
Unravelling the Complex Interplay of Transcription Factors Orchestrating Seed Oil Content in Brassica napus L.

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
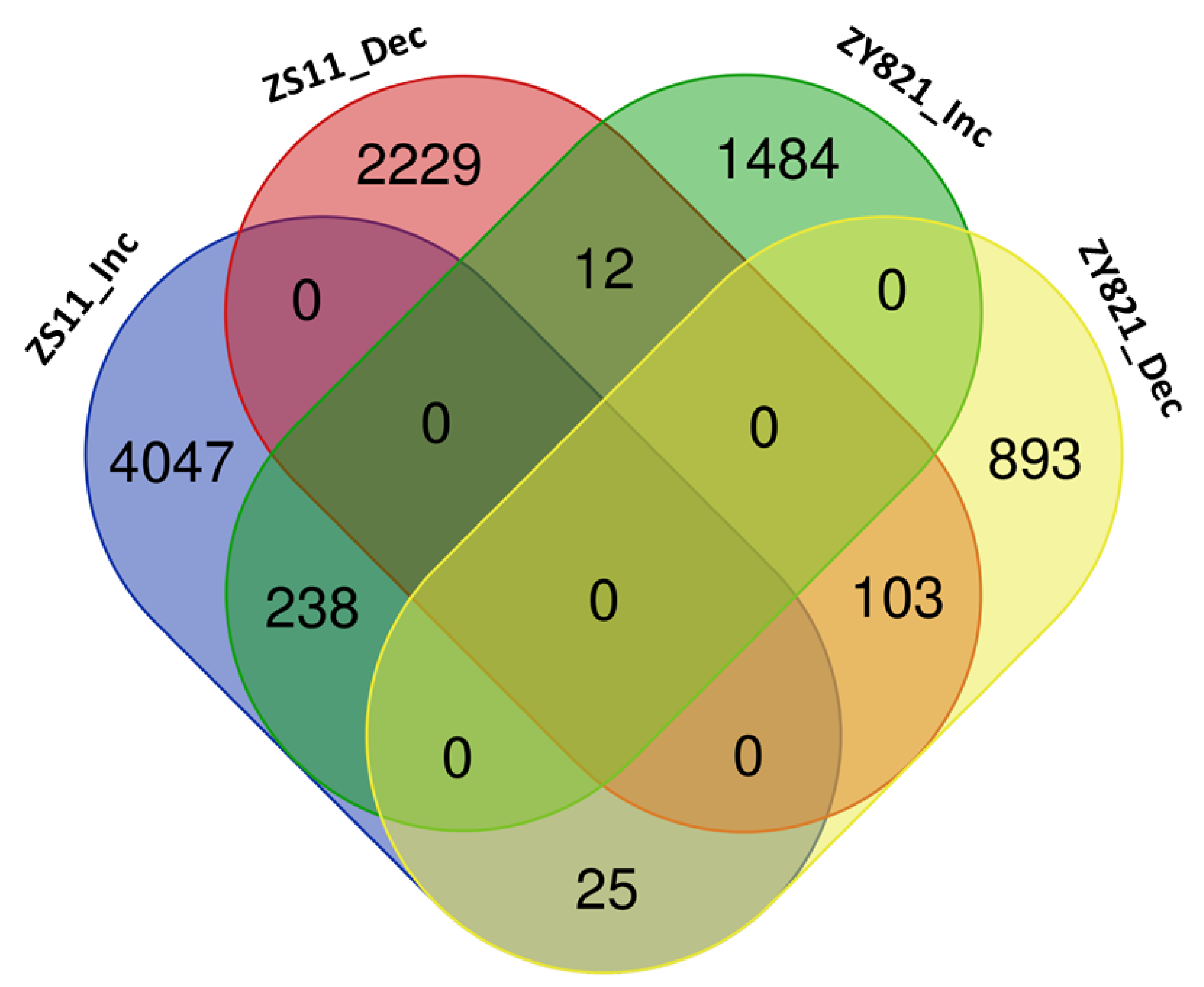
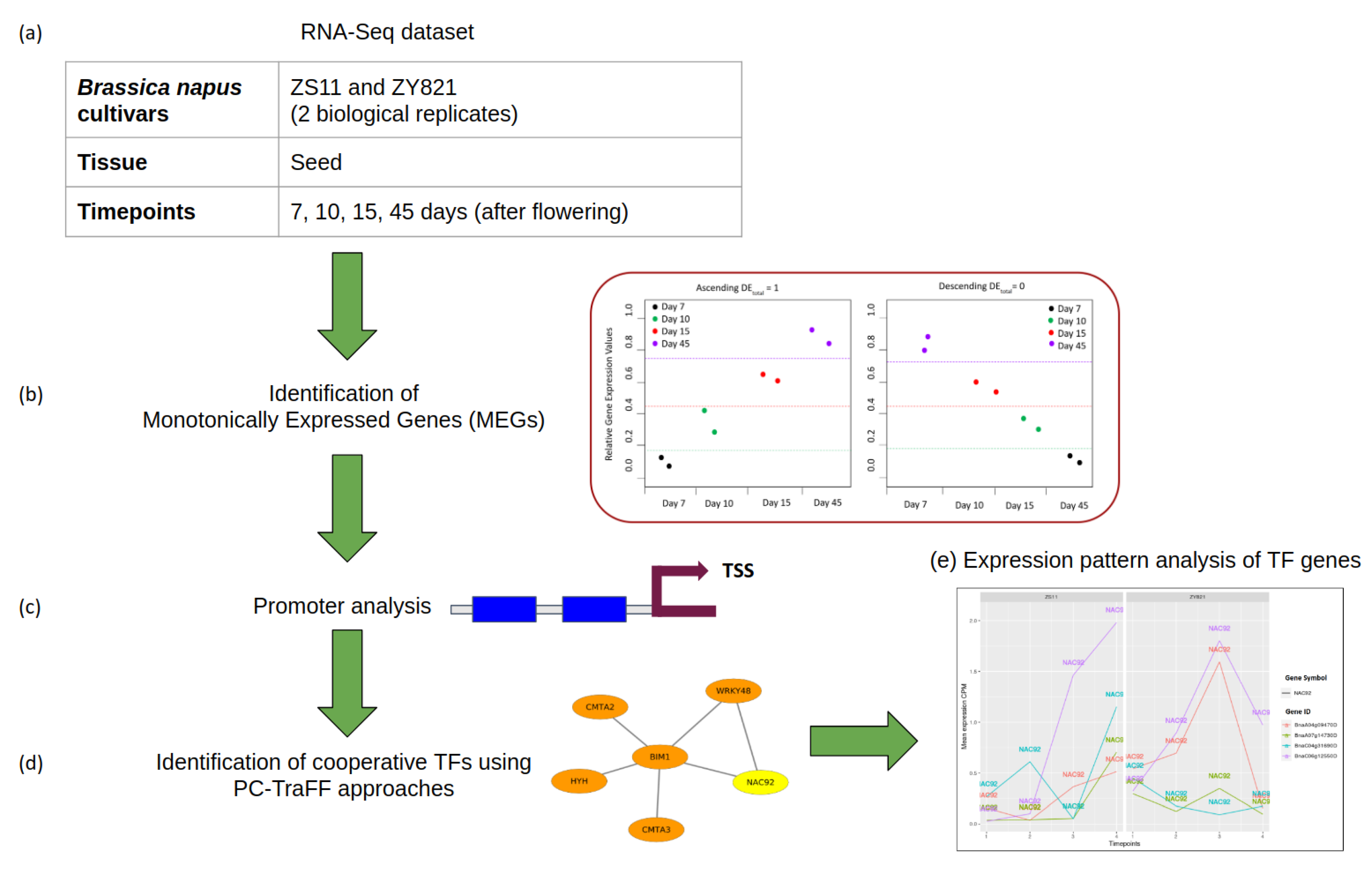
2.1. Identification and Analysis of MEGs
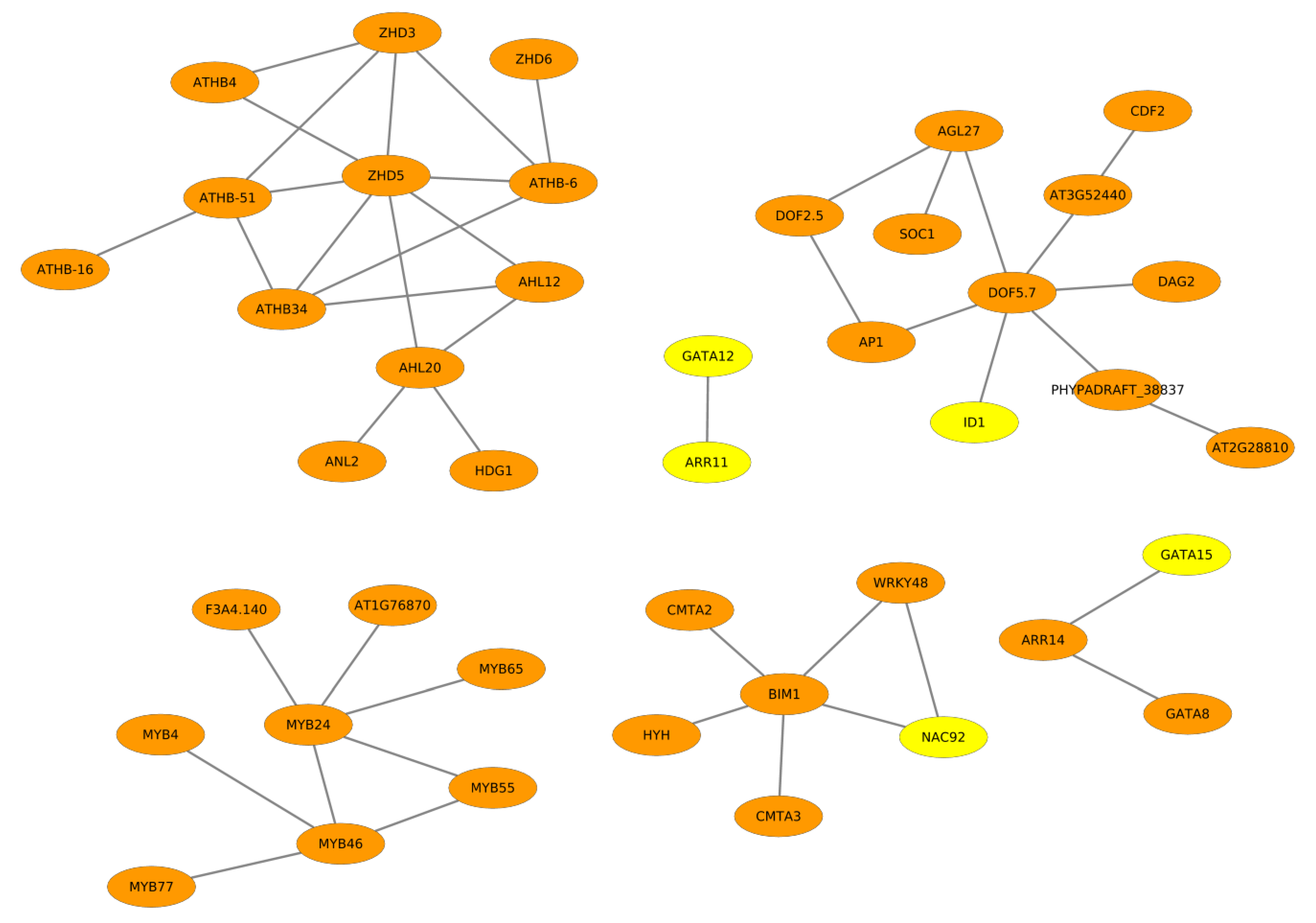
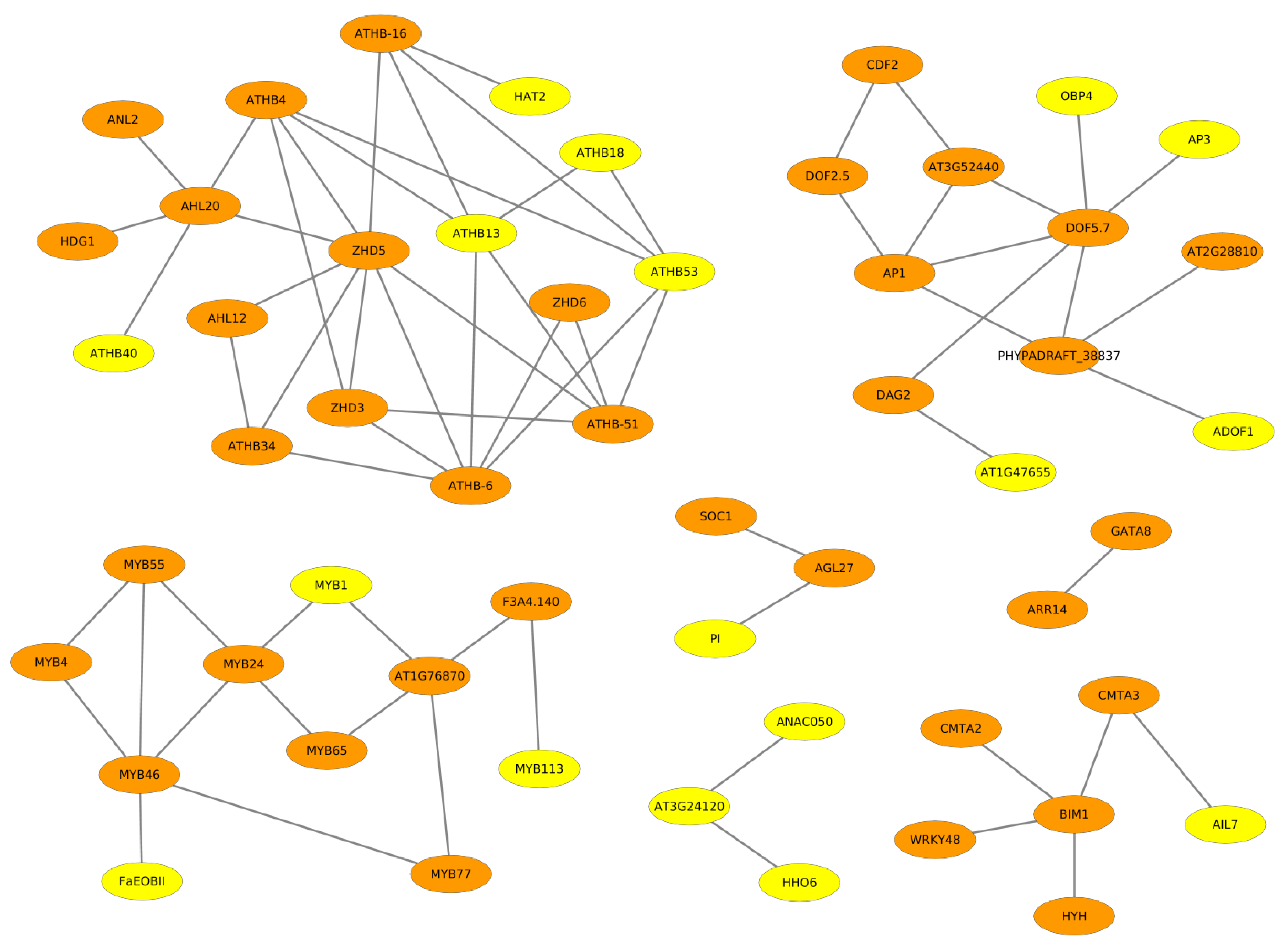
