Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia



1
Division of Biological and Biomedical Sciences, College of Health & Life Sciences, Hamad Bin Khalifa University, Qatar Foundation, Education City, Doha, Qatar
2
Department of Pediatric Medicine, Division of Endocrinology, Sidra Medicine, Doha, Qatar
3
Department of Pediatric Medicine, Saida Governmental University Hospital, Beirut Arab University, Beirut 115020, Lebanon
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(17), 6286; https://doi.org/10.3390/ijms21176286
Submission received: 13 July 2020
/
Revised: 30 July 2020
/
Accepted: 2 August 2020
/
Published: 31 August 2020
(This article belongs to the Section Molecular Endocrinology and Metabolism)
Abstract
:Accumulation of glycogen in the kidney and liver is the main feature of Fanconi–Bickel Syndrome (FBS), a rare disorder of carbohydrate metabolism inherited in an autosomal recessive manner due to SLC2A2 gene mutations. Missense, nonsense, frame-shift (fs), in-frame indels, splice site, and compound heterozygous variants have all been identified in SLC2A2 gene of FBS cases. Approximately 144 FBS cases with 70 different SLC2A2 gene variants have been reported so far. SLC2A2 encodes for glucose transporter 2 (GLUT2) a low affinity facilitative transporter of glucose mainly expressed in tissues playing important roles in glucose homeostasis, such as renal tubular cells, enterocytes, pancreatic β-cells, hepatocytes and discrete regions of the brain. Dysfunctional mutations and decreased GLUT2 expression leads to dysglycaemia (fasting hypoglycemia, postprandial hyperglycemia, glucose intolerance, and rarely diabetes mellitus), hepatomegaly, galactose intolerance, rickets, and poor growth. The molecular mechanisms of dysglycaemia in FBS are still not clearly understood. In this review, we discuss the physiological roles of GLUT2 and the pathophysiology of mutants, highlight all of the previously reported SLC2A2 mutations associated with dysglycaemia, and review the potential molecular mechanisms leading to dysglycaemia and diabetes mellitus in FBS patients.
1. Introduction
Fanconi–Bickel syndrome (OMIM# 227810), a carbohydrate metabolism disorder due to glucose transporter 2 (GLUT2) transporter defect was first described by Fanconi and Bickel in 1949 [1]. In 1987, Fanconi–Bickel Syndrome (FBS) was identified as a defect in a sodium glucose secondary active transporter responsible for galactose and glucose transport in many tissues, including kidney and liver [2]. Studies in 1989, using Xenopus oocytes injected with human liver type glucose transporter synthetic mRNA construct, identified the role of the plasma membrane glucose transporter in sensing glucose, uptake of glucose, and its possible role in non-insulin-dependent diabetes mellitus [3]. In 1994, a study in Xenopus oocytes proved that a highly conserved GLUT2 missense mutation in one allele of the gene (substituted Val197 to Ile197) leads to GLUT2 dysfunction, and might be expected to play an important role in pathogenicity of non-insulin dependent diabetes mellitus [4,5]. Then in 1997, Santer et. al. (1997) for first time described the role of GLUT2 (SLC2A2) gene mutations in three FBS affected families, that includes the original patient reported by Fanconi and Bickel in 1949 [6].
FBS is a rare disorder of carbohydrate metabolism with phenotypic variability in patients and is inherited in an autosomal recessive manner. FBS is due to abnormal accumulation of glycogen in liver and kidneys leading to impairment of glucose and galactose utilization, and proximal renal tubular dysfunction [2]. The clinical features in FBS patients include short stature, rickets, failure to thrive, hepatomegaly, polyuria, proximal renal tubular dysfunction, and osteoporosis [2,7,8,9,10]. Biochemically, in glucose physiology, FBS patients have dysglycaemia characterized by fasting hypoglycemia, post-prandial hyperglycemia, glucose intolerance, and rarely diabetes mellitus. The pathology of FBS is due to compound heterozygous mutation (each allele with different mutation) or homozygous mutations in the GLUT2 (SLC2A2) gene [4,6,11,12]. This coding gene contains 11 exons and 10 introns, located on chromosome 3q26.1–q26.3. Human GLUT2 consists of 524 amino acids (aa) and the transmembrane segment 9 to 12 play a vital role in the characteristic affinity for glucose [13,14,15]. As discussed above, GLUT2 is mainly expressed in hepatocytes, enterocytes [16], proximal tubules in the kidney [17], pancreatic β-cells [15,18,19], discrete neuronal cells [20], and in astrocytes [21].
Dysglycaemia (fasting hypoglycemia, postprandial hyperglycemia, glucose intolerance, and diabetes mellitus) is commonly observed in many patients with FBS, however the mechanisms underlying dysglycaemia are not well understood. In this review, we briefly summarize the role of GLUT2 in glucose physiology, then, review all the reported mutations in SLC2A2, which have been reported with dysglycaemia and we finally discuss the potential molecular mechanisms of dysglycaemia associated with FBS.
2. Physiological Roles of GLUT2
2.1. Overview
GLUT2 has a low affinity (high KM) for glucose (15–20 mM) and plays a role in organs that have an important function in controlling glucose homeostasis such as the pancreatic β-cells, kidney, intestine, and liver. GLUT2 maintains glucose homeostasis by regulating the transepithelial uptake of glucose in the epithelial cells of basolateral membrane, glucose reabsorption in the kidney proximal tubule, glucose uptake and release from the sinusoidal membrane of the liver cells, and glucose regulated insulin secretion from pancreatic β-cells [22]. For more comprehensive reviews on the physiological roles of GLUT2, please see references [23,24,25].
2.2. Role of GLUT2 in β-Cells
GLUT2 function in human pancreatic β-cells is not clearly understood and is still controversial. However, the role of GLUT2 in rodent pancreatic β-cells is well established and plays a major role in glucose transport [26]. Expression of glucose transporters in rodent β-cells is different from human β-cells, with human β-cells showing predominantly GLUT1 and GLUT3 instead of GLUT2 expression [27]. Rat and human pancreatic β-cells have similar gene expression of glucokinase, but different expression of glucose transporters [27]. There is an abundance of GLUT1 transcript and protein expression in human β-cells and the glucose transporter kinetic characteristics are similar to GLUT1 [15]. During human pancreatic development, both GLUT1 and GLUT2 are expressed in the fetal islets [28], with GLUT1 reaching adult transcriptional levels by 18 weeks of gestational age and GLUT2 mRNA is detectable as early as 13 weeks of gestational age. However, both GLUT1 and GLUT2 expression remains low throughout human pancreatic development in the β-cells [29].
In rodents, GLUT2 might also have a role in pancreatic β-cell glucose sensing independent from its role as a glucose transporter. One study, using liver cells which were transfected with GLUT2-intracellular loop amino acids (aa) 237–301 fused with GFP (green fluorescent protein) cDNA shows translocation of the GLUT2-GFP fused protein to the nucleus from the cytoplasm at high concentration of glucose. This suggests that, via protein interactions with its large loop, GLUT2 may transduce a glucose signal from the plasma membrane to the nucleus [30]. In another study, transgenic mice expressing the same fusion protein had multiple defects of glucose metabolism [31]. These mice also had greater amounts of urine glucose, which might play an important role in protecting the mice from developing hyperglycemia despite the reduced production of insulin from the β-cells.
