
Cytotoxic Activities and Molecular Mechanisms of the Beauvericin and Beauvericin G1 Microbial Products against Melanoma Cells

 , and
, and 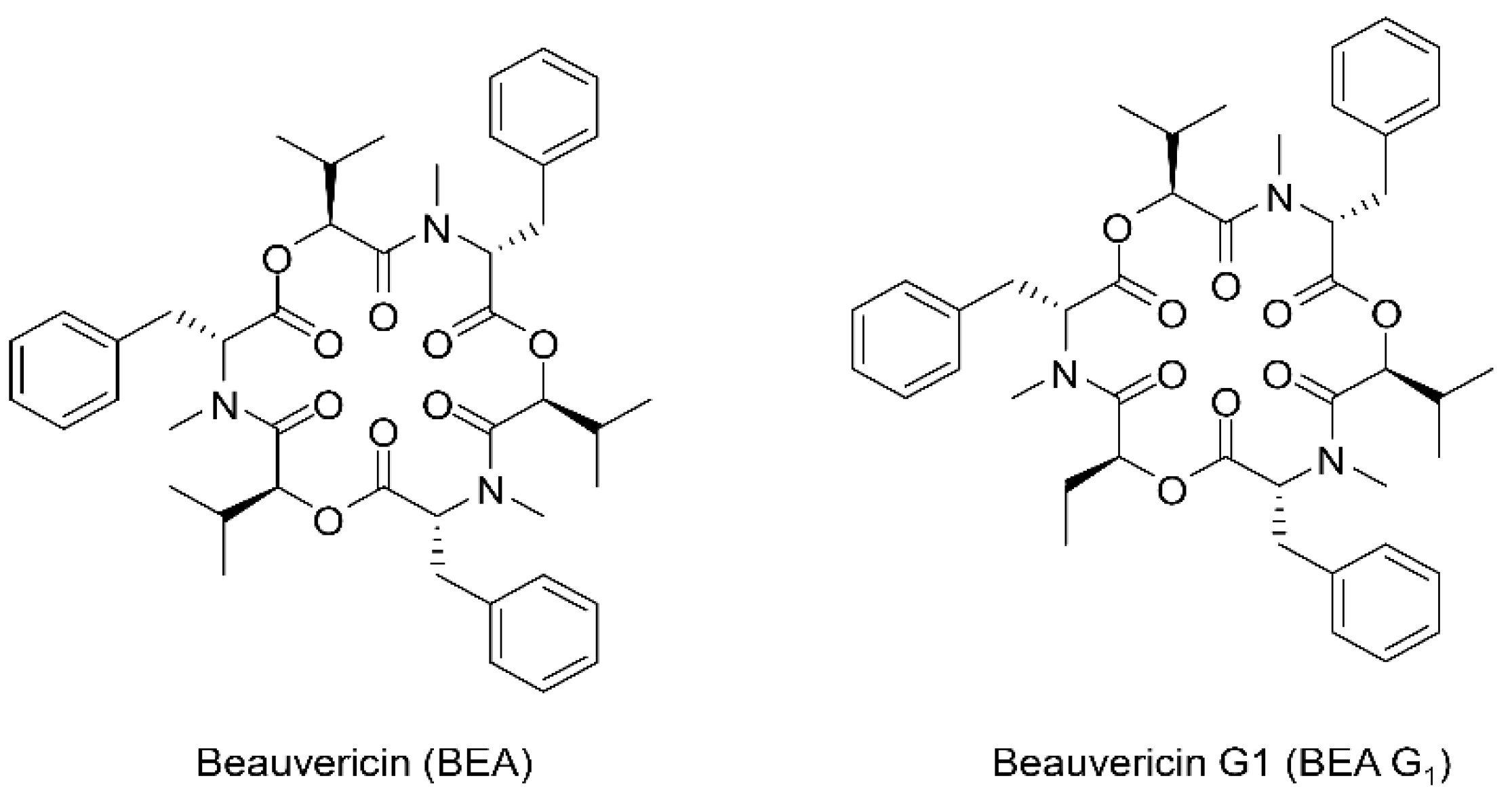{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
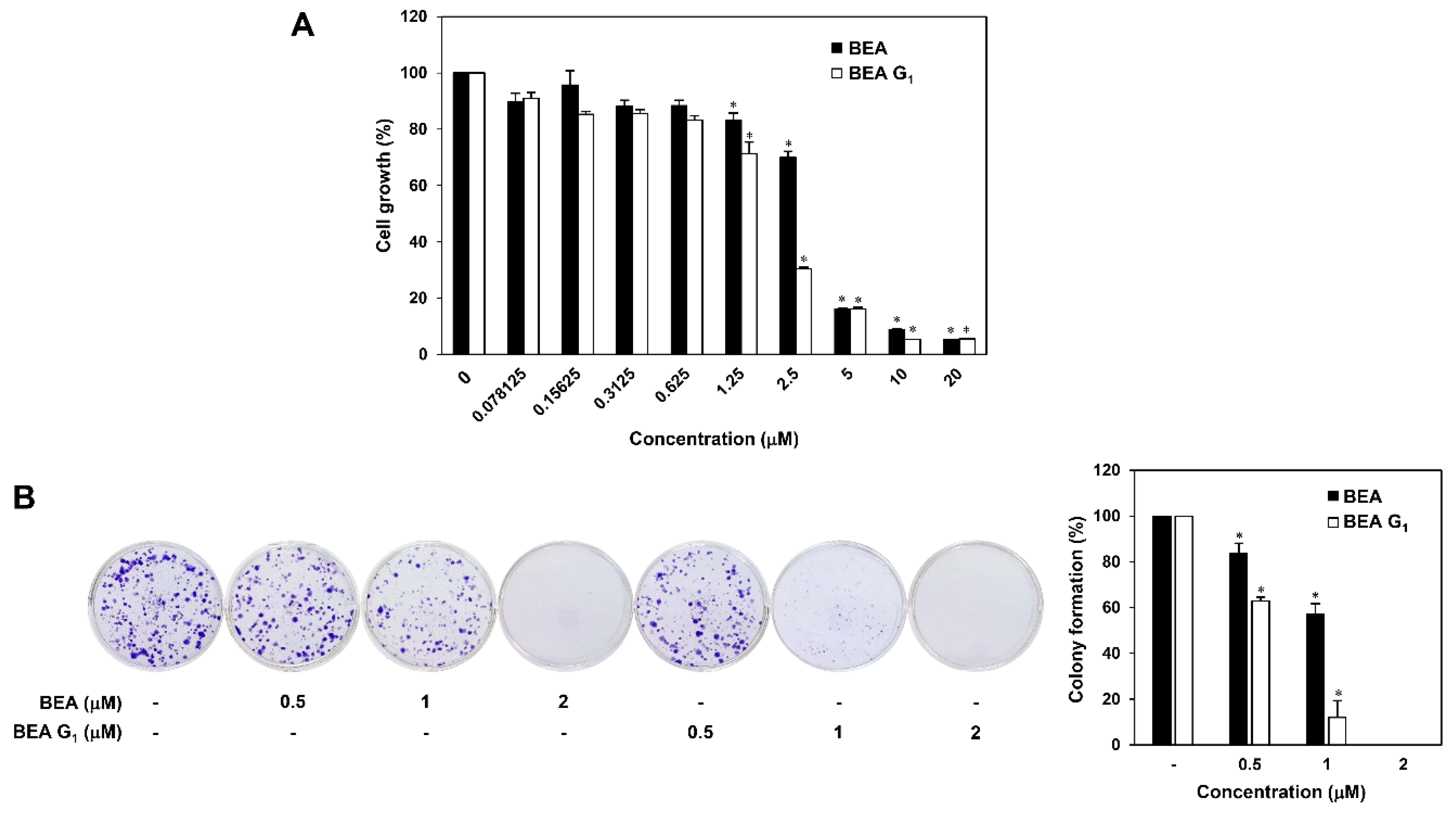
2.1. BEA and BEA G1 Inhibit the Growth of A375SM Melanoma Cells
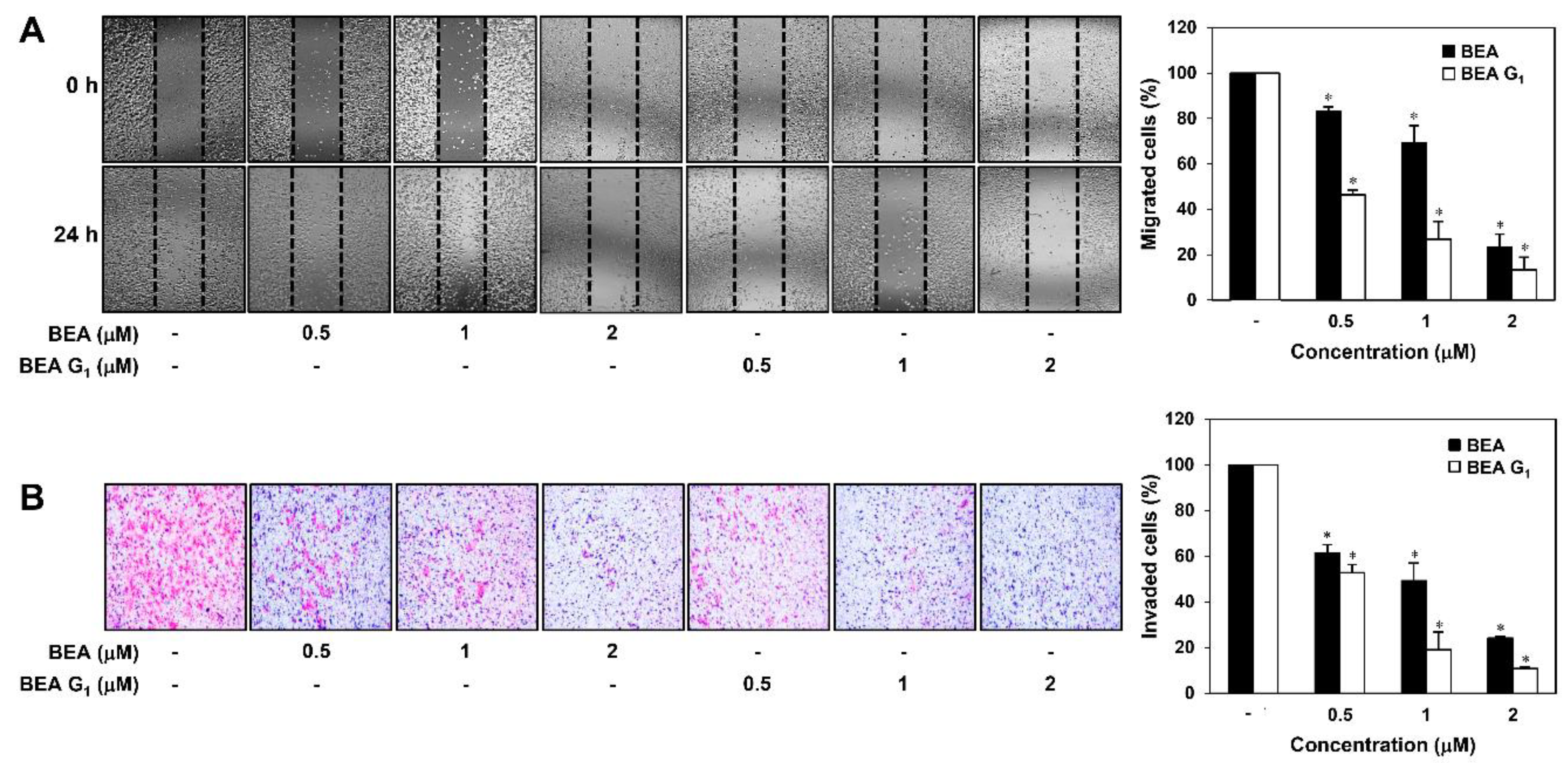
2.2. BEA and BEA G1 Inhibit the Migration of A375SM Melanoma Cells
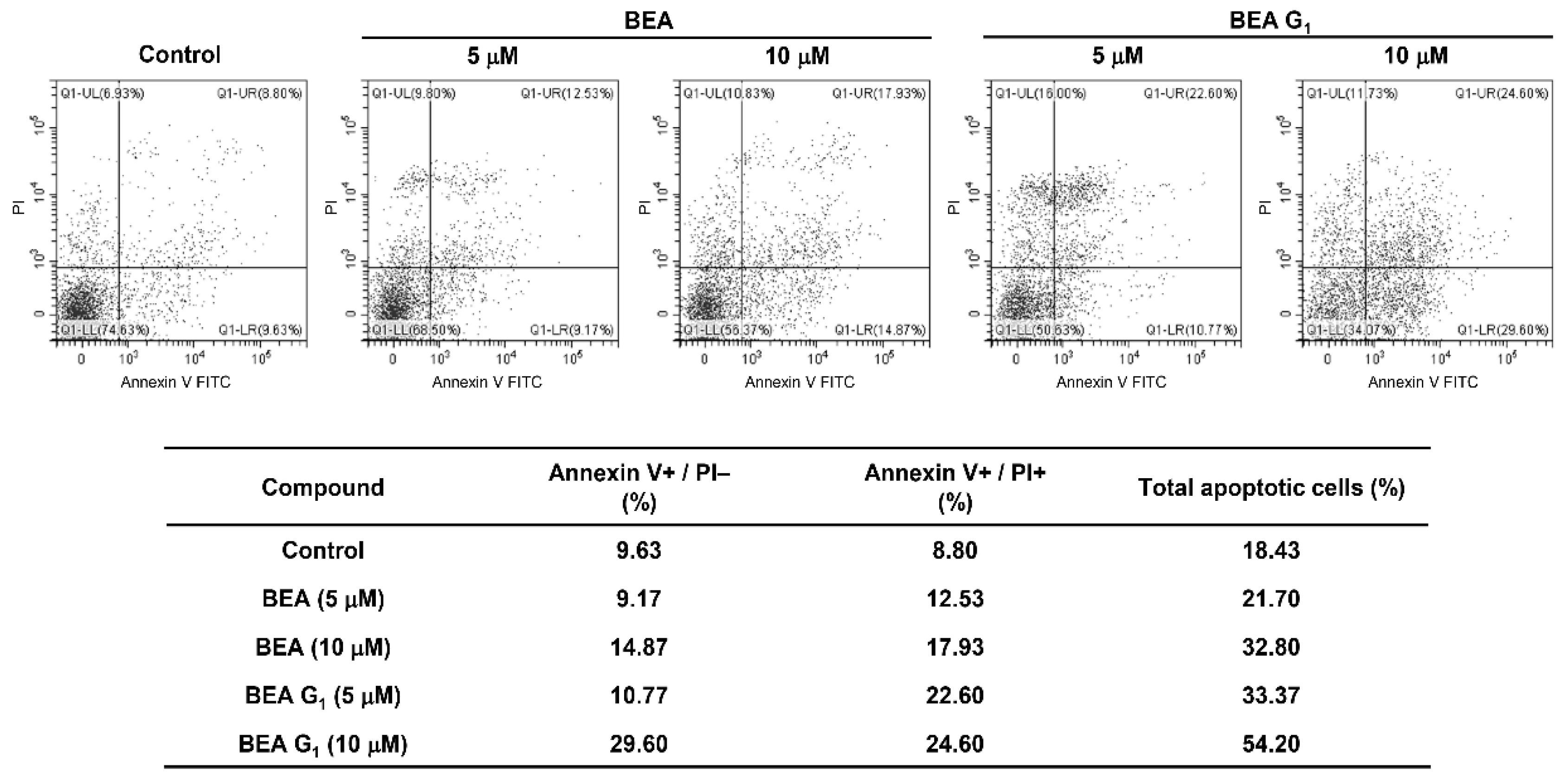
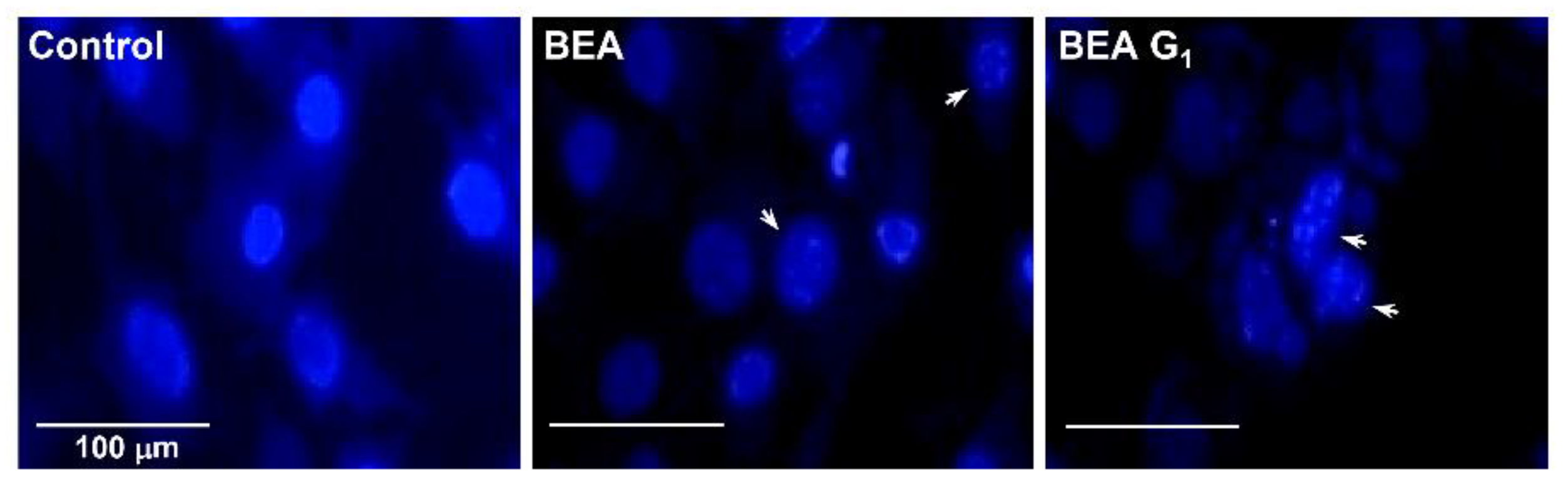
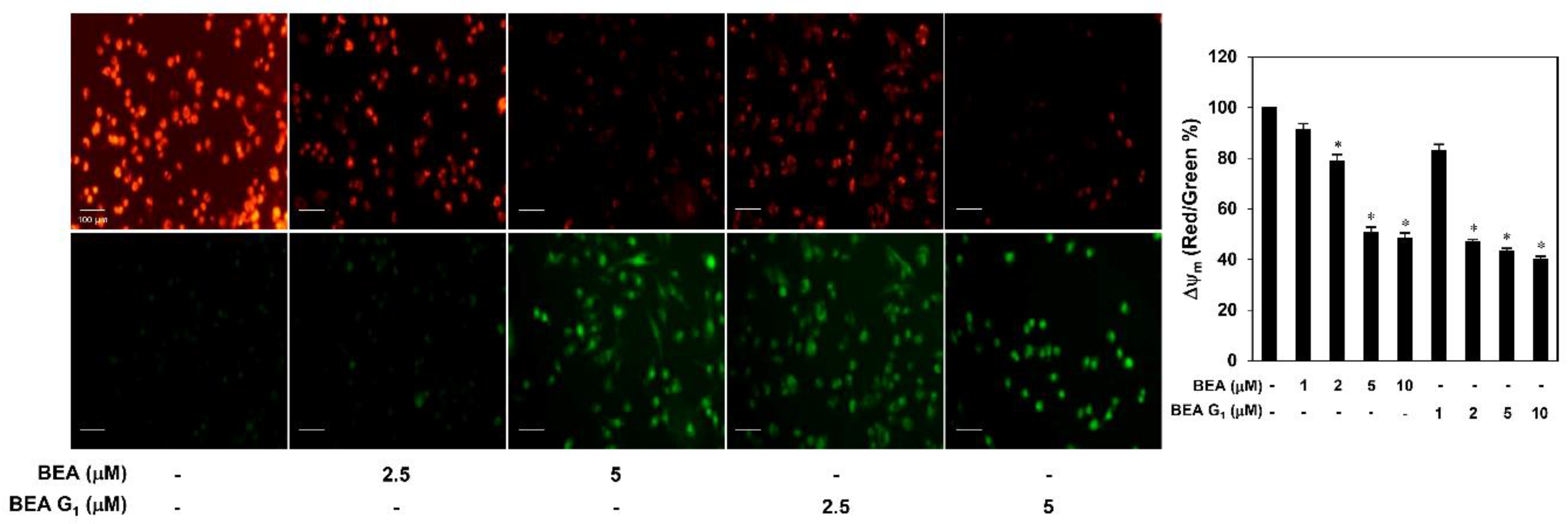
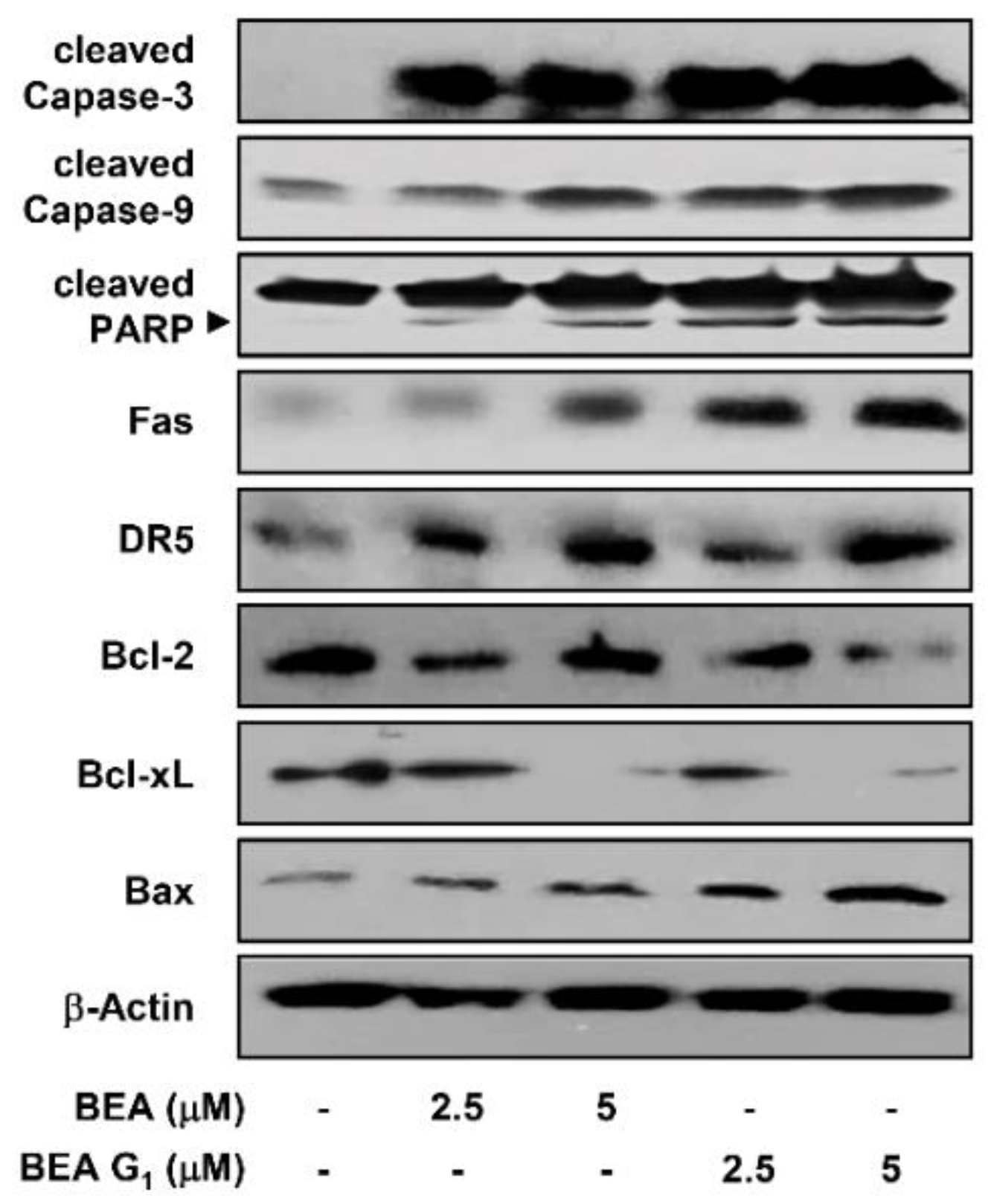
2.3. BEA and BEA G1 Induce Apoptosis in A375SM Melanoma Cells
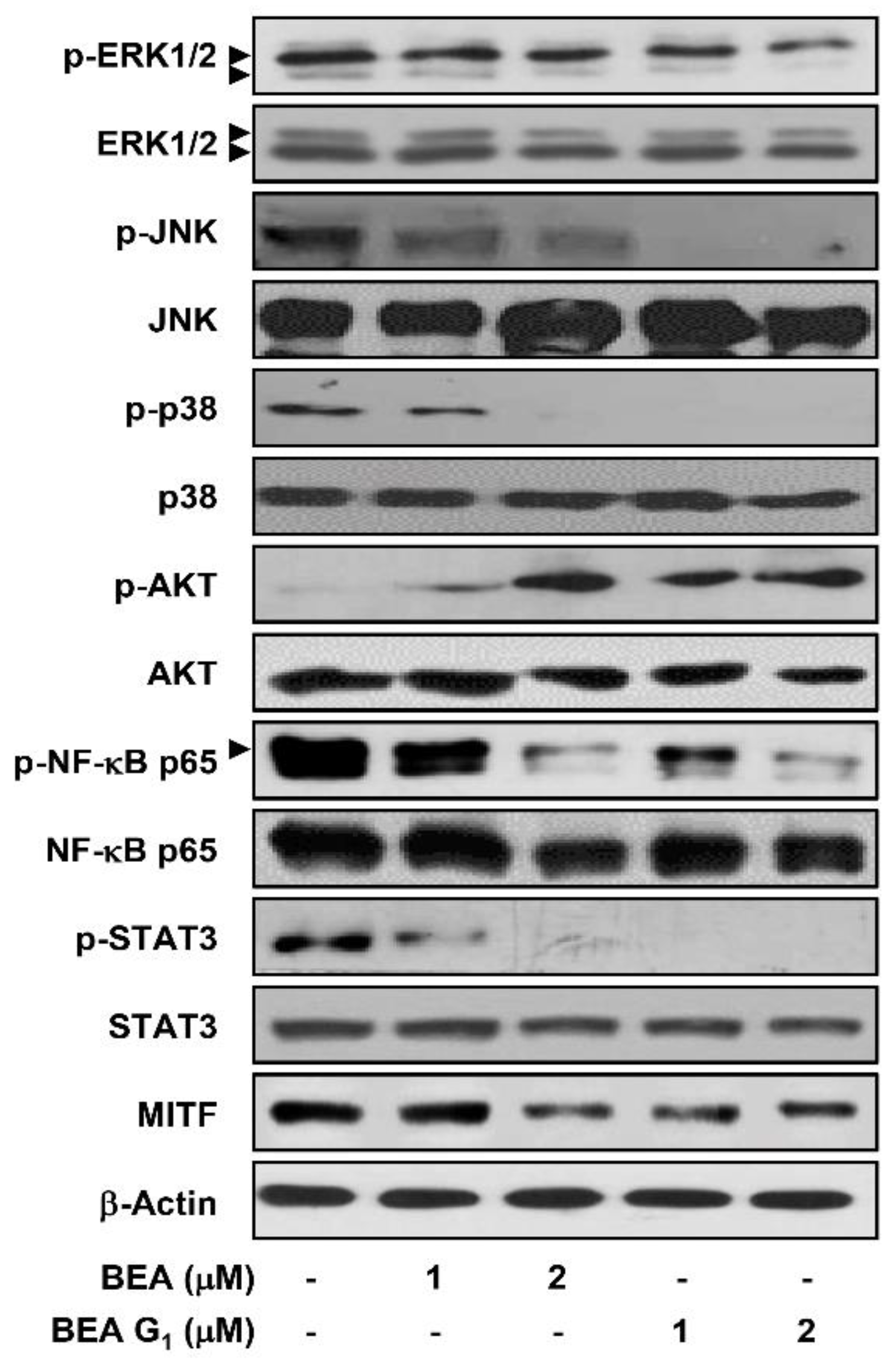
2.4. BEA and BEA G1 Downregulate Major Molecular Pathways Involved in Melanoma
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Culture Conditions for Fungus Strain
4.3. Isolation of BEA and BEA G1
4.4. Human Cell Culture
4.5. Cell Growth Assay
4.6. Colony Formation Assay
