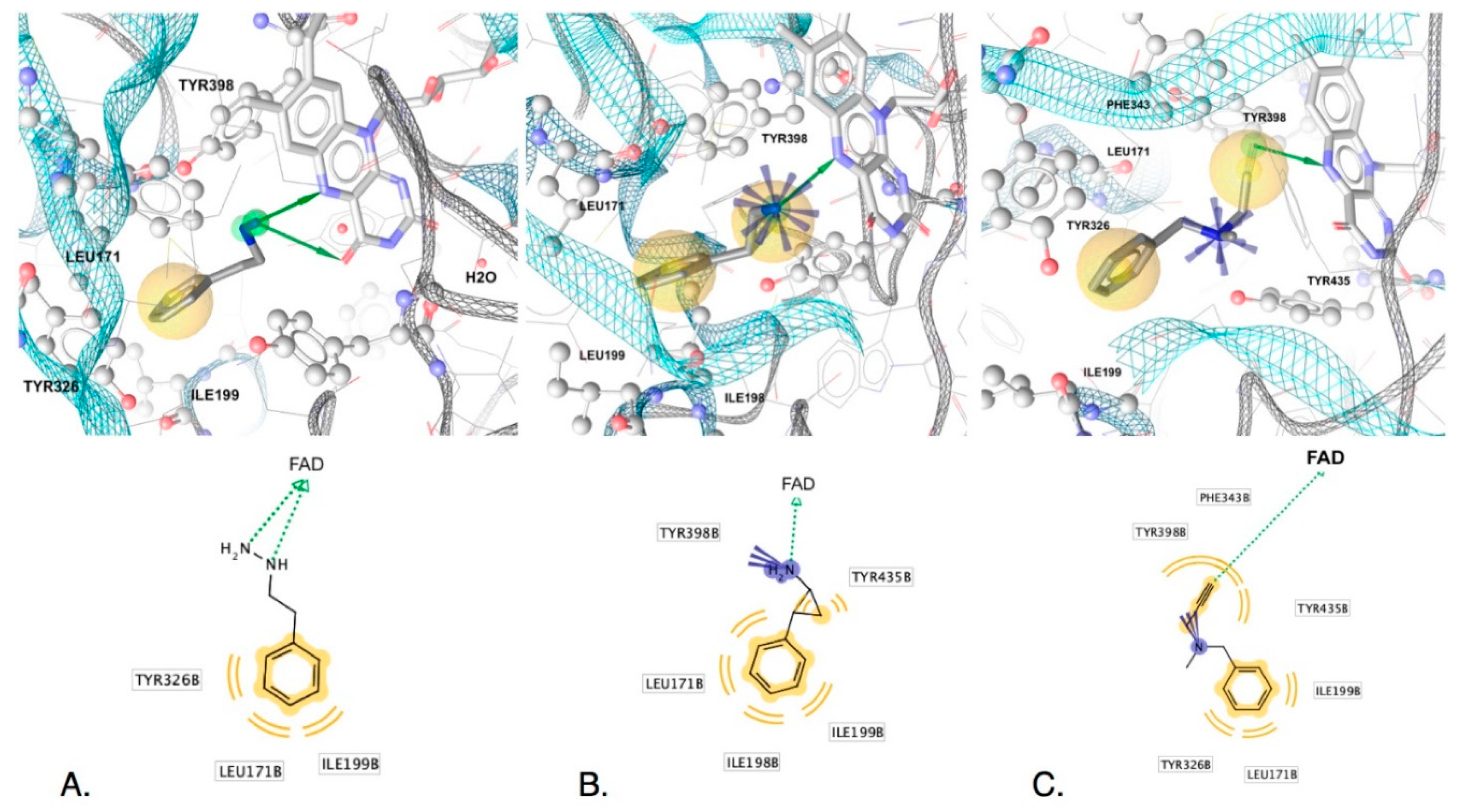
2.2.2. Structure-Based Modeling
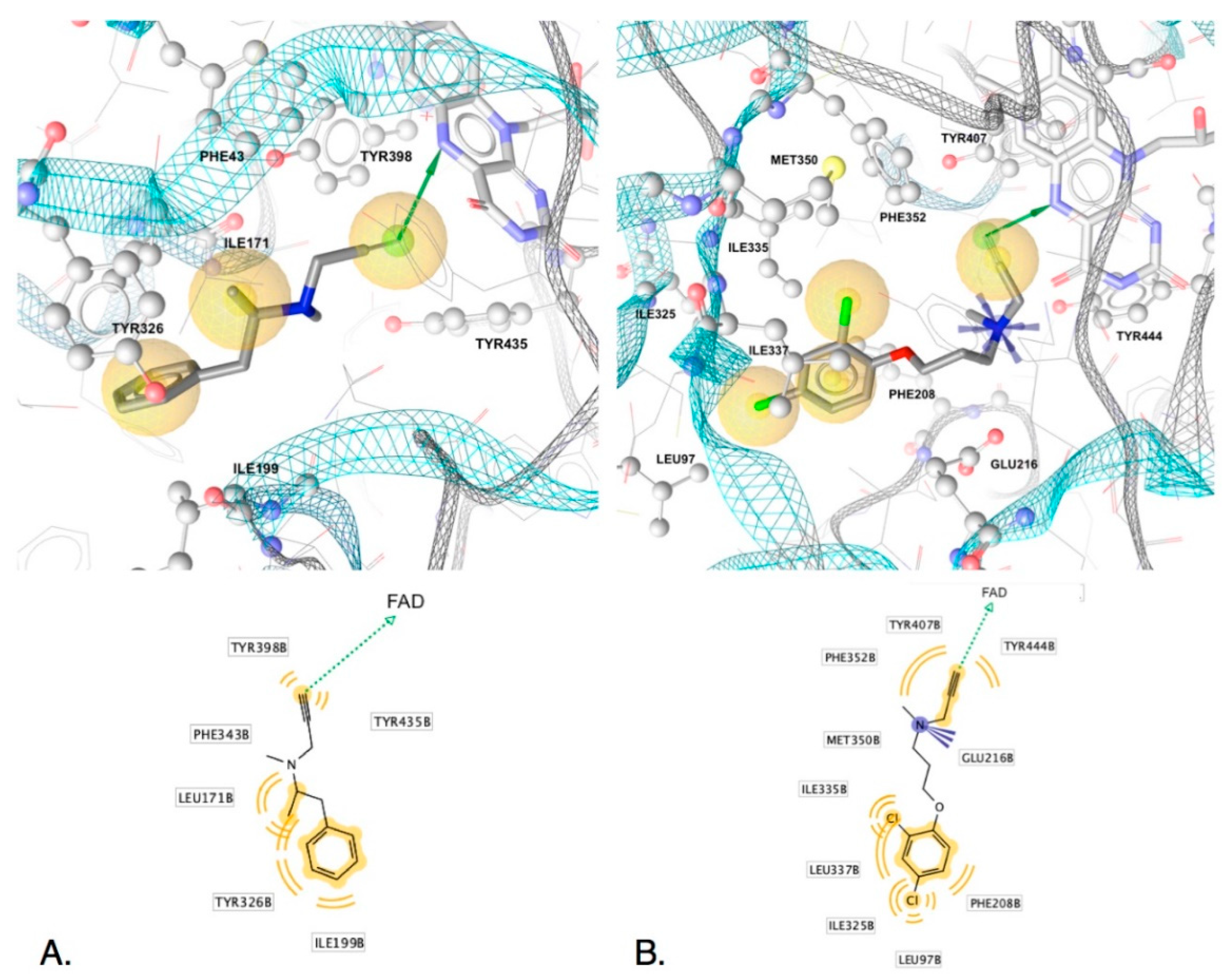
2.2.3. Interactions Conferring Selectivity



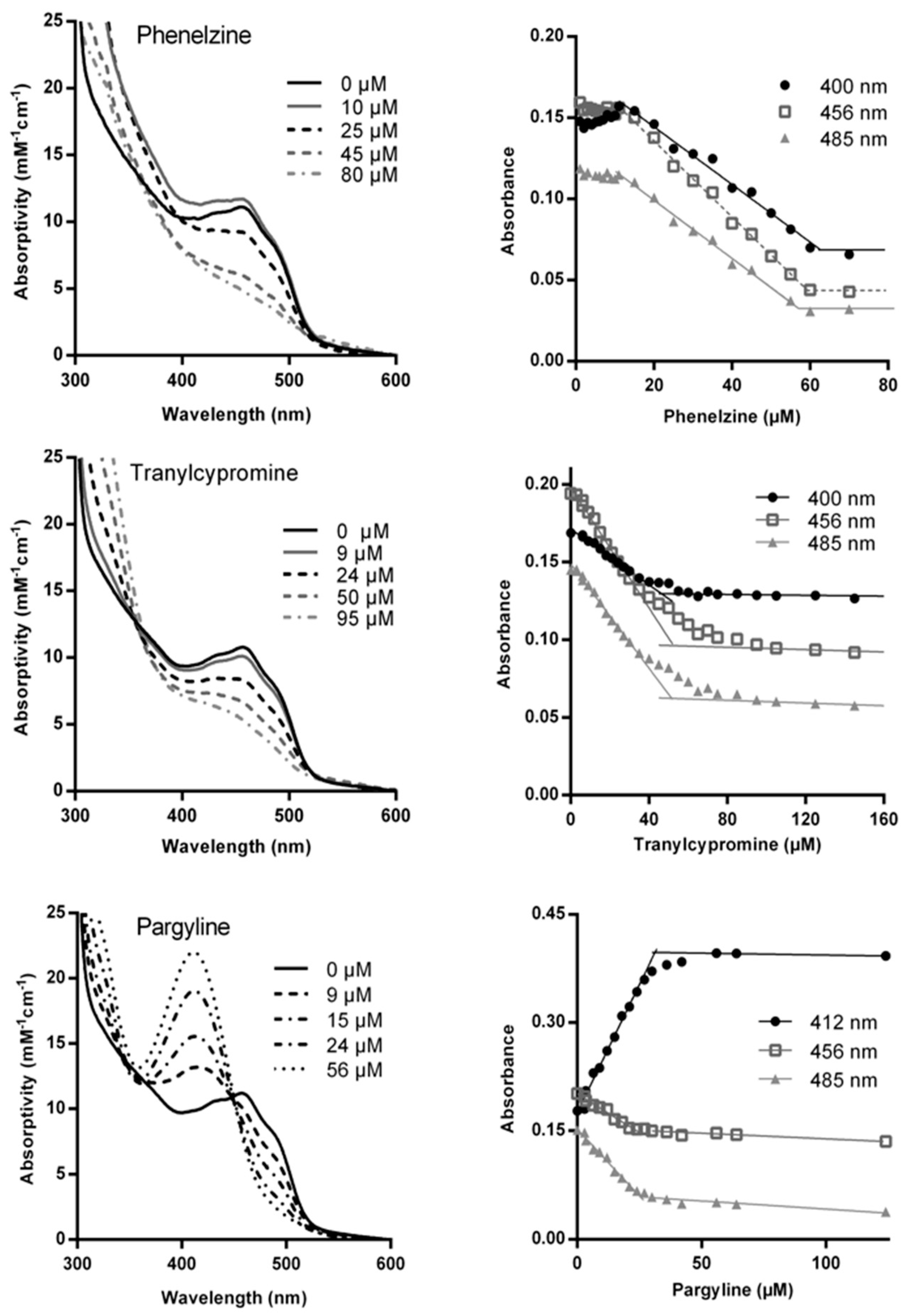
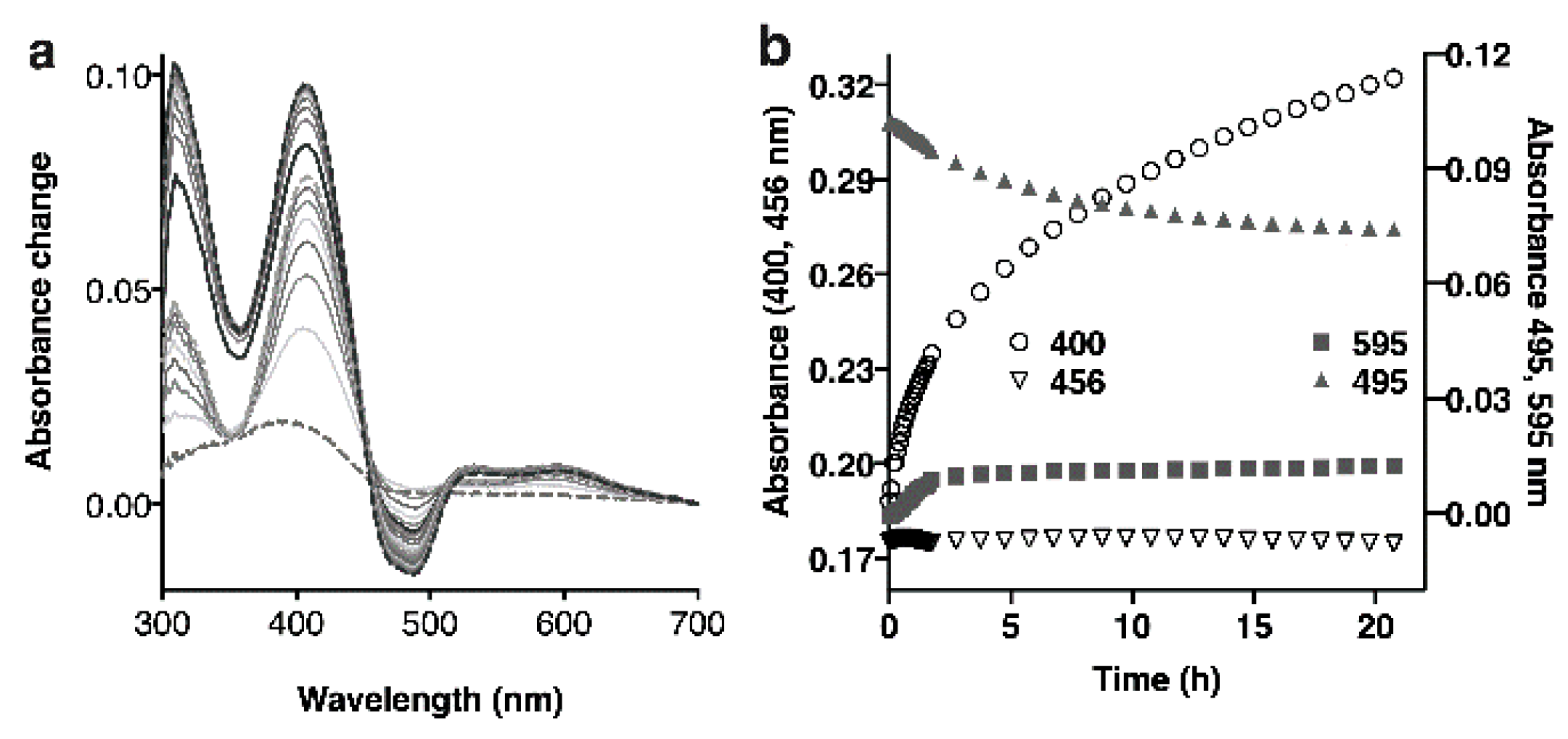
2.3. Using the Spectrum of MAO-A to Probe Inactivation
2.3.1. Titration of MAO-A with Inactivators
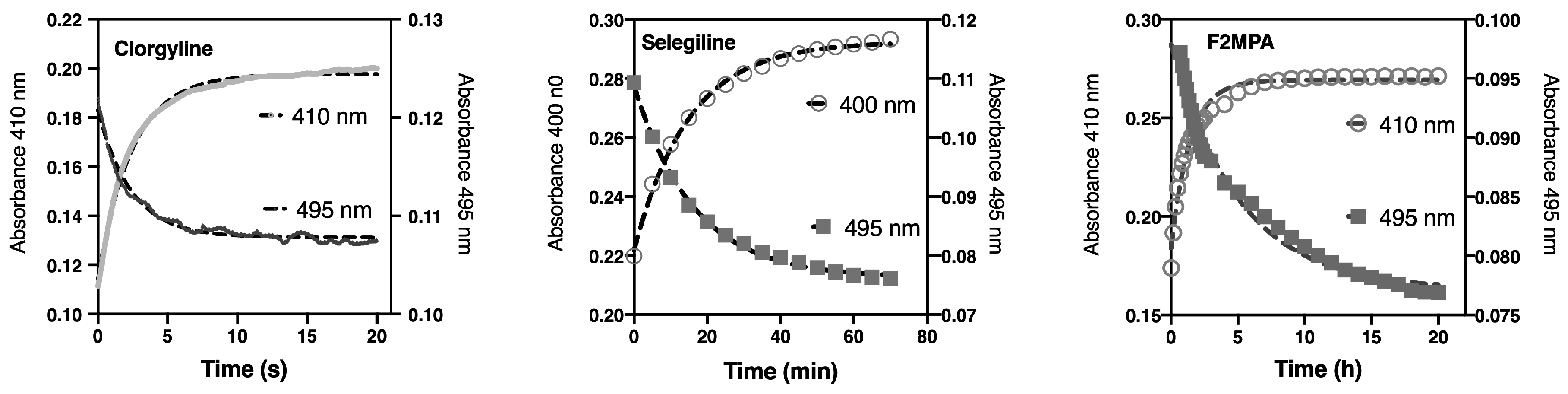
2.3.2. Spectral Changes Distinguish Flavin Reduction from Adduct Formation
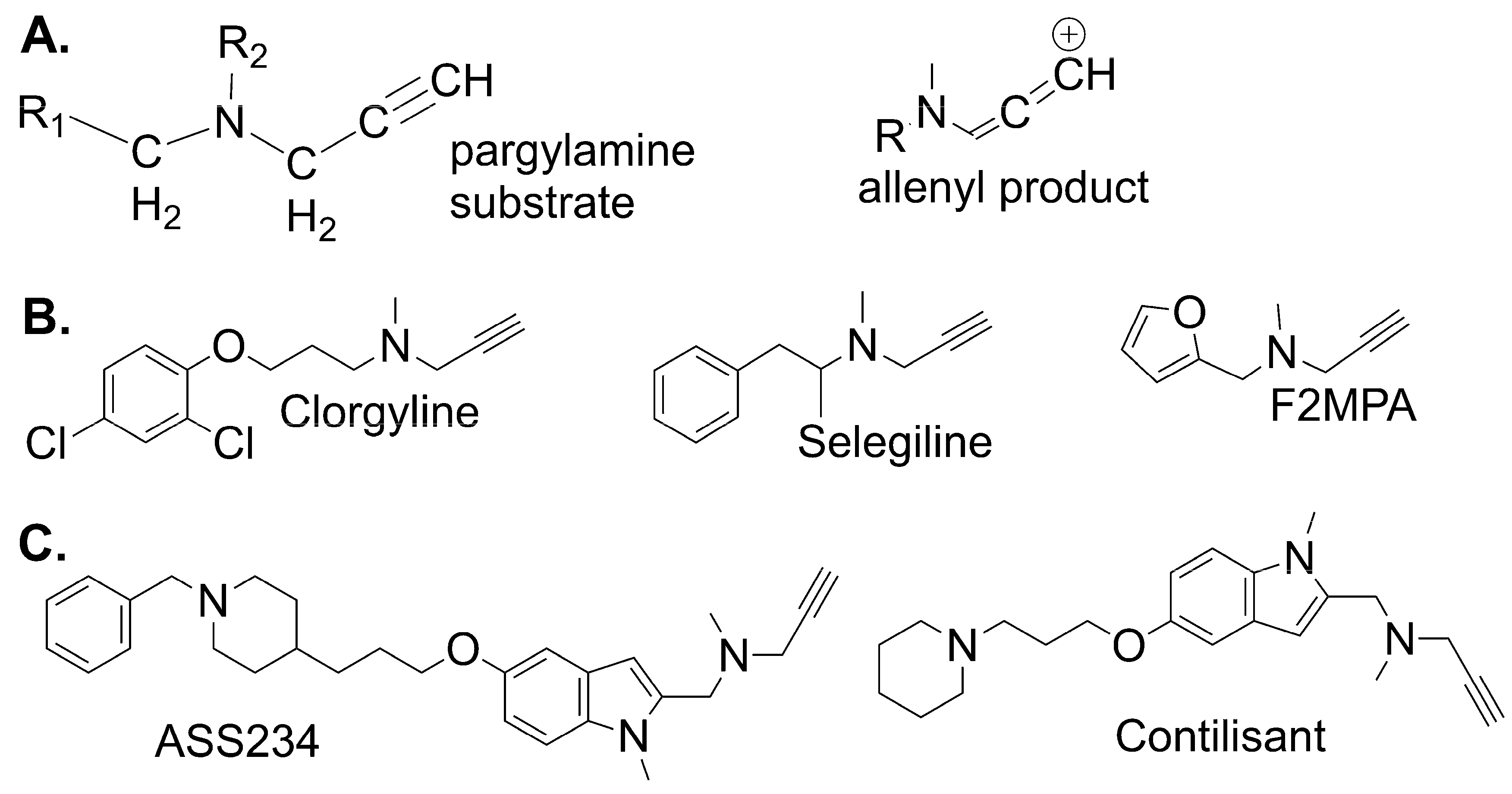
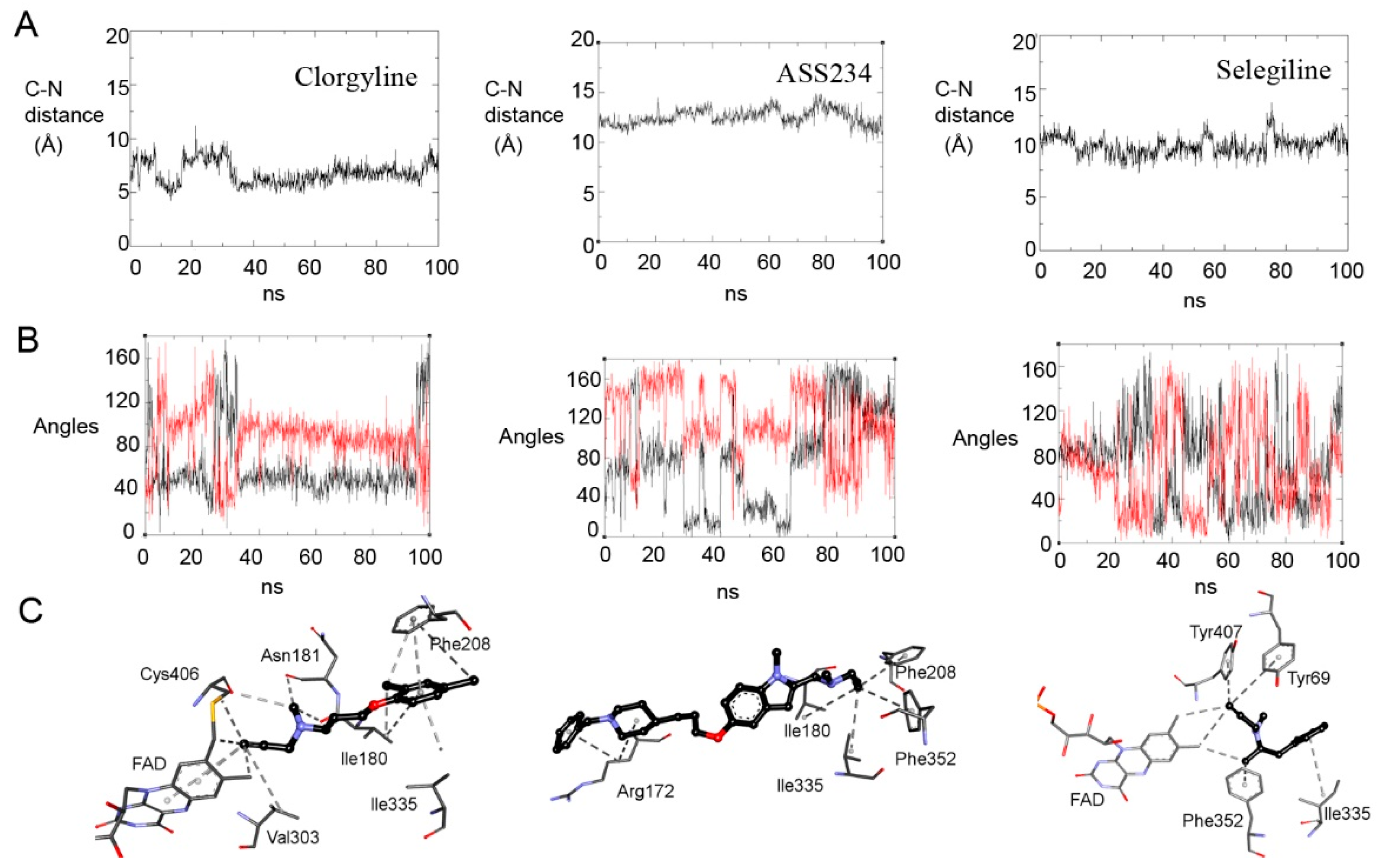
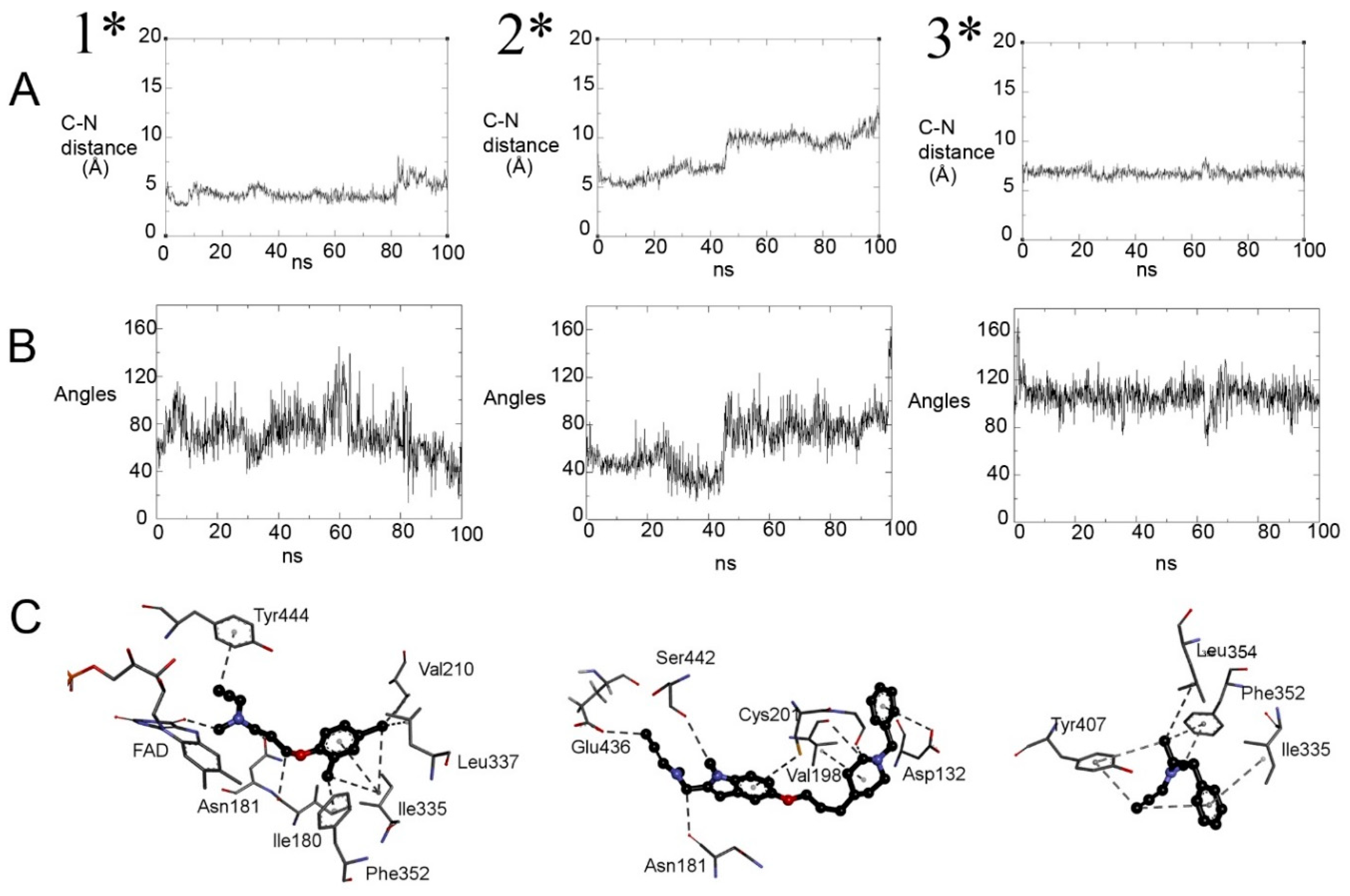
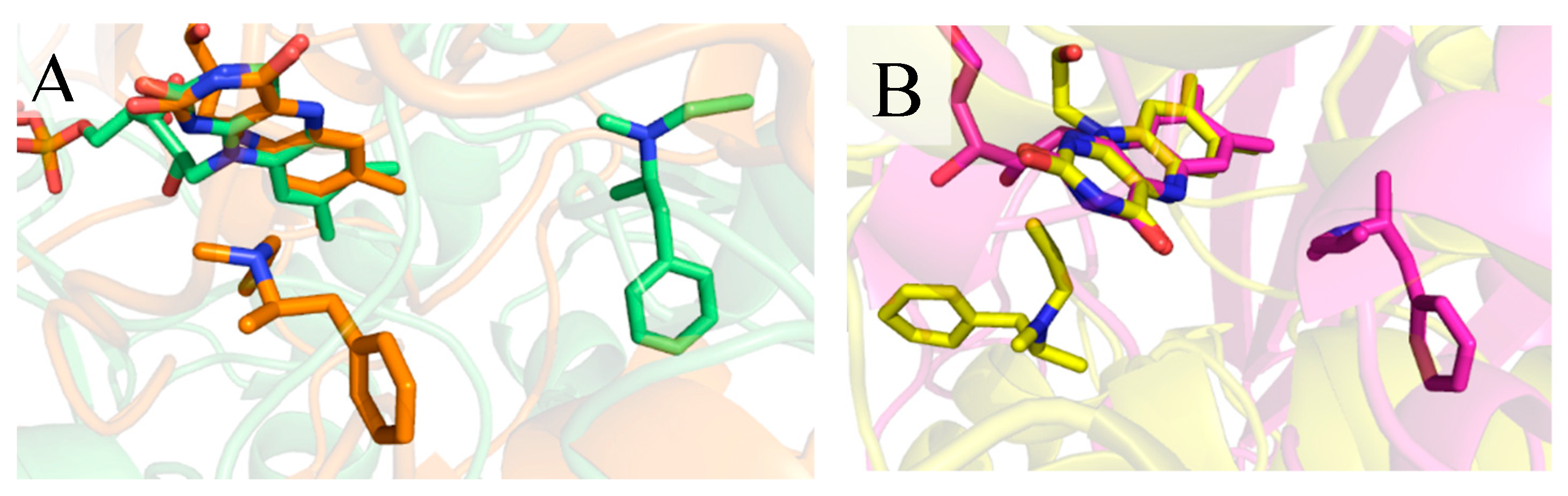
2.4. Exploring Propargylamine Selectivity and Efficacy by Molecular Dynamics
2.4.1. Initial Reversible Binding of Propargylamines to MAO-A
2.4.2. Distances from N5 to Cα and Orientation for Hydride Transfer
2.4.3. Retention of the Product Near the N5 of Reduced MAO-A (FADH−)
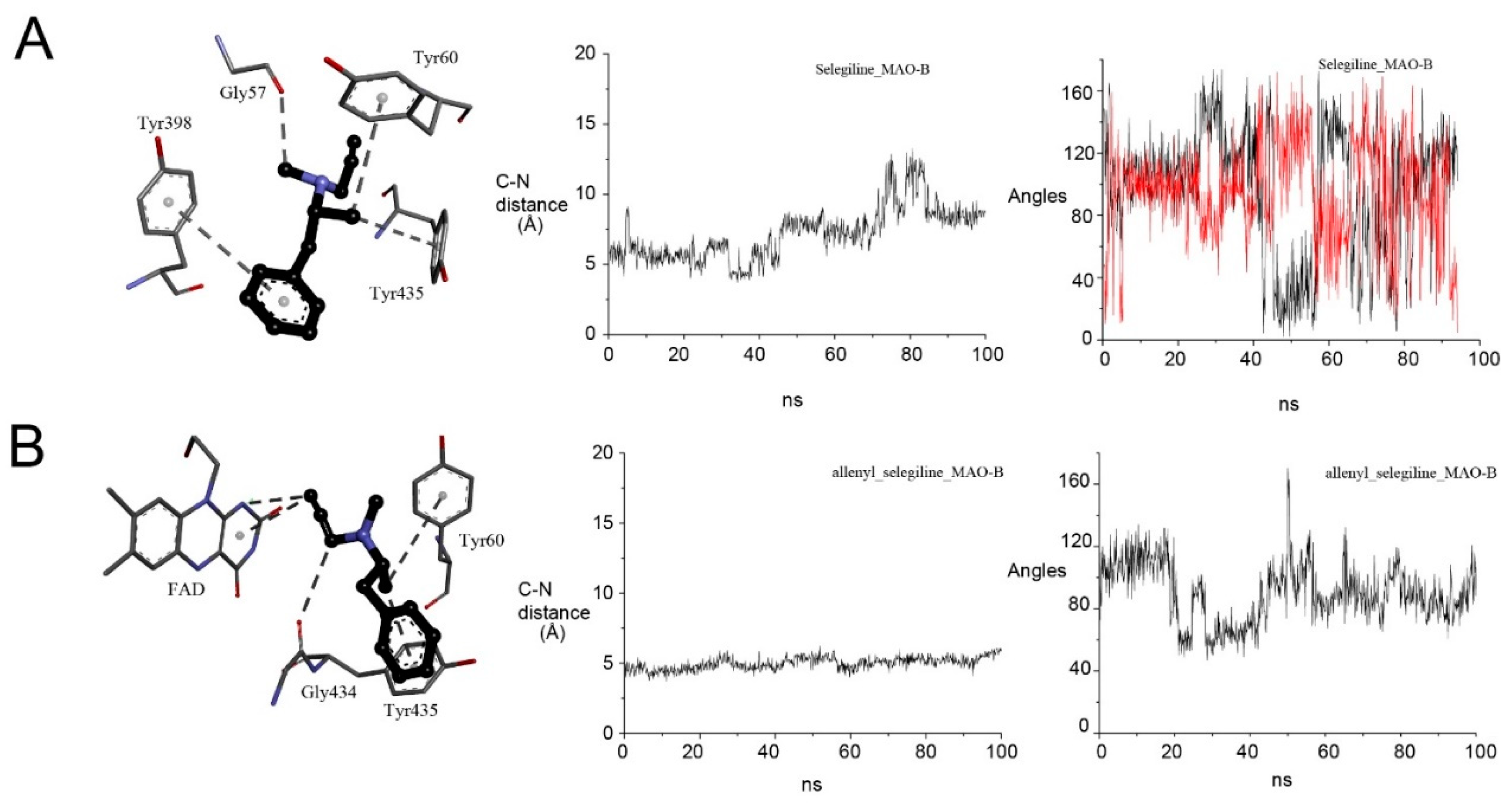
2.4.4. Selegiline with MAO-B
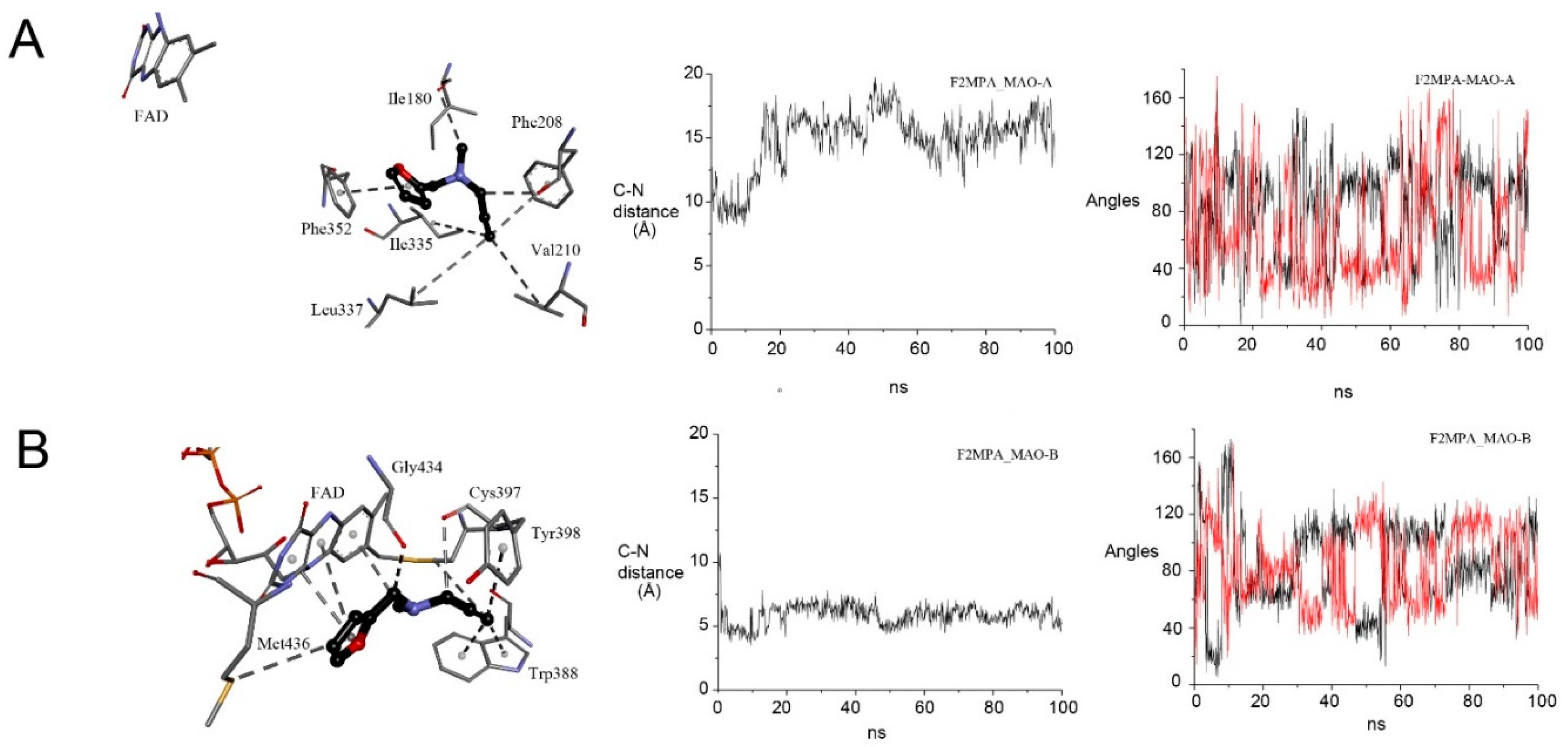
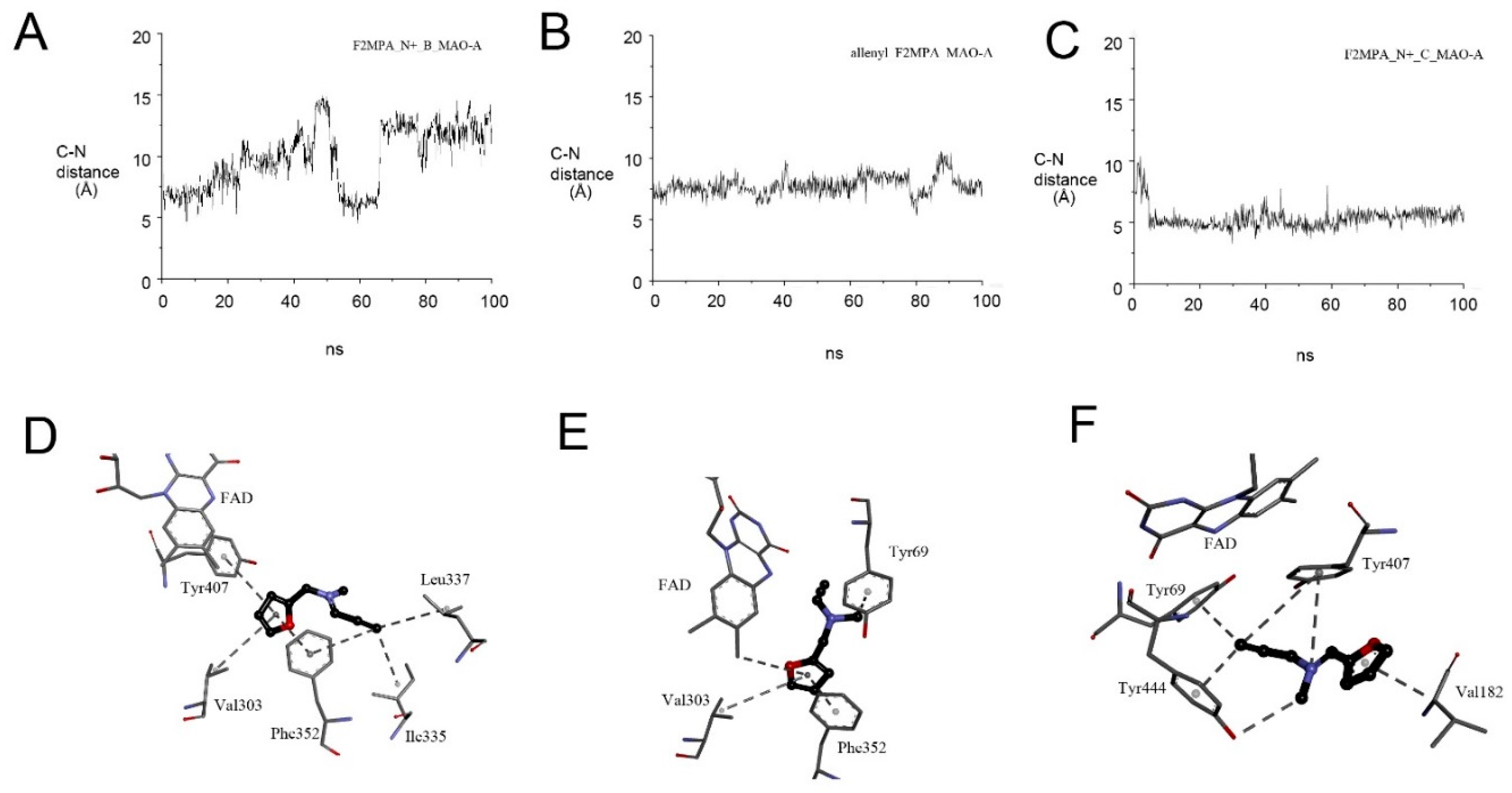
2.5. F2MPA Is Anomalous
2.5.1. F2MPA Interaction with MAO-A
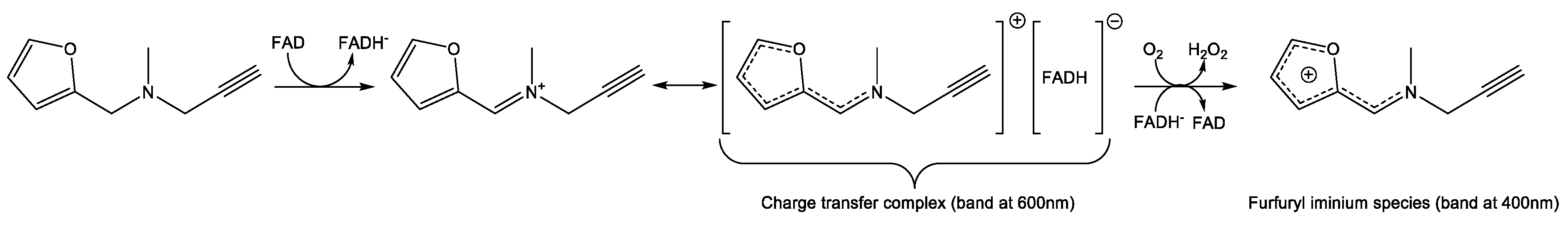
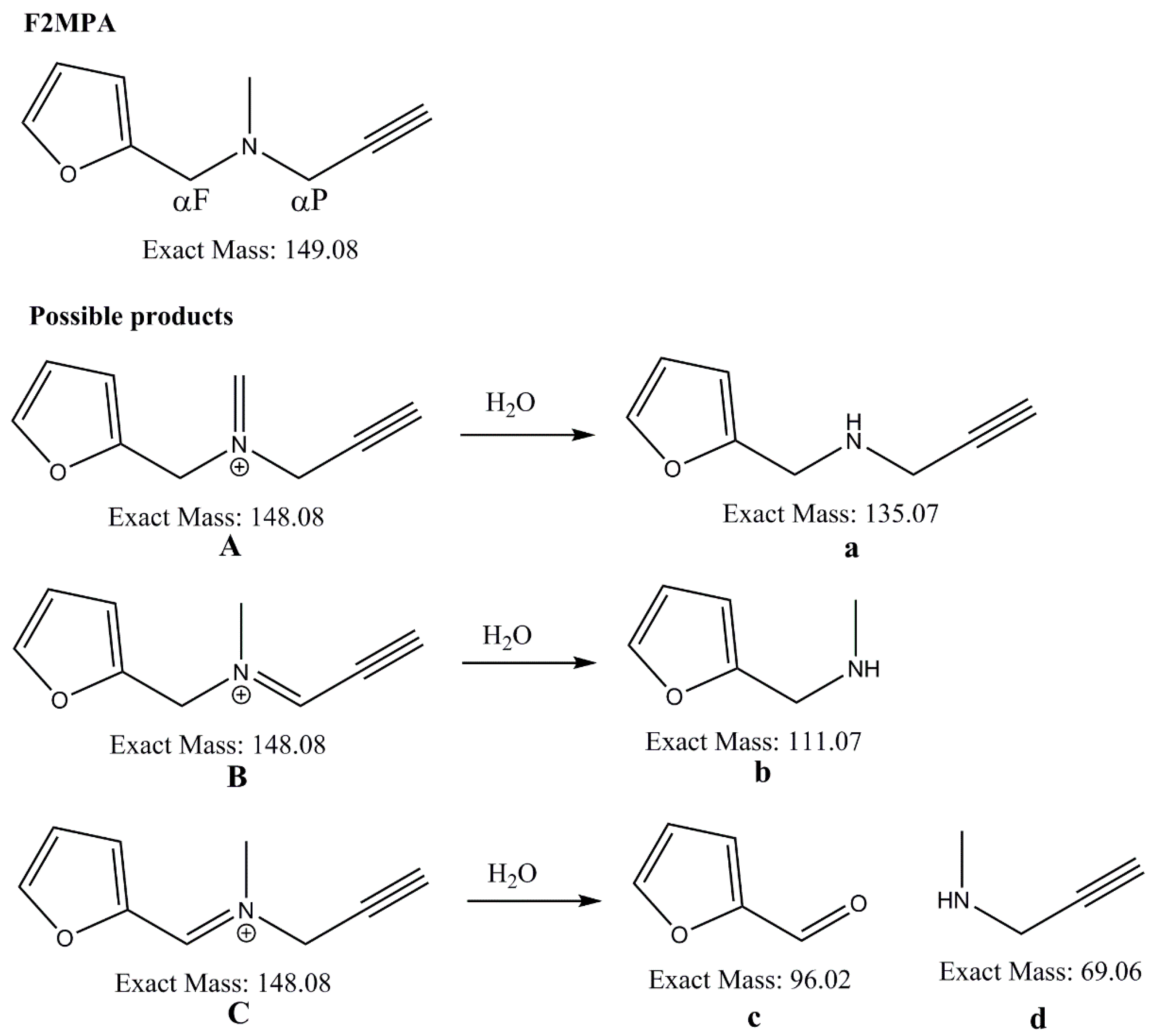
2.5.2. Detection of Alternative Products from MAO-A Oxidation of F2MPA
2.5.3. MD Provides Insight into the Differential Selectivity of F2MPA for MAO-A/B and the Spectral Anomalies with MAO-A
3. Conclusions
4. Materials and Methods
4.1. Kinetic Methods
4.1.1. Materials
4.1.2. MAO Assays
4.1.3. Reversible Inhibition
4.1.4. Parameters for Irreversible Inhibition
4.1.5. Partition Ratio
4.1.6. Visible Spectra of MAO-A with Inactivating Inhibitors
4.1.7. Stopped-Flow Kinetics
4.2. Computational Methods
4.2.1. 3D-Pharmacophores
4.2.2. Docking and Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nutt, D.J. The role of dopamine and norepinephrine in depression and antidepressant treatment. J. Clin. Psychiatry 2006, 67, 3–8. [Google Scholar] [PubMed]