2.2. Cooperative TFs Regulating Seed Developmental Processes of ZS11 and ZY821
2.3. Cooperation Network of ZS11 and ZY821
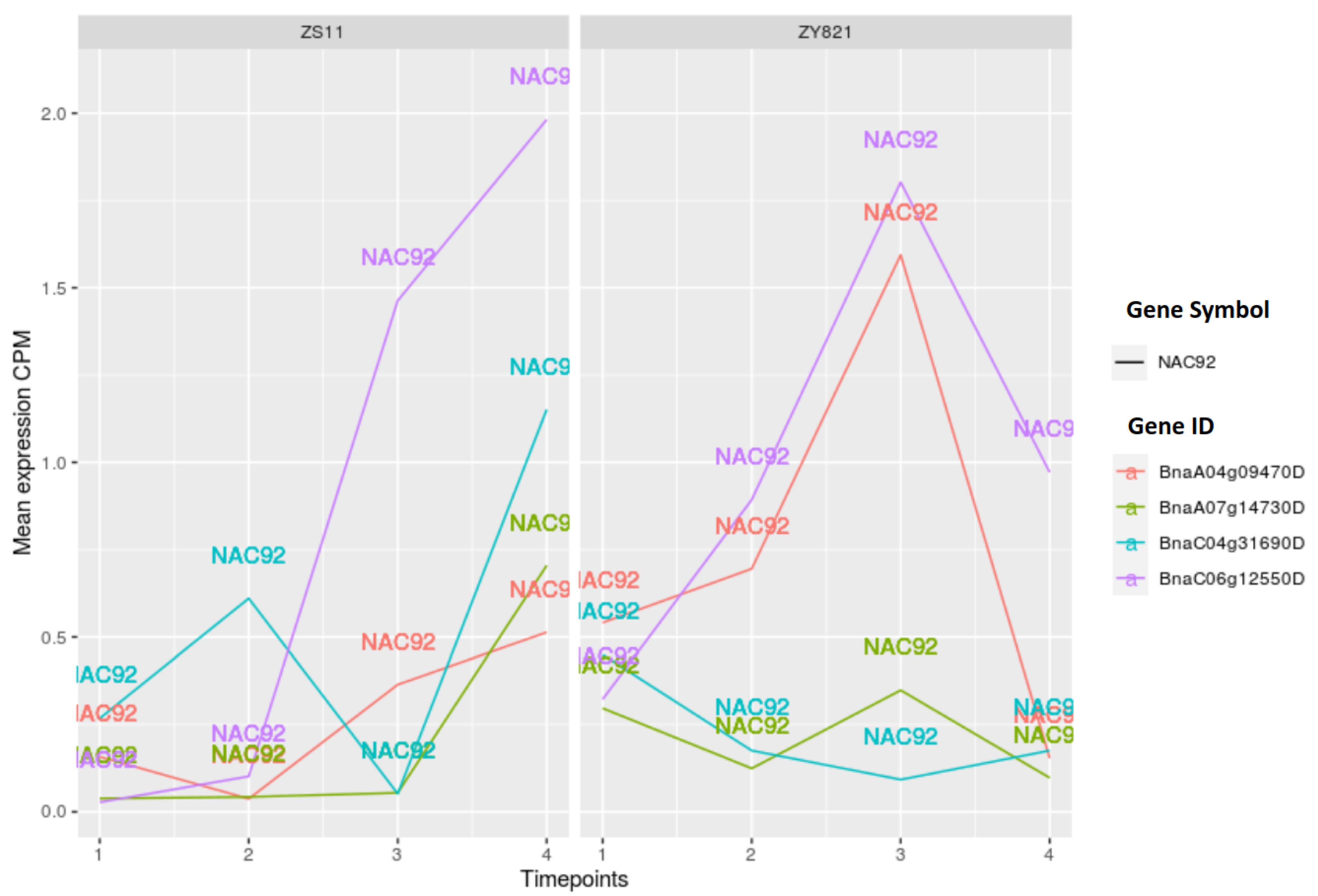
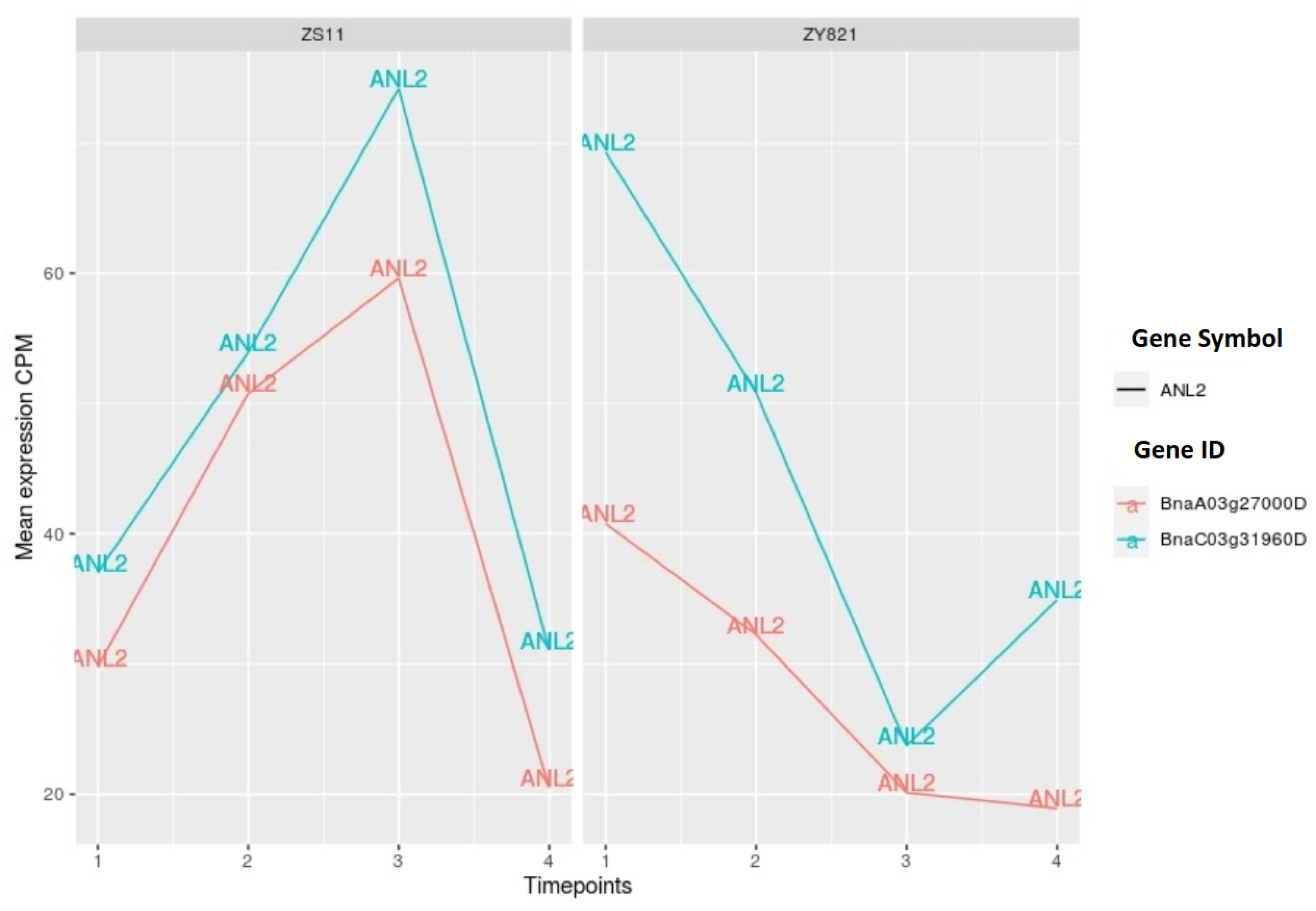
2.3.1. NAC Family of Transcription Factors
2.3.2. GATA Transcription Factors
2.3.3. DOF Family of Transcription Factors