4.7. Chemoinvasion Assay
4.8. Cell Migration Assay
4.9. Apoptosis Analysis
4.10. DAPI Fluorescent Staining
4.11. MMP Determination
4.12. Western Blot Analysis
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA A Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marghoob, A.A.; Kopf, A.W.; Rigel, D.S.; Bart, R.S.; Friedman, R.J.; Yadav, S.; Abadir, M.; Sanfilippo, L.; Silverman, M.K.; Vossaert, K.A. Risk of Cutaneous Malignant Melanoma in Patients with ’Classic’ Atypical-Mole Syndrome. Arch. Dermatol. 1994, 130, 993. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: from mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niezgoda, A.; Niezgoda, P.; Czajkowski, R. Novel Approaches to Treatment of Advanced Melanoma: A Review on Targeted Therapy and Immunotherapy. BioMed Res. Int. 2015, 2015, 851387. [Google Scholar] [CrossRef] [Green Version]
- Shtivelman, E.; Davies, M.A.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef] [Green Version]
- Chinembiri, T.N.; Du Plessis, L.; Gerber, M.; Hamman, J.H.; Du Plessis, J. Review of Natural Compounds for Potential Skin Cancer Treatment. Molecules 2014, 19, 11679–11721. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gong, X.; Li, P.; Lai, D.; Zhou, L. Structural Diversity and Biological Activities of Cyclic Depsipeptides from Fungi. Molecules 2018, 23, 169. [Google Scholar] [CrossRef] [Green Version]
- Sivanathan, S.; Scherkenbeck, J. Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research. Molecules 2014, 19, 12368–12420. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, L. Beauvericin, a Bioactive Compound Produced by Fungi: A Short Review. Molecules 2012, 17, 2367–2377. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Nepovimova, E.; Ramalho, T.C. A Review on the Synthesis and Bioactivity Aspects of Beauvericin, a Fusarium Mycotoxin. Front. Pharmacol. 2018, 9, 1338. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Park, S.