- Alborghetti, M.; Nicoletti, F. Different generations of type-B monoamine oxidase inhibitors in Parkinson’s disease: From bench to bedside. Curr. Neuropharmacol. 2019, 17, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Bar-Am, O.; Weinreb, O.; Amit, T.; Youdim, M.B.H. The novel Cholinesterase-Monoamine Oxidase Inhibitor and antioxidant, Ladostigil, confers neuroprotection in neuroblastoma cells and aged rats. J. Mol. Neurosci. 2009, 37, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Inaba-Hasegawa, K.; Shamoto-Nagai, M.; Maruyama, W.; Naoi, M. Type B and A monoamine oxidase and their inhibitors regulate the gene expression of Bcl-2 and neurotrophic factors in human glioblastoma U118MG cells: Different signal pathways for neuroprotection by selegiline and rasagiline. J. Neural Transm. 2017, 124, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Magyar, K.; Palfi, M.; Jenei, V.; Szoko, E. Deprenyl: From chemical synthesis to neuroprotection. J. Neural Transm. Suppl. 2006, 71, 143–156. [Google Scholar]
- Zindo, F.T.; Joubert, J.; Malan, S.F. Propargylamine as functional moiety in the design of multifunctional drugs for neurodegenerative disorders: MAO inhibition and beyond. Future Med. Chem. 2015, 7, 609–629. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Van der Schyf, C.J. Rationally designed multi-targeted agents against neurodegenerative diseases. Curr. Med. Chem. 2013, 20, 1662–1672. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martnez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234 as a new multi-target directed propargylamine for Alzheimer’s Disease therapy. Front. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Unzeta, M.; Esteban, G.; Bolea, I.; Fogel, W.A.; Ramsay, R.R.; Youdim, M.B.H.; Tipton, K.F.; Marco-Contelles, J. Multi-target directed donepezil-like ligands for Alzheimer’s Disease. Front. Neurosci. 2016, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Bar-Am, O.; Amit, T.; Youdim, M.B.; Weinreb, O. Neuroprotective and neurorestorative potential of propargylamine derivatives in ageing: Focus on mitochondrial targets. J. Neural Transm. 2016, 123, 125–135. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Li, M.; Hubalek, F.; Mattevi, A.; Edmondson, D.E. Structural and mechanistic studies of arylalkylhydrazine inhibition of human monoamine oxidases A and B. Biochemistry 2008, 47, 5616–5625. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Bonivento, D.; Milczek, E.M.; McDonald, G.R.; Binda, C.; Holt, A.; Edmondson, D.E.; Mattevi, A. Potentiation of ligand binding through cooperative effects in monoamine oxidase B. J. Biol. Chem. 2010, 285, 36849–36856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, S.; Ricken, R.; Adli, M. Tranylcypromine in mind (Part I): Review of pharmacology. Eur. Neuropsychopharmacol. 2017, 27, 697–713. [Google Scholar] [CrossRef]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef] [Green Version]
- Binda, C.; Hubalek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D.E.; Mattevi, A. Crystal structures of monoamine oxidase B in complex with four inhibitors of the N-propargylaminoindan class. J. Med. Chem. 2004, 47, 1767–1774. [Google Scholar] [CrossRef]
- Esteban, G.; Allan, J.; Samadi, A.; Mattevi, A.; Unzeta, M.; Marco-Contelles, J.; Binda, C.; Ramsay, R.R. Kinetic and structural analysis of the irreversible inhibition of human monoamine oxidases by ASS234, a multi-target compound designed for use in Alzheimer’s disease. BBA-Proteins Proteom. 2014, 1844, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- De Deurwaerdere, P.; Binda, C.; Corne, R.; Leone, C.; Valeri, A.; Valoti, M.; Ramsay, R.R.; Fall, Y.; Marco-Contelles, J. Comparative Analysis of the Neurochemical Profile and MAO Inhibition Properties of N-(Furan-2-ylmethyl)-N-methylprop-2-yn-1-amine. ACS Chem. Neurosci. 2017, 8, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.G.; Tipton, K.F. Enzymes: Irreversible inhibition. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2012. [Google Scholar]
- Gartner, B.; Hemmerich, P. Inhibition of Monoamine-Oxidase by Propargylamine—Structure of Inhibitor Complex. Angew. Chem. Int. Ed. Engl. 1975, 14, 110–111. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Breakefield, X.O. Differences in the structures of monoamine oxidase-A and oxidase-B in rat clonal cell-lines. Biochem. Pharmacol. 1983, 32, 441–448. [Google Scholar] [CrossRef]