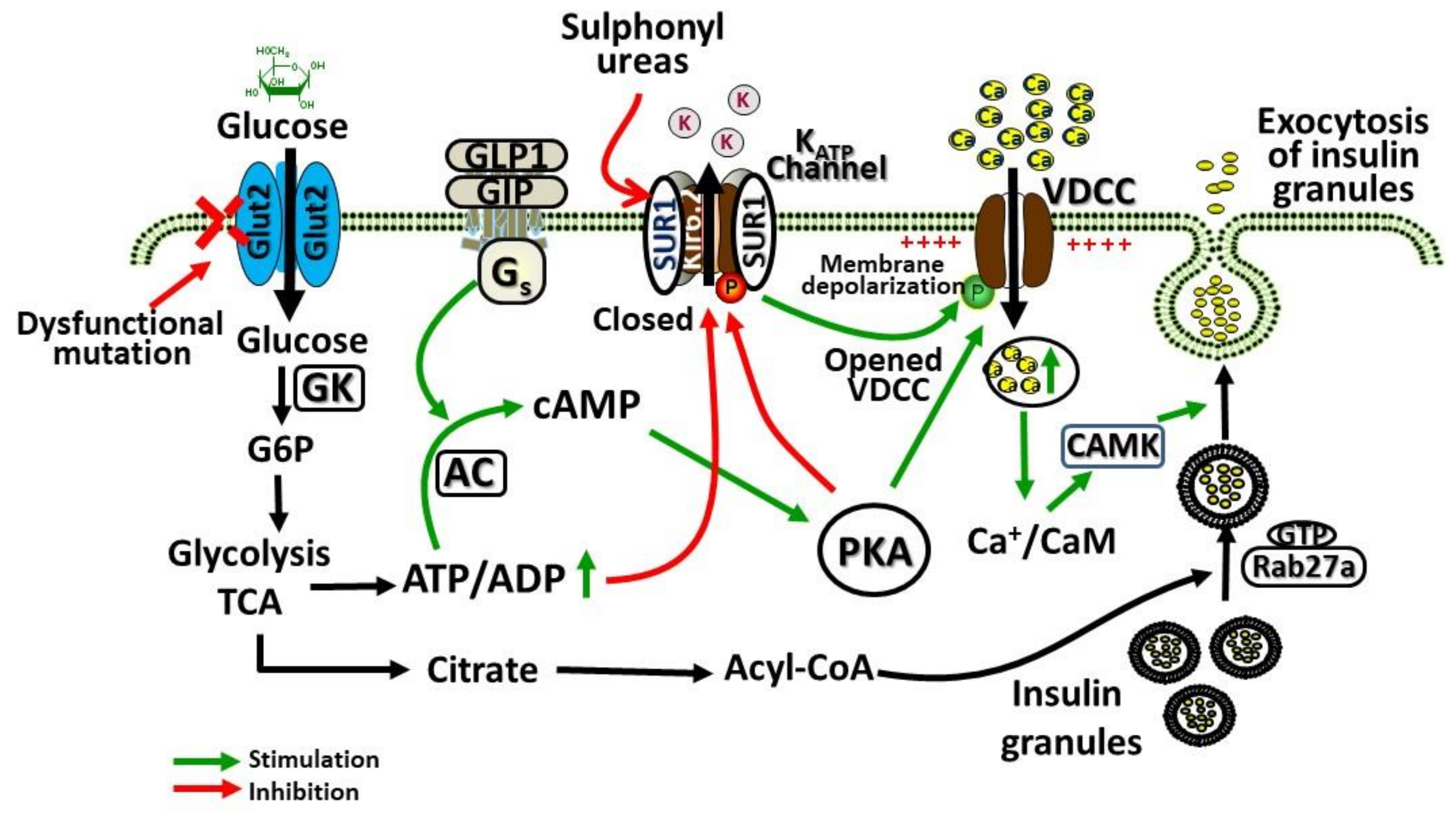
GLUT2 is a high KM (15–20 mM) and low affinity facilitative transporter of glucose allowing the glucose transport in proportion to the circulating blood glucose levels. This ensures the sensing and appropriate responses by the liver and pancreas for controlling the normal blood glucose levels [32]. GLUT2 is presumed to play a key role in regulating the molecular mechanisms of glucose-stimulated secretion of insulin. This molecular mechanism involves the transport of glucose through GLUT2 in β-cells, phosphorylation of glucose by glucokinase leading to increased glycolysis and production of ATP. An increased ratio of ATP/ADP leads to the potassium channel (KATP) closure and inhibition of potassium efflux, leading to β-cell membrane depolarization, opening of the calcium channel, facilitating calcium entry inside the cell, and causing insulin secretion. Figure 1 shows the regulation of pancreatic β cell glucose-stimulated insulin secretion by GLUT2.
2.3. Role of GLUT2 in Liver
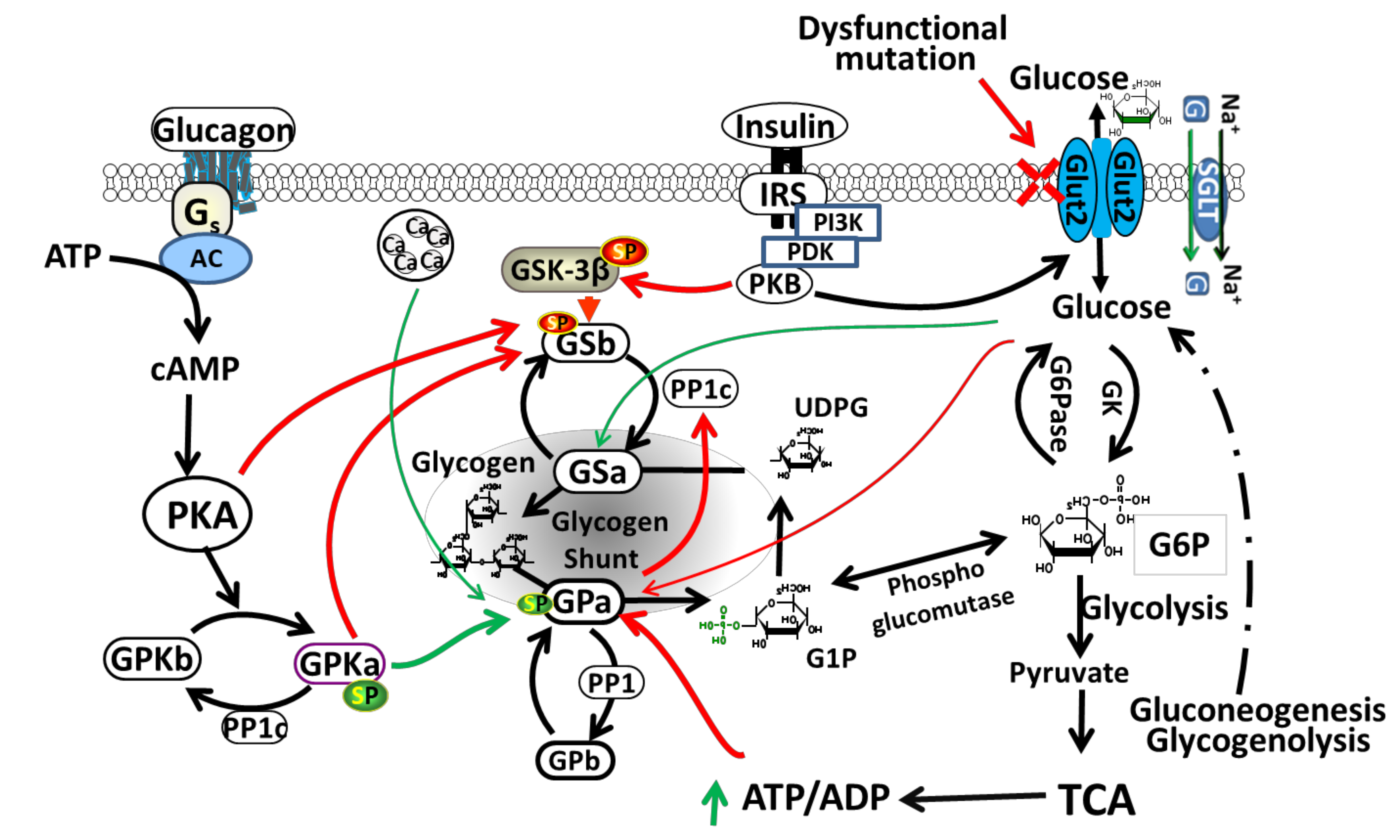
Liver GLUT2 plays an important role in homeostasis of glucose by regulating glucose uptake and release, depending on a fasting or feeding state [24,33]. In rodents and humans, GLUT2 is considered the major transporter of glucose in liver [11,32,33]. It is located in the basolateral (sinusoidal) plasma membrane domain of hepatocytes [18]. GLUT2 in the liver regulates glucose release (produced either from gluconeogenesis or glycogenolysis) during the fasting state and glucose uptake during the feeding state leading to glycogen synthesis in the liver [24,34]. Glycogen synthesis occurs through glycogen synthase (GS) (glycogen synthesis key enzyme) activation by allosteric stimulator phosphorylated glucose (glucose-6-phosphate) and through inactivation of glycogen synthase kinase-3 (GSK-3) with insulin, however glycogenolysis is inhibited by activation of glycogen phosphorylase kinase [35]. Figure 2 shows the regulation of glycogen metabolism in liver by GLUT2. During hyperglycemia, GLUT2 facilitates glucose transport across the cell membranes leading to activation of glycogen synthesis pathway. However, in hypoglycemia, glycogen phosphorylase (GP) catalyzes glycogenolysis, activated by AMP or protein kinase A (PKA).
The inactivation of GLUT2 in adult mice hepatocytes provides some interesting information about the role of GLUT2 in the physiology of glucose metabolism [36]. In these mice, the liver size is 40% larger due to increased glycogen storage, and fasting did not reverse the phenotype [37]. The loss of GLUT2 leads to reduced hepatic glucose uptake without any change in glucose output in these mice with normal hepatic glucose production suggesting glucose being produced in hepatocytes may use an alternative pathway to be released independent of GLUT2 or any facilitative diffusion at the plasma membrane [38,39]. When these mice were fed, there was an elevated expression of lipogenic and glycolytic target genes and ChREBP (Carbohydrate-Response Element-Binding Protein), which regulates transcription of genes related to lipid and glucose metabolism. More interestingly, both GLUT2 hepatocyte knockout mice and control mice had normal expenditure of energy, feeding behavior, and sensitivity of insulin. There was an increased decline in glucose-stimulated insulin secretion even without any changes in the contents of insulin and β-cell mass in the GLUT2 knockout mice hepatocyte.