-H.; Oh, S.W.; Yoo, J.A.; Kwon, K.; Park, S.J.; Kim, J.; Lee, H.S.; Cho, J.; Lee, J. Beauvericin inhibits melanogenesis by regulating cAMP/PKA/CREB and LXR-α/p38 MAPK–mediated pathways. Sci. Rep. 2018, 8, 14958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jow, G.-M.; Chou, C.-J.; Chen, B.-F.; Tsai, J.-H. Beauvericin induces cytotoxic effects in human acute lymphoblastic leukemia cells through cytochrome c release, caspase 3 activation: the causative role of calcium. Cancer Lett. 2004, 216, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-I.; Lee, Y.-J.; Chen, B.-F.; Tsai, M.-C.; Lu, J.-L.; Chou, C.-J.; Jow, G.-M. Involvement of Bcl-2 family, cytochrome and caspase 3 in induction of apoptosis by beauvericin in human non-small cell lung cancer cells. Cancer Lett. 2005, 230, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Mallebrera, B.; Prosperini, A.; Font, G.; Ruiz, M.-J. In vitro mechanisms of Beauvericin toxicity: A review. Food Chem. Toxicol. 2017, 111, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Martinou, J.-C.; Youle, R.J. Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Ombra, M.; Colombino, M.; Casula, M.; Sini, M.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A.G.M. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef]
- Smalley, K.S.; Haass, N.K.; Brafford, P.; Lioni, M.; Flaherty, K.T.; Herlyn, M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer Ther. 2006, 5, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Kalal, B.; Upadhya, D.; Pai, V.R. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol. Rev. 2017, 11, 326. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta (BBA) Bioenerg. 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.A.; McGaw, L.J. Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef] [Green Version]
- Jan, R.; Chaudhry, G.-E.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, S.; Seevasant, I.; Mohamad, J.; Mukheem, A.; Huri, H.Z.; Kamarul, T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis. 2016, 