- Silverman, R.B. Radical ideas about monoamine-oxidase. Acc. Chem. Res. 1995, 28, 335–342. [Google Scholar] [CrossRef]
- Binda, C.; Hubalek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D.E.; Mattevi, A. Binding of rasagiline-related inhibitors to human monoamine oxidases. A kinetic and crystallographic analysis. J. Med. Chem. 2005, 48, 8148–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borstnar, R.; Repic, M.; Krzan, M.; Mavri, J.; Vianello, R. Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines rasagiline and selegiline: A computational study. Eur. J. Org. Chem. 2011, 2011, 6419–6433. [Google Scholar] [CrossRef]
- Albreht, A.; Vovk, I.; Mavri, J.; Marco-Contelles, J.; Ramsay, R.R. Evidence for a Cyanine Link Between Propargylamine Drugs and Monoamine Oxidase Clarifies the Inactivation Mechanism. Front. Chem. 2018, 6, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, C.; Milczek, E.M.; Bonivento, D.; Wang, J.; Mattevi, A.; Edmondson, D.E. Lights and shadows on Monoamine Oxidase inhibition in neuroprotective pharmacological therapies. Curr. Top. Med. Chem. 2011, 11, 2788–2796. [Google Scholar] [CrossRef]
- Carradori, S.; Silvestri, R. New frontiers in selective human MAO-B inhibitors. J. Med. Chem. 2015, 58, 6717–6732. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; Petzer, J.P. MAO inhibitors and their wider applications: A patent review. Expert Ther. Pat. 2018, 28, 211–226. [Google Scholar] [CrossRef]
- Yu, P.H.; Tipton, K.F. Deuterium-Isotope Effect of Phenelzine on the Inhibition of Rat-Liver Mitochondrial Monoamine-Oxidase Activity. Biochem. Pharmacol. 1989, 38, 4245–4251. [Google Scholar] [CrossRef]
- Walker, M.C.; Edmondson, D.E. Structure-Activity-Relationships in the Oxidation of Benzylamine Analogs by Bovine Liver Mitochondrial Monoamine- Oxidase-B. Biochemistry 1994, 33, 7088–7098. [Google Scholar] [CrossRef]