GLUT2 mRNA and protein expression in hepatocytes is known to be modulated by thyroid hormones. In crude liver membranes, GLUT2 protein expression was found to be significantly increased in chronically hyperthyroid compared to hypothyroid animals [40]. However, total GLUT2 protein was similarly expressed when assayed in detergent extracts of liver. These observations suggest that glycolysis and gluconeogenesis enzymes together with the GLUT2 glucose transporter expression and functions are target for hormonal regulation that control hepatic glucose metabolism. GLUT2 also forms a complex with the insulin receptor forming a receptor-transporter complex providing a direct pathway between insulin binding and glucose transport regulation [41]. Protein Tyrosine Phosphatase-1B (PTP-1B) deficient mice studies suggested that GLUT2 and insulin receptor complex plays a vital role and modulate glucose uptake in neonatal mice hepatocytes [42]. Given the key role of GLUT2 in the liver, it explains some of the abnormalities observed in glucose metabolism in FBS patients (fasting hypoglycemia, postprandial hyperglycemia, and glycogen storage).
2.4. Role of GLUT2 in Kidney
Three glucose transporters (sodium-glucose cotransporter 1 (SGLT1), SGLT2, and GLUT2) play important roles for reabsorption of glucose in the proximal tubule of the kidney from the glomerular filtrate. In the kidney, glucose transporters GLUT2 is localized in the basolateral membrane; SGLT1 (sodium−glucose cotransporter-1) and SGLT2 are localized in the apical membrane [43]. Bulk of the filtered glucose is reabsorbed by SGLT2, and SGLT1 plays a role as reserve capacity for the filtration of glucose. In the basolateral membrane, GLUT2 plays an essential role for completing the glucose reabsorption. In FBS patients, GLUT2 dysfunction causes glycogen accumulation in the proximal tubules of the kidney [44]. GLUT2 dysfunction in proximal renal tubules leads to glycosuria, metabolic acidosis, phosphaturia, hypercalciuria, generalized aminoaciduria, bicarbonaturia, hypophosphatemia, and nephropathy [8,45,46,47]. Recent reports suggest that the expression of GLUT2 is upregulated in the renal proximal tubules during diabetes [48].
2.5. Role of GLUT2 in Intestine
The rate of movement of the contents of stomach (gastric emptying) and the absorption of glucose in the intestine regulate the glucose appearance rate in the circulation. Enterocytes in the small intestine are polarized epithelial cells and play important roles in the absorption of nutrients and uptake of glucose using various transporters to the blood vessels. This is the primary mechanism for the transport of glucose and other nutrients from intestine enter into the blood circulation. Two glucose transporters are expressed on the enterocytes, namely SGLT1 (sodium glucose co-transporter 1) and GLUT [49]. The GLUT2 transporters are situated in the enterocyte basolateral membrane, while SGLT1 is located in the intestinal membrane brush border. SGLT1 transports luminal glucose across the basolateral membrane and into the enterocyte by an active process. Once inside the enterocytes, glucose is phosphorylated to glucose-6-phosphate (G6P) by hexokinase and accumulates, G6P is dephosphorylated by glucose-6-phospatase to glucose for transport to portal vein. Dephosphorylated glucose is then passively transported out of the enterocytes by GLUT2 in the basolateral membrane. In conditions of high glucose load in the intestine endosomal GLUT2 can be rapidly translocated to the apical membrane to facilitate increased glucose absorption [34]. Interestingly, glucose absorption from the rat intestine can be enhanced by artificial sweeteners due to increased expression of apical GLUT2 [50]. Intestinal GLUT2 is a facilitative glucose transporter and also has the ability to transport galactose, mannose, glucosamine, and fructose [51].