2.3.4. HD-ZIP Family of Transcription Factors
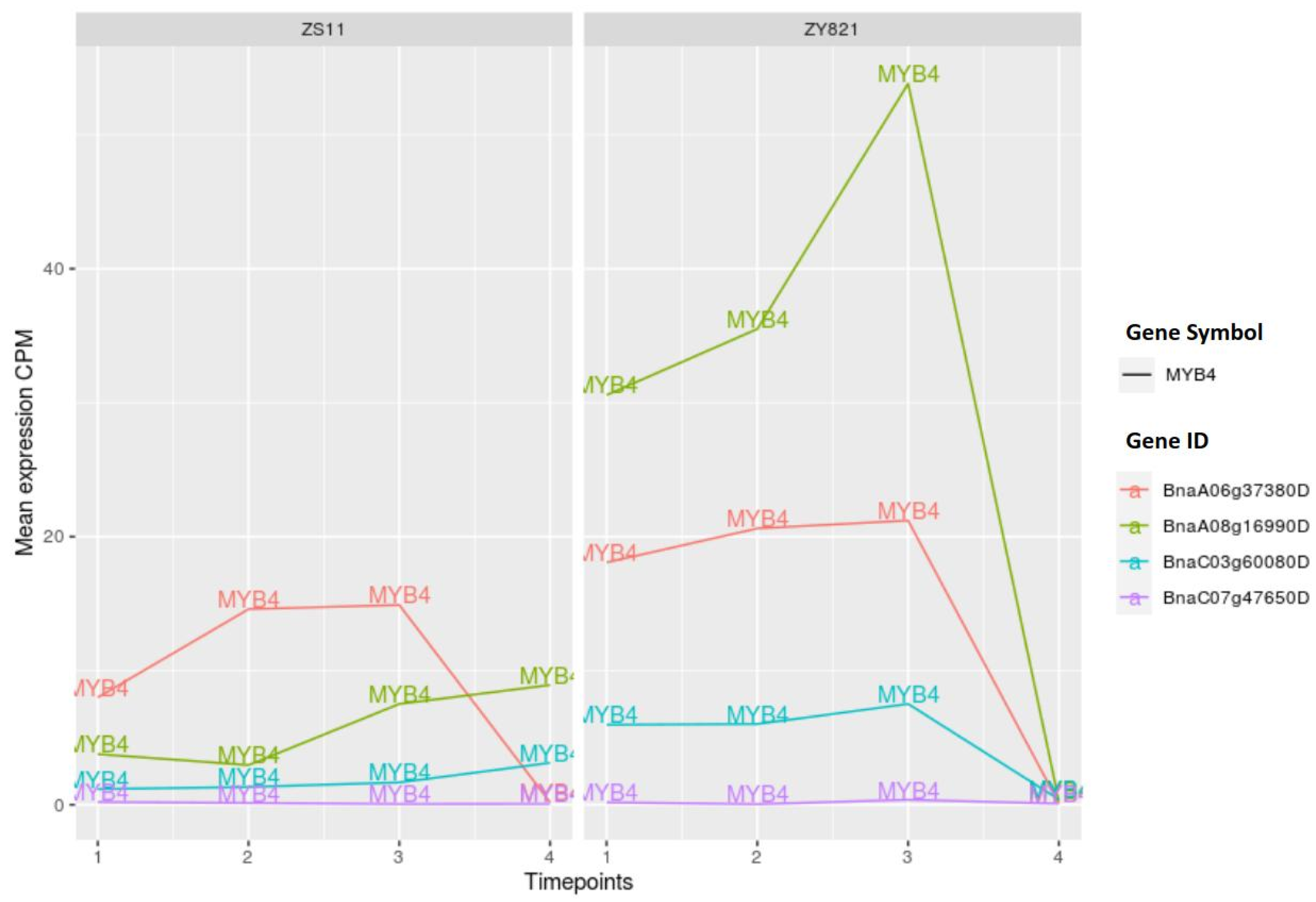
2.3.5. MYB Family of Transcription Factors
2.3.6. Other Transcription Factors
3. Materials and Methods
3.1. B. napus Data Set and Data Preparation
3.2. Identification of Monotonically Expressed Genes
3.3. Gene Ontology Enrichment Analysis
3.4. Identification of Transcription Factor Cooperations
- Pre-defined distances: For the identification of the regular cooperative binding pattern of TFs, the underlying PC-TraFF algorithm requires the pre-defined minimum and maximum distance thresholds between TFBSs. In our analysis, we used the recommended values of a distance ≤20 for the maximum and ≥5 for the minimum distance.