- Miller, J.R.; Edmondson, D.E. Structure-activity relationships in the oxidation of para- substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 1999, 38, 13670–13683. [Google Scholar] [CrossRef] [PubMed]
- Dunn, R.V.; Marshall, K.R.; Munro, A.W.; Scrutton, N.S. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS J. 2008, 275, 3850–3858. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J.; Mantle, T.J.; Tipton, K.F. The nature of the inhibition of rat-liver Monoamine-Oxidase type-A and Type-B by the acetylenic inhibitors Clorgyline, L-Deprenyl and Pargyline. Biochem. Pharmacol. 1982, 31, 3555–3561. [Google Scholar] [CrossRef]
- Vintem, A.; Price, N.; Silverman, R.; Ramsay, R. Mutation of surface cysteine 374 to alanine in monoamine oxidase A alters substrate turnover and inactivation by cyclopropylamines. Bioorg. Med. Chem. 2005, 13, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, O.M.; Hagenow, S.; Palomino-Antolin, A.; Farré-Alins, V.; Ismaili, L.; Joffrin, P.-L.; Jimeno, M.L.; .Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-directed ligands combining cholinesterase and monoamine oxidase Inhibition with histamine H3R antagonism for neurodegenerative diseases. Angew. Chem. Int. Ed. 2017, 56, 12765–12769. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; Volume 46. [Google Scholar]
- Ramsay, R.R.; Tipton, K.F. Assessment of Enzyme Inhibition: A review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules 2017, 22, 1192. [Google Scholar] [CrossRef] [Green Version]
- Binda, C.; Valente, S.; Romanenghi, M.; Pilotto, S.; Cirilli, R.; Karytinos, A.; Ciossani, G.; Botrugno, O.A.; Forneris, F.; Tardugno, M.; et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. [Google Scholar] [CrossRef]
- Malcomson, T.; Yelekci, K.; Borrello, M.T.; Ganesan, A.; Semina, E.; De Kimpe, N.; Mangelinckx, S.; Ramsay, R.R. cis-Cyclopropylamines as mechanism-based inhibitors of monoamine oxidases. FEBS J. 2015, 282, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Kitz, R.; Wilson, I.B. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [Google Scholar]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Davis, M.R.; Dougherty, D.A. Cation-π interactions: Computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation. Phys. Chem. Chem. Phys. PCCP 2015, 17, 29262–29270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Binda, C.; Mattevi, A.; Edmondson, D.E. Functional role of the “aromatic cage” in human monoamine oxidase B: Structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 2006, 45, 4775–4784. [Google Scholar] [CrossRef] [PubMed]
- .Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E. High-level expression of human liver monoamine oxidase B in Pichia pastoris. Protein Expr. Purif. 2000, 20, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hubalek, F.; Newton-Vinson, P.; Edmondson, D.E. High-level expression of human liver monoamine oxidase A in Pichia pastoris: Comparison with the enzyme expressed in Saccharomyces cerevisiae. Protein Expr. Purif. 2002, 24, 152–162. [Google Scholar] [CrossRef]
- Tandaric, T.; Vianello, R. Computational insight into the mechanism of the irreversible inhibition of monoamine oxidase enzymes by the antiparkinsonian propargylamine inhibitors rasagiline and selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Prah, A.; Franciskovic, E.; Mavri, J.; Stare, J. Electrostatics as the driving force behind the catalytic function of the monoamine oxidase a enzyme confirmed by quantum computations. ACS Catal. 2019, 9, 1231–1240. [Google Scholar] [CrossRef]
- Carreiras, M.D.; Ismaili, L.; Marco-Contelles, J. Propargylamine-derived multi-target directed ligands for Alzheimer’s disease therapy. Bioorg. Med. Chem. Lett. 2020, 30, 126880. [Google Scholar] [CrossRef]
- Bolea, I.; Juarez-Jimenez, J.; de los Rios, C.; Chioua, M.; Pouplana, R.; Javier Luque, F.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s Disease. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar]
- Hynson, R.M.G.; Wouters, J.; Ramsay, R.R. Monoamine oxidase A inhibitory potency and flavin perturbation are influenced by different aspects of pirlindole inhibitor structure. Biochem. Pharmacol. 2003, 65, 1867–1874. [Google Scholar] [CrossRef]