2.6. Role of GLUT2 in Brain
Glucose is transported across the blood brain barrier facilitated by glucose transporters (GLUTs), since neurons in the brain are not able to store or synthesize glucose. GLUT1 and GLUT3 are the main glucose transporters in the brain. GLUT1 is highly expressed in the blood brain barrier endothelial cells and transports glucose to the brain extracellular space from the blood. GLUT3 is the main glucose transporter into the neuronal cells from the extracellular space, and in astrocytes GLUT2 is main glucose transporter. Apart from transporting glucose into the astrocytes, GLUT2 plays a role as a glucose sensor in the brain. In GLUT2 knockout mice, studies have shown that GLUT2 elimination leads to suppression of glucose sensing in the brain and this impacts feeding behavior probably by regulating the hypothalamic melanocortin pathways [52]. GLUT2, via the autonomic nervous system also links brain glucose sensing with pancreatic β-cell mass and function. Nervous system GLUT2 inactivation in mice leads to the late onset glucose intolerance development with reduced secretion of insulin due to reduced β-cell mass and proliferation [53]. Both parasympathetic and sympathetic activity were reduced and not affected by glucose. Thus GLUT2 plays an important role in linking the endocrine pancreas with the central nervous system.
3. SLC2A2 (GLUT2) Mutations and Patterns of Dysglycaemia
From 1987 to the present, 144 clinical reports of FBS have been published. All the FBS published cases have been summarized in Table 1, including those with and without mutations in the SLC2A2 gene. Table 2 and Figure 3 list all of the different types of mutations that are reported in the SLC2A2 gene. These mutations included missense, nonsense, fs/indel, intronic and compound heterozygous mutations. We analyzed the pattern and severity of dysglycaemia associated with different types of SLC2A2 gene mutations. The patterns of dysglycaemia and the mutations are shown in Table 3. In this table, we only included those cases where there was a clear clinical description of the patient having blood glucose measurements to document either fasting hypoglycemia, post-prandial hyperglycemia, glucose intolerance or frank diabetes. The pattern of dysglycaemia ranged from fasting hypoglycemia, post-prandial hyperglycemia, glucose intolerance, transient neonatal diabetes to gestational diabetes and frank diabetes mellitus. There was no correlation between the mutation type and the pattern of dysglycaemia. We also analyzed the birth weight of the FBS patients with GLUT2 mutations and dysglycaemia, and these are shown in Table 4. Virtually all patients with GLUT2 mutations had low birth weights.
3.1. Potential Biochemical Mechanisms Leading to Dysglycaemia in Patients with FBS
Dysglycaemia is defined as any abnormality in the level of blood glucose including hypoglycemia or hyperglycemia. Patients with FBS show several different patterns in the blood glucose levels, including fasting hypoglycemia, post-prandial hyperglycemia, impaired glucose tolerance, transient neonatal diabetes, and very rarely late onset diabetes mellitus. The underlying biochemical and molecular mechanisms that lead to these blood glucose profiles are not well understood. Homozygous GLUT2 knockout mice demonstrate hyperglycemia with relative hypoinsulinism, develop frank diabetes mellitus and die two to three weeks after birth [105]. These mice lack the first-phase secretion of insulin which is pancreatic KATP channels dependent, but the second-phase secretion of insulin is preserved which is dependent on amplifying pathways [105]. During hyperglycemia, pancreatic β-cells are exposed to higher glucose concentrations which amplifies the glucose enhanced signal of insulin secretion through exocytosis of insulin granules in a time-dependent manner. This observation indicated that GLUT2 is required for the first phase of insulin secretion. In addition, the mice have elevated serum glucagon levels and ketone bodies (β-hydroxybutyrate). At a histological level, GLUT2 deletion in these mice leads to the postnatal pancreatic islets developmental changes as shown by a change in pancreatic alpha- to β-cell ratio.
3.2. Birth Weight in FBS
In contrast to homozygous GLUT2 knockout mice, frank diabetes mellitus is rare in FBS patients. Sansbury et al. [74,106] first reported transient neonatal diabetes mellitus (TNDM) in four FBS patients due to homozygous SLC2A2 mutation with loss of function which resolved at a median 18 months of age, and fifth patient was still on the treatment of insulin at the age of 28 months. All five of these patients had low birth weight which suggests a defect in β-cell insulin secretion in the last trimester of pregnancy, as insulin is a growth factor. The low birth weight is an interesting observation as we found that virtually all FBS patients are born with low birth weights (Table 4). Two of the reported patients by Sansbury et al. had a relatively low C-peptide level at the time of diagnosis, again suggesting defects in β-cell function. All patients needed treatment with subcutaneous insulin to control their diabetes mellitus.
3.3. Neonatal Diabetes in FBS
Since the initial report of transient neonatal diabetes by Sansbury et al. [74,106], two more cases have been described with homozygous SLC2A2 gene mutations. Yoo et al. (2002) reported a newborn with transient neonatal diabetes and galactosemia with a novel SLC2A2 homozygous gene mutation [83], and Khandelwal et al. (2018) reported phenotypic heterogeneity in siblings with one of the siblings having transient neonatal diabetes [55]. The findings of transient neonatal diabetes in some cases of FBS suggest an important role of GLUT2 in human β-cell physiology. The loss of function mutations in SLC2A2 presumably leads to reduced function or expression of GLUT2 in the pancreatic β-cell impairing insulin secretion, leading to hyperglycemia. After birth, there must be some compensatory mechanism(s) which allow the diabetes to remit, possibly involving GLUT1 or GLUT3. The intriguing fact is that not all cases of FBS patients develop transient neonatal diabetes mellitus and this suggests that there are other molecular mechanisms associated with the onset of the neonatal diabetes that may not be directly related to GLUT2. These studies suggest that in humans GLUT2 play important roles more likely in the neonatal and developmental period.
To the best of our knowledge there are no cases of FBS patients reported with neonatal diabetes where pancreatic histology has been studied at post-mortem, so it is not possible to say if there are any morphological changes as observed in the GLUT2 knockout mice. All these FBS patients with transient neonatal diabetes should be followed up in the long-term to see if the diabetes relapses (as occurs with other causes of transient neonatal diabetes). There is marked interindividual heterogeneity in the expression and regulation of SGLT2 (Sodium-glucose co-transporter-2) and this could account for some of the phenotypic differences observed between individuals even in the same family [107].
3.4. Frank Diabetes in FBS
Frank Diabetes is very rare in older patients with FBS. Seker-Yilmaz et al. [58] studied eight patients with FBS and found that only one had significant hypoglycemia, all patients had post-prandial hyperglycemia with two patients having blood glucose levels >200 mg/dL at 120 min after the oral glucose tolerance test (which by definition is diabetes) but their hemoglobin-A1c (HBA1c) value was normal (due to the low blood glucose level). Taha et al. (2008) [101] undertook the oral glucose testing (OGTT) in ten patients with FBS to evaluate the insulin and glucose levels in response to oral glucose load. They found that most but not all patients with FBS have impaired glucose tolerance/diabetes range hyperglycemia after the OGTT while maintaining normal HbA1c. Patients with FBS were also found to be relatively hypoinsulinemic. Ganesh R. et al. [102] evaluated twelve infants with infantile onset diabetes and found that one of the patients had FBS, and Pena et al. [78] have reported an adult female at the age of 31 years with FBS who developed gestational diabetes. In this case, it is unclear if the gestational diabetes is directly linked to the GLUT2 defect or is just a co-incidence. Interestingly one particular polymorphism in the GLUT2 gene (valine 197 to isoleucine) was found in a patient with gestational diabetes which showed a loss of function mutation of GLUT2 when tested in Xenopus oocytes [4].