3.5. Expression Pattern Analysis of TF Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gunstone, F.D.; Harwood, J.L.; Dijkstra, A.J. The Lipid Handbook with CD-ROM; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- USDA. Oilseeds: World Markets and Trade. Available online: https://www.fas.usda.gov/data/oilseeds-world-markets-and-trade (accessed on 19 January 2020).
- USDA. Unites States Department of Agriculture Foreign Agricultural Service, Global Agricultural Trade System (GATS), Forest Products Trade Statistics. 2011. Available online: http://www.fas.usda.gov/gats/default.aspx (accessed on 19 January 2020).
- Woodfield, H.; Harwood, J. Oilseed Crops: Linseed, Rapeseed, Soybean, and Sunflower. In Encyclopedia of Applied Plant Sciences, 2nd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Gracka, A.; Jeleń, H.H.; Majcher, M.; Siger, A.; Kaczmarek, A. Flavoromics approach in monitoring changes in volatile compounds of virgin rapeseed oil caused by seed roasting. J. Chromatogr. A 2016, 1428, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fan, C.; Li, J.; Cai, G.; Yang, Q.; Wu, J.; Yi, X.; Zhang, C.; Zhou, Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor. Appl. Genet. 2016, 129, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Velasco, L.; Möllers, C.; Becker, H.C. Estimation of seed weight, oil content and fatty acid composition in intact single seeds of rapeseed (Brassica napus L.) by near-infrared reflectance spectroscopy. Euphytica 1999, 106, 79–85. [Google Scholar] [CrossRef]
- Wittkop, B.; Snowdon, R.; Friedt, W. Status and perspectives of breeding for enhanced yield and quality of oilseed crops for Europe. Euphytica 2009, 170, 131. [Google Scholar] [CrossRef]
- Snowdon, R.; Lühs, W.; Friedt, W. Oilseed rape. In Oilseeds; Springer: Berlin/Heidelberg, Germany, 2007; pp. 55–114. [Google Scholar]
- Abbadi, A.; Leckband, G. Rapeseed breeding for oil content, quality, and sustainability. Eur. J. Lipid Sci. Technol. 2011, 113, 1198–1206. [Google Scholar] [CrossRef]
- Hannoufa, A.; Pillai, B.V.; Chellamma, S. Genetic enhancement of Brassica napus seed quality. Transgenic Res. 2014, 23, 39–52. [Google Scholar] [CrossRef]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef]
- Knauer, S.; Holt, A.L.; Rubio-Somoza, I.; Tucker, E.J.; Hinze, A.; Pisch, M.; Javelle, M.; Timmermans, M.C.; Tucker, M.R.; Laux, T. A protodermal miR394 signal defines a region of stem cell competence in the Arabidopsis shoot meristem. Dev. Cell 2013, 24, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Yang, X.; Zhang, F.; Zheng, X.; Qu, C.; Mu, J.; Fu, F.; Li, J.; Guan, R.; Zhang, H.; et al. Enhanced seed oil production in canola by conditional expression of Brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in developing seeds. Plant Physiol. 2011, 156, 1577–1588. [Google Scholar] [CrossRef] [Green Version]
- Nesi, N.; Delourme, R.; Brégeon, M.; Falentin, C.; Renard, M. Genetic and molecular approaches to improve nutritional value of Brassica napus L. seed. Comptes Rendus Biol. 2008, 331, 763–771. [Google Scholar] [CrossRef]
- Murphy, D.J.; Cummins, I.; Kang, A.S. Synthesis of the major oil-body membrane protein in developing rapeseed (Brassica napus) embryos. Integration with storage-lipid and storage-protein synthesis and implications for the mechanism of oil-body formation. Biochem. J. 1989, 258, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, R.B.; De Paiva, G.; Yadegari, R. Plant embryogenesis: Zygote to seed. Science 1994, 266, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.H. Oil bodies and oleosins in seeds. Annu. Rev. Plant Biol. 1992, 43, 177–200. [Google Scholar] [CrossRef]
- Van Erp, H.; Kelly, A.A.; Menard, G.; Eastmond, P.J. Multigene engineering of triacylglycerol metabolism boosts seed oil content in Arabidopsis. Plant Physiol. 2014, 165, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Zhao, X.; Xu, Y.; Wang, Q.; Wang, H.; Wu, D.; Jiang, L. Effect of germination potential on storage lipids and transcriptome changes in premature developing seeds of oilseed rape (Brassica napus L.). Theor. Appl. Genet. 2020, 1–14. [Google Scholar] [CrossRef]
- Shahid, M.; Cai, G.; Zu, F.; Zhao, Q.; Qasim, M.U.; Hong, Y.; Fan, C.; Zhou, Y. Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica napus L. Int. J. Mol. Sci. 2019, 20, 1982. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zhu, S.; Yuan, Y.; Wang, Y.; Zeng, L.; Batley, J.; Wang, Y.P. Transcriptomic comparison between developing seeds of yellow-and black-seeded Brassica napus reveals that genes influence seed quality. BMC Plant Biol. 2019, 19, 203. [Google Scholar] [CrossRef]
- Ding, L.N.; Guo, X.J.; Li, M.; Fu, Z.L.; Yan, S.Z.; Zhu, K.M.; Wang, Z.; Tan, X.L. Improving seed germination and oil contents by regulating the GDSL transcriptional level in Brassica napus. Plant Cell Rep. 2019, 38, 243–253. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, C.; Tang, F.; Yang, B.; Zhang, L.; Liu, J.; Huo, Q.; Wang, S.; Li, S.; Wei, L.; et al. Identification of candidate genes controlling oil content by combination of genome-wide association and transcriptome analysis in the oilseed crop Brassica napus. Biotechnol. Biofuels 2019, 12, 216. [Google Scholar] [CrossRef]
- Qu, C.; Yin, N.; Chen, S.; Wang, S.; Chen, X.; Zhao, H.; Shen, S.; Fu, F.; Zhou, B.; Xu, X.; et al. Comparative Analysis of the Metabolic Profiles of Yellow-versus Black-Seeded Rapeseed Using UPLC–HESI–MS/MS and Transcriptome Analysis. J. Agric. Food Chem. 2020, 68, 3033–3049. [Google Scholar] [CrossRef]