7, e2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi-Sichani, M.; Moerke, N.J.; Niepel, M.; Zhang, T.; Gray, N.S.; Sorger, P.K. Systematic analysis of BRAF V 600E melanomas reveals a role for JNK/c-Jun pathway in adaptive resistance to drug-induced apoptosis. Mol. Syst. Boil. 2015, 11, 797. [Google Scholar] [CrossRef]
- Denkert, C.; Siegert, A.; LeClere, A.; Turzynski, A.; Hauptmann, S. An inhibitor of stress-activated MAP-kinases reduces invasion and MMP-2 expression of malignant melanoma cells. Clin. Exp. Metastasis 2002, 19, 79–85. [Google Scholar] [CrossRef]
- Davies, M.A. The Role of the PI3K-AKT Pathway in Melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef]
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Cappellini, G.C.A.; D’Atri, S. Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating effect of dabrafenib on the invasive behavior of melanoma cells with acquired resistance to the BRAF inhibitor. Int. J. Oncol. 2016, 49, 1164–1174. [Google Scholar] [CrossRef]
- Madonna, G.; Ullman, C.D.; Gentilcore, G.; Palmieri, G.; Ascierto, P.A. NF-κB as potential target in the treatment of melanoma. J. Transl. Med. 2012, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Lesinski, G.B. The potential for targeting the STAT3 pathway as a novel therapy for melanoma. Futur. Oncol. 2013, 9, 925–927. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.J.; Huang, J.M.; Zhong, H.J.; Dong, Z.Z.; Vellaisamy, K.; Lu, J.J.; Chen, X.P.; Chiu, P.; Kwong, D.W.J.; Han, Q.B.; et al. A natural product-like JAK2/STAT3 inhibitor induces apoptosis of malignant melanoma cells. PLoS ONE 2017, 12, e0177123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Aida, S.; Sonobe, Y.; Tanimura, H.; Oikawa, N.; Yuhki, M.; Sakamoto, H.; Mizuno, T. MITF suppression improves the sensitivity of melanoma cells to a BRAF inhibitor. Cancer Lett. 2017, 409, 116–124. [Google Scholar] [CrossRef]