In some of the previous studies, no associations were found between GLUT2 gene polymorphisms and diabetes mellitus [108,109]. More recent Genome Wide Associations studies (GWAS) have found associations between GLUT2 variants and fasting hyperglycemia, glucose intolerance and type 2 diabetes mellitus [110,111], but the underlying mechanisms linking variants in GLUT2 and abnormalities in glucose physiology are not clear from these GWAS studies. Santer et al. [89] followed up the original patient with FBS who was described in 1949 after 50 years and did not report overt diabetes in that patient. This is probably the longest follow up of any FBS patient and suggests that late frank diabetes is not a clinically recognized feature of FBS.
3.5. Glycogen Storage in FBS
The glycogen accumulation mechanism in FBS patients is presumably due to GLUT2 deficiency and defective glucose transport during glycogenolysis to the extracellular compartment. This causes an accumulation of intracellular glucose, leading to reduced glycogenolysis and increased glycogen storage. Impaired glucose export due to GLUT2 deficiency in hepatocytes causes fasting hypoglycemia when sources of peripheral glucose are diminished. The hypoglycemia is exacerbated by the glycosuria from the renal tubular leak. In the FBS patients due to nonfunctional GLUT2 mutation, defective uptake of monosaccharides and reduced glucose-stimulated secretion of insulin from pancreatic β-cells in the fed state causes hyperglycemia and hypergalactosemia [6]. FBS patients have relative hypoinsulinemia for the levels of blood glucose [2,101], as expected due to a defective secretion of first phase of insulin.
In addition, GLUT2 dysfunction in the liver can lead to hyperglycemia due to decreased response and sensitivity of hepatocytes to insulin signaling, causing a decreased inhibition of glucose production [112]. In the FBS patients, due to repeated episodes of hypoglycemia, glycated HbA1c (haemoglobin-A1c) levels are usually in the normal range. Hyperglycemia in the fed state and hypoglycemia in the fasting state appear to improve over time [101]. Glucose absorption is normal from the intestine of FBS patients suggesting that GLUT2 involvement in the facilitative glucose transport in the enterocytes must be involved at the basolateral membrane [113]. These observations are supported by studies in GLUT2 knockout mice which demonstrate the role of glucose transepithelial transport system in the mice intestine that requires phosphorylation of glucose [114]. Thus, dysglycaemia in FBS patients is not due to abnormalities in glucose absorption from the intestine. Figure 4 summarizes the expected pathophysiology of GLUT2 dysfunction based on the clinical profile of FBS: Postprandial hyperglycemia is due to an impairment of glucose transportation from the enterocytes and decreased glucose uptake by the liver due to impaired insulin secretion. Hypoglycaemia in the fasting state is due to the lack of renal glucose reabsorption from the kidney, as well as the accumulation of glucose in the liver leading to reduced glycogen breakdown.
3.6. Structure Function Relationship of GLUT2 in FBS
The structure-function relationships of several naturally occurring FBS SLC2A2 mutations and several engineered mutations and their effects on kinetic parameters, protein expression, differentiation of β-cell, and secretion of insulin has been examined using Xenopus oocytes, hepatocytes, and pancreatic β cells [115]. The naturally occurring FBS SLC2A2 mutations showed loss of transport function despite the similar to wild-type targeting of the protein on the plasma membrane and similar or reduced expression of protein. These functional changes in GLUT2 could be due to reduced capacity of glucose transport despite the proper targeting of the transport protein at the plasma membrane or due to loss of protein expression. These findings point to the vital role of GLUT2 in glucose transport. All the engineered mutants of GLUT2 are found to be localized at the plasma membrane and able to transport glucose, with modified kinetic properties and characteristics of glucose transport. In addition, they show increased β-cell differentiation and secretion of insulin. An important finding from three engineered GLUT2 transporter mutants show increased numbers of pancreatic β-cell, suggesting an important GLUT2 role in the development of pancreatic β-cells. These observations suggested a functional role for GLUT2 as an important glucose transporter and extracellular receptor, which have an effect on pancreatic β cell differentiation and secretion of insulin.
4. Conclusions and Future Perspective
A large number of the reported patients with FBS have dysglycaemia characterized by post-prandial hyperglycemia, fasting hypoglycemia, glucose intolerance, and rarely diabetes mellitus. The role of human GLUT2 in pancreatic β-cell physiology is still unclear, although the increasing number of cases of transient neonatal diabetes suggests that it might have a role in the newborn period. The low birth weights of virtually all the reported FBS cases suggests that GLUT2 might have a role in human fetal insulin physiology. Currently there are no drugs in clinical practice which target the underlying molecular defect in GLUT2 in patients with FBS and management of these patients is clinically challenging. Future GLUT2 functional studies in human pancreatic β-cells, liver, enterocytes, kidney, and brain cells, will be required for a better understanding of underlying molecular mechanisms of dysglycaemia in FBS that will facilitate the development of precision medicine based on the molecular mechanisms of GLUT2.
Author Contributions
Conceptualization: S.S., K.H., and F.A.K.; writing—original draft preparation: S.S., M.A.-A., and F.A.K.; writing—review and editing: K.H. and F.A.K.; All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the support of Hamad Bin Khalifa University (Doha, Qatar) for funding the article processing fee for this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GLUT2 | Glucose transporter 2 |
| cAMP | Cyclic adenosine monophosphate |
| FBS | Fanconi–Bickel Syndrome |
| HbA1c | Haemoglobin-A1c |
| GP | Glycogen phosphorylase |
| PKA | Protein kinase A |
| G6P | Glucose-6-phosphate |
| GK | Glucokinase |
| G | Glucose |
| NR | Not Reported |
| aa | amino acid |
References
- Fanconi, G.; Bickel, H. [Chronic aminoaciduria (amino acid diabetes or nephrotic-glucosuric dwarfism) in glycogen storage and cystine disease]. Helv. Paediatr. Acta 1949, 4, 359–396. [Google Scholar] [PubMed]