- Niu, Y.; Wu, L.; Li, Y.; Huang, H.; Qian, M.; Sun, W.; Zhu, H.; Xu, Y.; Fan, Y.; Mahmood, U.; et al. Deciphering the transcriptional regulatory networks that control size, color, and oil content in Brassica rapa seeds. Biotechnol. Biofuels 2020, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klees, S.; Lange, T.M.; Bertram, H.; Rajavel, A.; Schlüter, J.-S.; Lu, K.; Schmitt, A.O.; Gültas, M. In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data. Int. J. Mol. Sci. 2020, 22, 789. [Google Scholar] [CrossRef]
- Kaufmann, K.; Wellmer, F.; Muiño, J.M.; Ferrier, T.; Wuest, S.E.; Kumar, V.; Serrano-Mislata, A.; Madueno, F.; Krajewski, P.; Meyerowitz, E.M.; et al. Orchestration of floral initiation by APETALA1. Science 2010, 328, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Mena, C.; de Folter, S.; Costa, M.M.R.; Angenent, G.C.; Sablowski, R. Transcriptional program controlled by the floral homeotic gene AGAMOUS during early organogenesis. Development 2005, 132, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerowitz, E.M. Plants compared to animals: The broadest comparative study of development. Science 2002, 295, 1482–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, S. Transcription factors in plants: Physiological functions and regulation of expression. J. Plant Res. 1998, 111, 363–371. [Google Scholar] [CrossRef]
- Singh, K.B. Transcriptional regulation in plants: The importance of combinatorial control. Plant Physiol. 1998, 118, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Guilfoyle, T.J. The structure of plant gene promoters. In Genetic Engineering; Springer: Boston, MA, USA, 1997; pp. 15–47. [Google Scholar]
- Kaufmann, K.; Pajoro, A.; Angenent, G.C. Regulation of transcription in plants: Mechanisms controlling developmental switches. Nat. Rev. Genet. 2010, 11, 830–842. [Google Scholar] [CrossRef]
- Rajavel, A.; Heinrich, F.; Schmitt, A.O.; Gültas, M. Identifying Cattle Breed-Specific Partner Choice of Transcription Factors during the African Trypanosomiasis Disease Progression Using Bioinformatics Analysis. Vaccines 2020, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Du, Z.; Su, Z. PlantGSEA: A gene set enrichment analysis toolkit for plant community. Nucleic Acids Res. 2013, 41, W98–W103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambhampati, S.; Aznar-Moreno, J.A.; Hostetler, C.; Caso, T.; Bailey, S.R.; Hubbard, A.H.; Durrett, T.P.; Allen, D.K. On the Inverse Correlation of Protein and Oil: Examining the Effects of Altered Central Carbon Metabolism on Seed Composition Using Soybean Fast Neutron Mutants. Metabolites 2020, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapala-Kozik, M.; Wolak, N.; Kujda, M.; Banas, A.K. The upregulation of thiamine (vitamin B 1) biosynthesis in Arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol. 2012, 12, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuernagel, L.; Meckbach, C.; Heinrich, F.; Zeidler, S.; Schmitt, A.O.; Gültas, M. Computational identification of tissue-specific transcription factor cooperation in ten cattle tissues. PLoS ONE 2019, 14, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meckbach, C.; Wingender, E.; Gültas, M. Removing background co-occurrences of transcription factor binding sites greatly improves the prediction of specific transcription factor cooperations. Front. Genet. 2018, 9, 189. [Google Scholar] [CrossRef]
- Meckbach, C.; Tacke, R.; Hua, X.; Waack, S.; Wingender, E.; Gültas, M. PC-TraFF: Identification of potentially collaborating transcription factors using pointwise mutual information. BMC Bioinform. 2015, 16, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidler, S.; Meckbach, C.; Tacke, R.; Raad, F.S.; Roa, A.; Uchida, S.; Zimmermann, W.H.; Wingender, E.; Gültas, M. Computational Detection of Stage-Specific Transcription Factor Clusters during Heart Development. Front. Genet. 2016, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Collinge, M.; Boller, T. Differential induction of two potato genes, Stprx2 and StNAC, in response to infection by Phytophthora infestans and to wounding. Plant Mol. Biol. 2001, 46, 521–529. [Google Scholar] [CrossRef]
- Hegedus, D.; Yu, M.; Baldwin, D.; Gruber, M.; Sharpe, A.; Parkin, I.; Whitwill, S.; Lydiate, D. Molecular characterization of Brassica napus NAC domain transcriptional activators induced in response to biotic and abiotic stress. Plant Mol. Biol. 2003, 53, 383–397. [Google Scholar] [CrossRef]
- Tran, L.S.P.; Nakashima, K.; Sakuma, Y.; Simpson, S.D.; Fujita, Y.; Maruyama, K.; Fujita, M.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 2004, 16, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Fujita, Y.; Maruyama, K.; Seki, M.; Hiratsu, K.; Ohme-Takagi, M.; Tran, L.S.P.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A dehydration-induced NAC protein, RD26, is involved in a novel ABA-dependent stress-signaling pathway. Plant J. 2004, 39, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Balazadeh, S.; Siddiqui, H.; Allu, A.D.; Matallana-Ramirez, L.P.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Köhler, B.; Mueller-Roeber, B. A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J. 2010, 62, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.Q.; Ma, Z.Y.; Huang, H.W.; Mo, H.; Zhao, T.t.; Li, L.; Cai, T.; Chen, S.; Ma, L.; He, X.J. Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic Acids Res. 2015, 43, 1469–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Bolser, D.M.; Staines, D.M.; Perry, E.; Kersey, P.J. Ensembl plants: Integrating tools for visualizing, mining, and analyzing plant genomic data. In Plant Genomics Databases; Springer: New York, NY, USA, 2017; pp. 1–31. [Google Scholar]