- Xu, Y.; Zhan, J.; Wijeratne, E.M.K.; Burns, A.M.; Gunatilaka, A.A.L.; Molnar, I. Cytotoxic and Antihaptotactic Beauvericin Analogues from Precursor-Directed Biosynthesis with the Insect PathogenBeauveria bassianaATCC 7159. J. Nat. Prod. 2007, 70, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.N.; Jang, J.-P.; Shin, H.J.; Jang, J.-H.; Ahn, J.S.; Jung, H.J. Cytotoxic Activities and Molecular Mechanisms of the Beauvericin and Beauvericin G1 Microbial Products against Melanoma Cells. Molecules 2020, 25, 1974. https://doi.org/10.3390/molecules25081974
Lim HN, Jang J-P, Shin HJ, Jang J-H, Ahn JS, Jung HJ. Cytotoxic Activities and Molecular Mechanisms of the Beauvericin and Beauvericin G1 Microbial Products against Melanoma Cells. Molecules. 2020; 25(8):1974. https://doi.org/10.3390/molecules25081974
Chicago/Turabian StyleLim, Haet Nim, Jun-Pil Jang, Hee Jeong Shin, Jae-Hyuk Jang, Jong Seog Ahn, and Hye Jin Jung. 2020. "Cytotoxic Activities and Molecular Mechanisms of the Beauvericin and Beauvericin G1 Microbial Products against Melanoma Cells" Molecules 25, no. 8: 1974. https://doi.org/10.3390/molecules25081974