- Manz, F.; Bickel, H.; Brodehl, J.; Feist, D.; Gellissen, K.; Gescholl-Bauer, B.; Gilli, G.; Harms, E.; Helwig, H.; Nutzenadel, W.; et al. Fanconi–Bickel syndrome. Pediatr. Nephrol. 1987, 1, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Permutt, M.A.; Kornyi, L.; Keller, K.; Lacy, P.E.; Scharp, D.W.; Mueckler, M. Cloning and functional expression of a human pancreatic islet glucose-transporter cDNA. Proc. Natl. Acad. Sci. USA 1989, 86, 8688–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueckler, M.; Kruse, M.; Strube, M.; Riggs, A.C.; Chiu, K.C.; Permutt, M.A. A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity. J. Biol. Chem. 1994, 269, 17765–17767. [Google Scholar]
- Tanizawa, Y.; Riggs, A.C.; Chiu, K.C.; Janssen, R.C.; Bell, D.S.; Go, R.P.; Roseman, J.M.; Acton, R.T.; Permutt, M.A. Variability of the pancreatic islet beta cell/liver (GLUT 2) glucose transporter gene in NIDDM patients. Diabetologia 1994, 37, 420–427. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi–Bickel syndrome. Nat. Genet. 1997, 17, 324–326. [Google Scholar] [CrossRef]
- Mohandas Nair, K.; Sakamoto, O.; Jagadeesh, S.; Nampoothiri, S. Fanconi–Bickel syndrome. Indian J. Pediatr. 2012, 79, 112–114. [Google Scholar] [CrossRef]
- Al-Haggar, M. Fanconi–Bickel syndrome as an example of marked allelic heterogeneity. World J. Nephrol. 2012, 1, 63–68. [Google Scholar] [CrossRef]
- Batool, H.; Zubaida, B.; Hashmi, M.A.; Naeem, M. Genetic testing of two Pakistani patients affected with rare autosomal recessive Fanconi–Bickel syndrome and identification of a novel SLC2A2 splice site variant. J. Pediatr. Endocrinol. Metab. 2019, 32, 1229–1233. [Google Scholar] [CrossRef]
- Kehar, M.; Bijarnia, S.; Ellard, S.; Houghton, J.; Saxena, R.; Verma, I.C.; Wadhwa, N. Fanconi–Bickel syndrome–mutation in SLC2A2 gene. Indian J. Pediatr. 2014, 81, 1237–1239. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Eddy, R.L.; Fukushima, Y.; Byers, M.G.; Shows, T.B.; Bell, G.I. Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc. Natl. Acad. Sci. USA 1988, 85, 5434–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, J.; Kayano, T.; Fukomoto, H.; Bell, G.I. Organization of the human GLUT2 (pancreatic beta-cell and hepatocyte) glucose transporter gene. Diabetes 1993, 42, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fritz, J.D.; Powers, A.C. Different functional domains of GLUT2 glucose transporter are required for glucose affinity and substrate specificity. Endocrinology 1998, 139, 4205–4212. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J.; Benito, C.; Gomis, R. Pancreatic islet GLUT2 glucose transporter mRNA and protein expression in humans with and without NIDDM. Diabetes 1995, 44, 1369–1374. [Google Scholar] [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A major pathway of intestinal sugar absorption. Diabetes 2005, 54, 3056–3062. [Google Scholar] [CrossRef] [Green Version]
- Freitas, H.S.; Schaan, B.D.; Seraphim, P.M.; Nunes, M.T.; Machado, U.F. Acute and short-term insulin-induced molecular adaptations of GLUT2 gene expression in the renal cortex of diabetic rats. Mol. Cell Endocrinol. 2005, 237, 49–57. [Google Scholar] [CrossRef]
- Thorens, B.; Cheng, Z.Q.; Brown, D.; Lodish, H.F. Liver glucose transporter: A basolateral protein in hepatocytes and intestine and kidney cells. Am. J. Physiol. 1990, 259, C279–C285. [Google Scholar] [CrossRef]
- Leturque, A.; Brot-Laroche, E.; Le Gall, M.; Stolarczyk, E.; Tobin, V. The role of GLUT2 in dietary sugar handling. J. Physiol. Biochem. 2005, 61, 529–537. [Google Scholar] [CrossRef]
- Mounien, L.; Marty, N.; Tarussio, D.; Metref, S.; Genoux, D.; Preitner, F.; Foretz, M.; Thorens, B. Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J. 2010, 24, 1747–1758. [Google Scholar] [CrossRef]
- Garcia, M.; Millan, C.; Balmaceda-Aguilera, C.; Castro, T.; Pastor, P.; Montecinos, H.; Reinicke, K.; Zuniga, F.; Vera, J.C.; Onate, S.A.; et al. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J. Neurochem. 2003, 86, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Molecular and cellular physiology of GLUT-2, a high-Km facilitated diffusion glucose transporter. Int. Rev. Cytol. 1992, 137, 209–238. [Google Scholar] [PubMed]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflug. Arch. 2020, 1–24. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Johnson, J.H.; Newgard, C.B.; Milburn, J.L.; Lodish, H.F.; Thorens, B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J. Biol. Chem. 1990, 265, 6548–6551. [Google Scholar]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Investig. 1995, 96, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.C.; Hussain, K.; Jones, P.M.; Persaud, S.; Löbner, K.; Boehm, A.; Clark, A.; Christie, M.R. Low levels of glucose transporters and K+ATP channels in human pancreatic beta cells early in development. Diabetologia 2007, 50, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Mally, M.I.; Otonkoski, T.; Lopez, A.D.; Hayek, A. Developmental gene expression in the human fetal pancreas. Pediatr. Res. 1994, 36, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Guillemain, G.; Loizeau, M.; Pinçon-Raymond, M.; Girard, J.; Leturque, A. The large intracytoplasmic loop of the glucose transporter GLUT2 is involved in glucose signaling in hepatic cells. J. Cell Sci. 2000, 113 Pt 5, 841–847. [Google Scholar]
- Stolarczyk, E.; Le Gall, M.; Even, P.; Houllier, A.; Serradas, P.; Brot-Laroche, E.; Leturque, A. Loss of sugar detection by GLUT2 affects glucose homeostasis in mice. PLoS ONE 2007, 2, e1288. [Google Scholar] [CrossRef] [Green Version]
- Gould, G.W.; Thomas, H.M.; Jess, T.J.; Bell, G.I. Expression of human glucose transporters in Xenopus oocytes: Kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry 1991, 30, 5139–5145. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef]
- Leturque, A.; Brot-Laroche, E.; Le Gall, M. GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E985–E992. [Google Scholar] [CrossRef] [PubMed]
- Carabaza, A.; Ciudad, C.J.; Baque, S.; Guinovart, J.J. Glucose has to be phosphorylated to activate glycogen synthase, but not to inactivate glycogen phosphorylase in hepatocytes. FEBS Lett. 1992, 296, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Seyer, P.; Vallois, D.; Poitry-Yamate, C.; Schütz, F.; Metref, S.; Tarussio, D.; Maechler, P.; Staels, B.; Lanz, B.; Grueter, R.; et al. Hepatic glucose sensing is required to preserve β cell glucose competence. J. Clin. Investig. 2013, 123, 1662–1676. [Google Scholar] [CrossRef] [Green Version]