- Argüello-Astorga, G.; Herrera-Estrella, L. Evolution of light-regulated plant promoters. Annu. Rev. Plant Biol. 1998, 49, 525–555. [Google Scholar] [CrossRef]
- Koch, K. Carbohydrate-modulated gene expression in plants. Annu. Rev. Plant Biol. 1996, 47, 509–540. [Google Scholar] [CrossRef] [Green Version]
- Terzaghi, W.B.; Cashmore, A.R. Photomorphenesis: Seeing the light in plant development. Curr. Biol. 1995, 5, 466–468. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.; Bate, N.J.; Sivasankar, S.; Rothstein, S.J. Footprinting of the spinach nitrite reductase gene promoter reveals the preservation of nitrate regulatory elements between fungi and higher plants. Plant Mol. Biol. 1997, 34, 465–476. [Google Scholar] [CrossRef]
- Oliveira, I.C.; Coruzzi, G.M. Carbon and amino acids reciprocally modulate the expression of glutamine synthetase in Arabidopsis. Plant Physiol. 1999, 121, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Yeap, W.C.; Lee, F.C.; Shabari Shan, D.K.; Musa, H.; Appleton, D.R.; Kulaveerasingam, H. WRI 1-1, ABI 5, NF-YA 3 and NF-YC 2 increase oil biosynthesis in coordination with hormonal signaling during fruit development in oil palm. Plant J. 2017, 91, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hou, L.; Meng, J.; You, H.; Li, Z.; Gong, Z.; Yang, S.; Shi, Y. The antagonistic action of abscisic acid and cytokinin signaling mediates drought stress response in Arabidopsis. Mol. Plant 2018, 11, 970–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, S. The Dof family of plant transcription factors. Trends Plant Sci. 2002, 7, 555–560. [Google Scholar] [CrossRef]
- Ahmad, M.; Rim, Y.; Chen, H.; Kim, J. Functional characterization of Arabidopsis Dof transcription factor AtDof4. 1. Russ. J. Plant Physiol. 2013, 60, 116–123. [Google Scholar] [CrossRef]
- Santopolo, S.; Boccaccini, A.; Lorrai, R.; Ruta, V.; Capauto, D.; Minutello, E.; Serino, G.; Costantino, P.; Vittorioso, P. DOF AFFECTING GERMINATION 2 is a positive regulator of light-mediated seed germination and is repressed by DOF AFFECTING GERMINATION 1. BMC Plant Biol. 2015, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.W.; Zhang, B.; Hao, Y.J.; Huang, J.; Tian, A.G.; Liao, Y.; Zhang, J.S.; Chen, S.Y. The soybean Dof-type transcription factor genes, GmDof4 and GmDof11, enhance lipid content in the seeds of transgenic Arabidopsis plants. Plant J. 2007, 52, 716–729. [Google Scholar] [CrossRef]
- Fobert, P.R. Role of Transcription Factors in Storage Lipid Accumulation in Plants; AOCS: Urbana, IL, USA, 2012. [Google Scholar]
- Su, Y.; Liang, W.; Liu, Z.; Wang, Y.; Zhao, Y.; Ijaz, B.; Hua, J. Overexpression of GhDof1 improved salt and cold tolerance and seed oil content in Gossypium hirsutum. J. Plant Physiol. 2017, 218, 222–234. [Google Scholar] [CrossRef]
- Rerie, W.G.; Feldmann, K.A.; Marks, M.D. The GLABRA2 gene encodes a homeo domain protein required for normal trichome development in Arabidopsis. Genes Dev. 1994, 8, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Sinkevicius, K.W.; Selinger, D.A.; Tarczynski, M.C. The homeobox gene GLABRA2 affects seed oil content in Arabidopsis. Plant Mol. Biol. 2006, 60, 377–387. [Google Scholar] [CrossRef]
- Ariel, F.D.; Manavella, P.A.; Dezar, C.A.; Chan, R.L. The true story of the HD-Zip family. Trends Plant Sci. 2007, 12, 419–426. [Google Scholar] [CrossRef]
- Kubo, H.; Peeters, A.J.; Aarts, M.G.; Pereira, A.; Koornneef, M. ANTHOCYANINLESS2, a homeobox gene affecting anthocyanin distribution and root development in Arabidopsis. Plant Cell 1999, 11, 1217–1226. [Google Scholar] [PubMed] [Green Version]
- Yu, H.; Chen, X.; Hong, Y.Y.; Wang, Y.; Xu, P.; Ke, S.D.; Liu, H.Y.; Zhu, J.K.; Oliver, D.J.; Xiang, C.B. Activated expression of an Arabidopsis HD-START protein confers drought tolerance with improved root system and reduced stomatal density. Plant Cell 2008, 20, 1134–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javelle, M.; Vernoud, V.; Depege-Fargeix, N.; Arnould, C.; Oursel, D.; Domergue, F.; Sarda, X.; Rogowsky, P.M. Overexpression of the epidermis-specific homeodomain-leucine zipper IV transcription factor Outer Cell Layer1 in maize identifies target genes involved in lipid metabolism and cuticle biosynthesis. Plant Physiol. 2010, 154, 273–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimberg, Å.; Lager, I.; Street, N.R.; Robinson, K.M.; Marttila, S.; Mähler, N.; Ingvarsson, P.K.; Bhalerao, R.P. Storage lipid accumulation is controlled by photoperiodic signal acting via regulators of growth cessation and dormancy in hybrid aspen. New Phytol. 2018, 219, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, E.; Leonhardt, N.; Eisler, H.; Parmentier, Y.; Alioua, M.; Jacquet, H.; Leung, J.; Genschik, P. MATH/BTB CRL3 receptors target the homeodomain-leucine zipper ATHB6 to modulate abscisic acid signaling. Dev. Cell 2011, 21, 1116–1128. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.; Merlot, S.; Giraudat, J. The Arabidopsis ABSCISIC ACID-INSENSITIVE2 (ABI2) and ABI1 genes encode homologous protein phosphatases 2C involved in abscisic acid signal transduction. Plant Cell 1997, 9, 759–771. [Google Scholar]
- Son, O.; Cho, H.Y.; Kim, M.R.; Lee, H.; Lee, M.S.; Song, E.; Park, J.H.; Nam, K.H.; Chun, J.Y.; Kim, H.J.; et al. Induction of a homeodomain–leucine zipper gene by auxin is inhibited by cytokinin in Arabidopsis roots. Biochem. Biophys. Res. Commun. 2004, 326, 203–209. [Google Scholar] [CrossRef]