- Weyler, W.; Salach, J.I. Purification and properties of mitochondrial Monoamine-Oxidase type-A from human-placenta. J. Biol. Chem. 1985, 260, 3199–3207. [Google Scholar]
- Zhou, M.; Panchuk-Voloshina, N. A one-step fluorometric method for the continuous measurement of monoamine oxidase activity. Anal. Biochem. 1997, 253, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Jamei, M.; Yeo, K.R.; Tucker, G.T.; Rostami-Hodjegan, A. Kinetic values for mechanism-based enzyme inhibition: Assessing the bias introduced by the conventional experimental protocol. Eur. J. Pharm. Sci. 2005, 26, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-d pharmacophores derived from protein-bound Ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.M.; Meng, E.; Ferrin, T. UCSF chimera visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Da Sousa Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Armijo, L. Minimization of functions having lipschitz continuous first partial derivatives. Pac. J. Math. 1966, 16, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Hestenes, M.R.; Stiefel, E. Methods of conjugate gradients for solving linear systems. J. Res. Natl. Bur. Stand. 1952, 49, 409–436. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints—Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | MAO-A | MAO-B | ||
|---|---|---|---|---|
| IC50 (μM) AR Assay | Ki (μM) Direct | IC50 (μM) AR Assay | Ki (μM) Direct | |
| Phenelzine | 112 ± 28 a 52.7 ± 5.1 | 47 ± 2 a | (substrate) | 15 ± 2 a (Km = 215 ± 48) |
| Tranylcypromine | 23.6 b 54.9 ± 6.2 | 19 c | 4.02 b | 16 c |
| Pargyline | 118 ± 21 | 15 d (rat) | 2.25 ± 0.13 | 1.8 d (rat) |
| Clorgyline | 0.042 ± 0.003 | 0.014 ± 0.001 e 0.054 d (rat) | 3.65 ± 0.39 | 58 d (rat) |
| Selegiline | 225 ± 31 | 75 ± 11 e 38 d (rat) | 0.283 ± 0.016 | 0.97 d (rat) |
| ASS234 | 0.033 ± 0.003 | 0.053 ± 0.014 e | 3.20 ± 0.41 | n.d. |
| Contilisant | 1.85 ± 0.21 f | 3.3 2.37 ± 0.41 | 1.94 ± 0.15 f | 0.406 ± 0.045 |
| Inhibitor | MAO-A | MAO-B | ||