- Burcelin, R.; del Carmen Muñoz, M.; Guillam, M.T.; Thorens, B. Liver hyperplasia and paradoxical regulation of glycogen metabolism and glucose-sensitive gene expression in GLUT2-null hepatocytes. Further evidence for the existence of a membrane-based glucose release pathway. J. Biol. Chem. 2000, 275, 10930–10936. [Google Scholar] [CrossRef] [Green Version]
- Guillam, M.T.; Burcelin, R.; Thorens, B. Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 12317–12321. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, M.; Thorens, B. Glucose release from GLUT2-null hepatocytes: Characterization of a major and a minor pathway. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E794–E801. [Google Scholar] [CrossRef]
- Weinstein, S.P.; O’Boyle, E.; Fisher, M.; Haber, R.S. Regulation of GLUT2 glucose transporter expression in liver by thyroid hormone: Evidence for hormonal regulation of the hepatic glucose transport system. Endocrinology 1994, 135, 649–654. [Google Scholar] [CrossRef]
- Eisenberg, M.L.; Maker, A.V.; Slezak, L.A.; Nathan, J.D.; Sritharan, K.C.; Jena, B.P.; Geibel, J.P.; Andersen, D.K. Insulin receptor (IR) and glucose transporter 2 (GLUT2) proteins form a complex on the rat hepatocyte membrane. Cell Physiol. Biochem. 2005, 15, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Nevado, C.; Escriva, F.; Sesti, G.; Rondinone, C.M.; Benito, M.; Valverde, A.M. PTP1B deficiency increases glucose uptake in neonatal hepatocytes: Involvement of IRA/GLUT2 complexes. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 295, G338–G347. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.A.; Forbes, J.M. Glucose and glycogen in the diabetic kidney: Heroes or villains? EBioMedicine 2019, 47, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannstadt, M.; Magen, D.; Segawa, H.; Stanley, T.; Sharma, A.; Sasaki, S.; Bergwitz, C.; Mounien, L.; Boepple, P.; Thorens, B.; et al. Fanconi–Bickel syndrome and autosomal recessive proximal tubulopathy with hypercalciuria (ARPTH) are allelic variants caused by GLUT2 mutations. J. Clin. Endocrinol. Metab. 2012, 97, E1978–E1986. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Fanconi–Bickel syndrome--a congenital defect of the liver-type facilitative glucose transporter. SSIEM Award. Society for the Study of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 1998, 21, 191–194. [Google Scholar] [CrossRef]
- Santer, R.; Steinmann, B.; Schaub, J. Fanconi–Bickel syndrome--a congenital defect of facilitative glucose transport. Curr. Mol. Med. 2002, 2, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pflugers Arch. 2020, 1–26. [Google Scholar] [CrossRef]
- Kellett, G.L.; Brot-Laroche, E.; Mace, O.J.; Leturque, A. Sugar absorption in the intestine: The role of GLUT2. Annu. Rev. Nutr. 2008, 28, 35–54. [Google Scholar] [CrossRef]
- Mace, O.J.; Affleck, J.; Patel, N.; Kellett, G.L. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J. Physiol. 2007, 582, 379–392. [Google Scholar] [CrossRef]
- Uldry, M.; Ibberson, M.; Hosokawa, M.; Thorens, B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002, 524, 199–203. [Google Scholar] [CrossRef]
- Bady, I.; Marty, N.; Dallaporta, M.; Emery, M.; Gyger, J.; Tarussio, D.; Foretz, M.; Thorens, B. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 2006, 55, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Tarussio, D.; Metref, S.; Seyer, P.; Mounien, L.; Vallois, D.; Magnan, C.; Foretz, M.; Thorens, B. Nervous glucose sensing regulates postnatal β cell proliferation and glucose homeostasis. J. Clin. Investig. 2014, 124, 413–424. [Google Scholar] [CrossRef]
- Gözmen, S.K.; Çelik, K.; Çalkavur, S.; Serdaroğlu, E. A Novel Mutation in Fanconi Bickel Syndrome Diagnosed in the Neonatal Period. J. Pediatr. Res. 2019, 6, 155–157. [Google Scholar] [CrossRef]
- Khandelwal, P.; Sinha, A.; Jain, V.; Houghton, J.; Hari, P.; Bagga, A. Fanconi syndrome and neonatal diabetes: Phenotypic heterogeneity in patients with GLUT2 defects. CEN Case Rep. 2018, 7, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Pogoriler, J.; O’Neill, A.F.; Voss, S.D.; Shamberger, R.C.; Perez-Atayde, A.R. Hepatocellular Carcinoma in Fanconi–Bickel Syndrome. Pediatr. Dev. Pathol. 2018, 21, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Tastemel-Ozturk, T.; Bilginer-Gurbuz, B.; Teksam, O.; Sivri, S. A Fanconi–Bickel syndrome patient with a novel mutation and accompanying situs inversus totalis. Turk. J. Pediatr. 2017, 59, 693–695. [Google Scholar] [CrossRef]
- Seker-Yilmaz, B.; Kor, D.; Bulut, F.D.; Yuksel, B.; Karabay-Bayazit, A.; Topaloglu, A.K.; Ceylaner, G.; Onenli-Mungan, N. Impaired glucose tolerance in Fanconi–Bickel syndrome: Eight patients with two novel mutations. Turk. J. Pediatr. 2017, 59, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Shafeghati, Y.; Sarkheil, P.; Baghdadi, T.; Hadipour, F.; Hadipour, Z.; Noruzinia, M. Osteogenesis Imperfecta or Fanconi–Bickel Syndrome? (Report of a Very Rare Disease Due to New Mutation on GLUT2 Gene). Sarem J. Reprod. Med. 2017, 1, 73–76. [Google Scholar] [CrossRef] [Green Version]
- In, N.S.; Amaral., V.; Tzun., R.C.; Marques, J.S. Fanconi Syndrome: Report of 2 Cases. EC Pediatrics 2017, 4.6, 165–169. [Google Scholar]
- Amita, M.; Srivastava, P.; Mandal, K.; De, S.; Phadke, S.R. Fanconi–Bickel Syndrome: Another Novel Mutation in SLC2A2. Indian J. Pediatr. 2017, 84, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Ahmad, I.; Ahmed, A.; Ahangar, A.A. Fanconi Bickel Syndrome: A Rare Entity. J. Clin. Case Rep. 2016, 6, 7. [Google Scholar] [CrossRef]
- Garg, M.; Gupta, A.; Mathur, P.; Sharma, M.; Kumar, R.; Gupta, V.; Manjunath, M. A rare case of Glycogen storage disease type XI Fanconi–Bickel Syndrome. J. Pediatric Crit. Care 2016, 3, 66–68. [Google Scholar] [CrossRef]
- Shah, R.; Rao, S.; Parikh, R.; Sophia, T.; Khalid, H. Fanconi Bickel Syndrome with Hypercalciuria due to GLUT 2 Mutation. Indian Pediatr. 2016, 53, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Nambam, B.; Weinstein, D.A.; Shoemaker, L.R. Late Diagnosis of Fanconi–Bickel Syndrome: Challenges With the Diagnosis and Literature Review. J. Inborn Errors Metab. Screen. 2016, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dweikat, I.M.; Alawneh, I.S.; Bahar, S.F.; Sultan, M.I. Fanconi–Bickel syndrome in two Palestinian children: Marked phenotypic variability with identical mutation. BMC Res. Notes 2016, 9, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afroze, B.; Chen, M. Fanconi–Bickel Syndrome: Two Pakistani Patients Presenting with Hypophosphatemic Rickets. J. Pediatr. Genet 2016, 5, 161–166. [Google Scholar]