- Jin, H.; Martin, C. Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Biol. 1999, 41, 577–585. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Mu, R.L.; Cao, Y.R.; Liu, Y.F.; Lei, G.; Zou, H.F.; Liao, Y.; Wang, H.W.; Zhang, W.K.; Ma, B.; Du, J.Z.; et al. An R2R3-type transcription factor gene AtMYB59 regulates root growth and cell cycle progression in Arabidopsis. Cell Res. 2009, 19, 1291–1304. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Zou, H.F.; Wang, H.W.; Zhang, W.K.; Ma, B.; Zhang, J.S.; Chen, S.Y. Soybean GmMYB76, GmMYB92, and GmMYB177 genes confer stress tolerance in transgenic Arabidopsis plants. Cell Res. 2008, 18, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Araki, S.; Ito, M.; Soyano, T.; Nishihama, R.; Machida, Y. Mitotic cyclins stimulate the activity of c-Myb-like factors for transactivation of G2/M phase-specific genes in tobacco. J. Biol. Chem. 2004, 279, 32979–32988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwinn, K.E.; Ngo, H.; Kenel, F.; Brummell, D.A.; Albert, N.W.; McCallum, J.A.; Pither-Joyce, M.; Crowhurst, R.N.; Eady, C.; Davies, K.M. The onion (Allium cepa L.) R2R3-MYB gene MYB1 regulates anthocyanin biosynthesis. Front. Plant Sci. 2016, 7, 1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, L.W.; Li, L. Characterization of the regulatory network of BoMYB2 in controlling anthocyanin biosynthesis in purple cauliflower. Planta 2012, 236, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Medina-Puche, L.; Molina-Hidalgo, F.J.; Boersma, M.; Schuurink, R.C.; López-Vidriero, I.; Solano, R.; Franco-Zorrilla, J.M.; Caballero, J.L.; Blanco-Portales, R.; Muñoz-Blanco, J. An R2R3-MYB transcription factor regulates eugenol production in ripe strawberry fruit receptacles. Plant Physiol. 2015, 168, 598–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.C.; Wu, J.; Guan, M.L.; Zhao, C.H.; Geng, P.; Zhao, Q. Arabidopsis MYB4 plays dual roles in flavonoid biosynthesis. Plant J. 2020, 101, 637–652. [Google Scholar] [CrossRef]
- Ko, J.H.; Jeon, H.W.; Kim, W.C.; Kim, J.Y.; Han, K.H. The MYB46/MYB83-mediated transcriptional regulatory programme is a gatekeeper of secondary wall biosynthesis. Ann. Bot. 2014, 114, 1099–1107. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, V.; García-Andrade, J.; Vera, P. Enhanced disease resistance to Botrytis cinerea in myb46 Arabidopsis plants is associated to an early down-regulation of CesA genes. Plant Signal. Behav. 2011, 6, 911–913. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, S.; Gilmour, S.J.; Thomashow, M.F. Roles of CAMTA transcription factors and salicylic acid in configuring the low-temperature transcriptome and freezing tolerance of A rabidopsis. Plant J. 2013, 75, 364–376. [Google Scholar] [CrossRef]
- Doherty, C.J.; Van Buskirk, H.A.; Myers, S.J.; Thomashow, M.F. Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell 2009, 21, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Ali, G.S.; Simons, K.A.; Hou, J.; Yang, T.; Reddy, A.; Poovaiah, B. Ca2+/calmodulin regulates salicylic-acid-mediated plant immunity. Nature 2009, 457, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Le, D.T.; Hwang, S.; Seo, P.J.; Kim, H.U. Role of the INDETERMINATE DOMAIN Genes in Plants. Int. J. Mol. Sci. 2019, 20, 2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colasanti, J.; Tremblay, R.; Wong, A.Y.; Coneva, V.; Kozaki, A.; Mable, B.K. The maize INDETERMINATE1 flowering time regulator defines a highly conserved zinc finger protein family in higher plants. BMC Genom. 2006, 7, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Xu, Y.; Wang, J.; Singer, S.D.; Chen, G. The Role of Triacylglycerol in Plant Stress Response. Plants 2020, 9, 472. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.W.; Sun, H.J.; Chang, T.Y.; Lo, H.H.; Cheng, W.C.; Tseng, G.C.; Lin, C.T.; Chang, S.J.; Pal, N.R.; Chung, I.F. Discovering monotonic stemness marker genes from time-series stem cell microarray data. BMC Genom. BioMed Cent. 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. bioRxiv 2014. Available online: http://xxx.lanl.gov/abs/https://www.biorxiv.org/content/early/2014/08/19/002824.full.pdf (accessed on 10 December 2020).
- Smyth, G.K.; Ritchie, M.; Thorne, N.; Wettenhall, J.; Shi, W.; Hu, Y. Limma: Linear Models for Microarray and RNA-Seq Data User’s Guide; Pennsylvania State University: University Park, PA, USA, 2002. [Google Scholar]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Kel, A.E.; Gossling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCHTM: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; Van Der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZS11 | ZY821 | |||
|---|---|---|---|---|
| Ascending | Descending | Ascending | Descending | |
| Genes | 4310 | 2344 | 1734 | 1021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajavel, A.; Klees, S.; Schlüter, J.-S.; Bertram, H.; Lu, K.; Schmitt, A.O.; Gültas, M. Unravelling the Complex Interplay of Transcription Factors Orchestrating Seed Oil Content in Brassica napus L. Int. J. Mol. Sci. 2021, 22, 1033. https://doi.org/10.3390/ijms22031033
Rajavel A, Klees S, Schlüter J-S, Bertram H, Lu K, Schmitt AO, Gültas M. Unravelling the Complex Interplay of Transcription Factors Orchestrating Seed Oil Content in Brassica napus L. International Journal of Molecular Sciences. 2021; 22(3):1033. https://doi.org/10.3390/ijms22031033
Chicago/Turabian StyleRajavel, Abirami, Selina Klees, Johanna-Sophie Schlüter, Hendrik Bertram, Kun Lu, Armin Otto Schmitt, and Mehmet Gültas. 2021. "Unravelling the Complex Interplay of Transcription Factors Orchestrating Seed Oil Content in Brassica napus L." International Journal of Molecular Sciences 22, no. 3: 1033. https://doi.org/10.3390/ijms22031033