|---|---|---|---|---|
| IC50 (μM) | Enhancement | IC50 (μM) | Enhancement | |
| Phenelzine | 4.50 ± 0.72 | 24.9 | Substrate | n/a |
| Tranylcypromine | 0.237 ± 0.061 a | 100 a | 0.0735 ± 0.0049 a | 54.7 a |
| Pargyline | 2.47 ± 0.28 | 48 | 0.0077 + 0.0007 | 292 |
| Clorgyline | 0.00042 ± 0.00008 | 100 | 3.57 ± 0.36 | 1.0 |
| Selegiline | 0.630 ± 0.06 b | 357 | 0.0040 ± 0.0009 | 13.3 |
| ASS234 | 0.00027 ± 0.00003 b | 122 | 0.12 ± 0.02 | 26.7 |
| Contilisant | 0.145 ± 0.010 b | 12.8 | 0.078 ± 0.006 b | 24.9 |
| Compound | MAO-A | MAO-B | ||||
|---|---|---|---|---|---|---|
| kinact (min−1) | KI (μM) | kinact/KI (min−1μM−1) | kinact (min−1) | KI (μM) | kinact/KI (min−1μM−1) | |
| Phenelzine | 0.12 | 3.13 | 0.0383 | 0.9 b | 50 b | 0.018 |
| Tranylcypromine | 0.776 | 7.7 | 0.1008 | 0.161 | 0.677 | 0.238 |
| Pargyline | 0.653 | 22 | 0.0297 | 0.35 | 0.40 | 0.875 |
| Clorgyline | 1.8 | 0.033 | 55 | 0.02 c | 1.8 c | 0.011 |
| Selegiline | 0.252 | 193 | 0.0013 | 0.53 | 0.103 | 5.145 |
| ASS234 | 0.133 | 0.045 | 2.956 | 0.52 | 1.74 | 0.299 |
| F2MPA c | 0.087 | 187 | 0.0005 | 0.61 | 135 | 0.0045 |
| Contilisant | 0.39 | 3 | 0.1300 | 0.19 | 4.6 | 0.041 |
| MBA364 | 0.5 | 4.6 | 0.1087 | 0.25 | 0.055 | 4.545 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsay, R.R.; Basile, L.; Maniquet, A.; Hagenow, S.; Pappalardo, M.; Saija, M.C.; Bryant, S.D.; Albreht, A.; Guccione, S. Parameters for Irreversible Inactivation of Monoamine Oxidase. Molecules 2020, 25, 5908. https://doi.org/10.3390/molecules25245908
Ramsay RR, Basile L, Maniquet A, Hagenow S, Pappalardo M, Saija MC, Bryant SD, Albreht A, Guccione S. Parameters for Irreversible Inactivation of Monoamine Oxidase. Molecules. 2020; 25(24):5908. https://doi.org/10.3390/molecules25245908
Chicago/Turabian StyleRamsay, Rona R., Livia Basile, Antonin Maniquet, Stefanie Hagenow, Matteo Pappalardo, Maria Chiara Saija, Sharon D. Bryant, Alen Albreht, and Salvatore Guccione. 2020. "Parameters for Irreversible Inactivation of Monoamine Oxidase" Molecules 25, no. 24: 5908. https://doi.org/10.3390/molecules25245908