- Wang, W.; Wei, M.; Song, H.M.; Qiu, Z.Q.; Zhang, L.J.; Li, Z.; Tang, X.Y. SLC2A2 gene analysis in three Chinese children with Fanconi–Bickel syndrome. Zhongguo Dang Dai Er Ke Za Zhi 2015, 17, 362–366. [Google Scholar]
- Abbasi, F.; Azizi, F.; Javaheri, M.; Mosallanejad, A.; Ebrahim-Habibi, A.; Ghafouri-Fard, S. Segregation of a novel homozygous 6 nucleotide deletion in GLUT2 gene in a Fanconi–Bickel syndrome family. Gene 2015, 557, 103–105. [Google Scholar] [CrossRef]
- Mihout, F.; Devuyst, O.; Bensman, A.; Brocheriou, I.; Ridel, C.; Wagner, C.A.; Mohebbi, N.; Boffa, J.J.; Plaisier, E.; Ronco, P. Acute metabolic acidosis in a GLUT2-deficient patient with Fanconi–Bickel syndrome: New pathophysiology insights. Nephrol. Dial. Transpl. 2014, 29 (Suppl. 4), iv113–iv116. [Google Scholar] [CrossRef]
- Hadipour, F.; Sarkheil, P.; Noruzinia, M.; Hadipour, Z.; Baghdadi, T.; Shafeghati, Y. Fanconi–Bickel syndrome versus osteogenesis imperfeeta: An Iranian case with a novel mutation in glucose transporter 2 gene, and review of literature. Indian J. Hum. Genet 2013, 19, 84–86. [Google Scholar] [PubMed] [Green Version]
- Jahnavi, S.; Poovazhagi, V.; Mohan, V.; Bodhini, D.; Raghupathy, P.; Amutha, A.; Suresh Kumar, P.; Adhikari, P.; Shriraam, M.; Kaur, T.; et al. Clinical and molecular characterization of neonatal diabetes and monogenic syndromic diabetes in Asian Indian children. Clin. Genet 2013, 83, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Dayal, D.; Dekate, P.; Sharda, S.; Das, A.; Attri, S. An Indian girl with Fanconi–Bickel syndrome without SLC2A2 gene mutation. J. Pediatr. Genet 2013, 2, 109–112. [Google Scholar]
- Sansbury, F.H.; Flanagan, S.E.; Houghton, J.A.; Shuixian Shen, F.L.; Al-Senani, A.M.; Habeb, A.M.; Abdullah, M.; Kariminejad, A.; Ellard, S.; Hattersley, A.T. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 2012, 55, 2381–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoodeh, A.; Rabbani, A. Transient neonatal diabetes as a presentation of Fanconi- Bickel Syndrome. Acta Med. Iran 2012, 50, 836–838. [Google Scholar] [PubMed]
- Al-Haggar, M.; Sakamoto, O.; Shaltout, A.; Al-Hawari, A.; Wahba, Y.; Abdel-Hadi, D. Mutation analysis of the GLUT2 gene in three unrelated Egyptian families with Fanconi–Bickel syndrome: Revisited gene atlas for renumbering. Clin. Exp. Nephrol. 2012, 16, 604–610. [Google Scholar] [CrossRef]
- Al-Haggar, M.; Sakamoto, O.; Shaltout, A.; El-Hawary, A.; Wahba, Y.; Abdel-Hadi, D. Fanconi Bickel Syndrome: Novel Mutations in GLUT 2 Gene Causing a Distinguished Form of Renal Tubular Acidosis in Two Unrelated Egyptian Families. Case Rep. Nephrol. 2011, 2011, 754369. [Google Scholar] [CrossRef]
- Pena, L.; Charrow, J. Fanconi–Bickel syndrome: Report of life history and successful pregnancy in an affected patient. Am. J. Med. Genet. A 2011, 155a, 415–417. [Google Scholar] [CrossRef]
- Simsek, E.; Savas-Erdeve, S.; Sakamoto, O.; Doganci, T.; Dallar, Y. A novel mutation of the GLUT2 gene in a Turkish patient with Fanconi–Bickel syndrome. Turk. J. Pediatr. 2009, 51, 166–168. [Google Scholar]
- Hoffman, T.L.; Blanco, E.; Lane, A.; Galvin-Parton, P.; Gadi, I.; Santer, R.; DeLeon, D.; Stanley, C.; Wilson, T.A. Glucose metabolism and insulin secretion in a patient with ABCC8 mutation and Fanconi–Bickel syndrome caused by maternal isodisomy of chromosome 3. Clin. Genet. 2007, 71, 551–557. [Google Scholar] [CrossRef]
- Saltik-Temizel, I.N.; Coskun, T.; Yuce, A.; Kocak, N. Fanconi–Bickel syndrome in three Turkish patients with different homozygous mutations. Turk. J. Pediatr. 2005, 47, 167–169. [Google Scholar] [PubMed]
- Peduto, A.; Spada, M.; Alluto, A.; La Dolcetta, M.; Ponzone, A.; Santer, R. A novel mutation in the GLUT2 gene in a patient with Fanconi–Bickel syndrome detected by neonatal screening for galactosaemia. J. Inherit. Metab. Dis. 2004, 27, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.W.; Shin, Y.L.; Seo, E.J.; Kim, G.H. Identification of a novel mutation in the GLUT2 gene in a patient with Fanconi–Bickel syndrome presenting with neonatal diabetes mellitus and galactosaemia. Eur. J. Pediatr. 2002, 161, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Tamura, T.; Chinen, Y.; Ohta, T. A novel mutation (N32K) of GLUT2 gene in a Japanese patient with Fanconi–Bickel syndrome. Clin. Genet 2002, 62, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Kitasawa, E.; Ida, H.; Eto, Y.; Owada, M. A newly recognized missense mutation in the GLUT2 gene in a patient with Fanconi–Bickel syndrome. Eur. J. Pediatr. 2000, 159, 867. [Google Scholar] [CrossRef]
- Sakamoto, O.; Ogawa, E.; Ohura, T.; Igarashi, Y.; Matsubara, Y.; Narisawa, K.; Iinuma, K. Mutation analysis of the GLUT2 gene in patients with Fanconi–Bickel syndrome. Pediatr. Res. 2000, 48, 586–589. [Google Scholar] [CrossRef] [Green Version]
- Akagi, M.; Inui, K.; Nakajima, S.; Shima, M.; Nishigaki, T.; Muramatsu, T.; Kokubu, C.; Tsukamoto, H.; Sakai, N.; Okada, S. Mutation analysis of two Japanese patients with Fanconi–Bickel syndrome. J. Hum. Genet 2000, 45, 60–62. [Google Scholar] [CrossRef]
- Burwinkel, B.; Sanjad, S.A.; Al-Sabban, E.; Al-Abbad, A.; Kilimann, M.W. A mutation in GLUT2, not in phosphorylase kinase subunits, in hepato-renal glycogenosis with Fanconi syndrome and low phosphorylase kinase activity. Hum. Genet 1999, 105, 240–243. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Suter, D.; Schaub, J.; Steinmann, B. Fanconi–Bickel syndrome--the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature. Eur. J. Pediatr. 1998, 157, 783–797. [Google Scholar] [CrossRef]
- Müller, D.; Santer, R.; Krawinkel, M.; Christiansen, B.; Schaub, J. Fanconi–Bickel syndrome presenting in neonatal screening for galactosaemia. J. Inherit. Metab. Dis. 1997, 20, 607–608. [Google Scholar] [CrossRef]
- Lee, P.J.; Van’t Hoff, W.G.; Leonard, J.V. Catch-up growth in Fanconi–Bickel syndrome with uncooked cornstarch. J. Inherit. Metab. Dis. 1995, 18, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Karamizadeh, Z.; Saki, F.; Imanieh, M.H.; Zahmatkeshan, M.; Fardaee, M. A new mutation of Fanconi–Bickel syndrome with liver failure and pseudotumour cerebri. J. Genet. 2012, 91, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Schwab, K.O.; Pohl, M.; Sass, J.O.; Santer, R. Fanconi–Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol. Genet. Metab. 2012, 105, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Du, M.L.; Chen, H.S.; Chen, Q.L.; Yu, C.S.; Mal, H.M. Two cases of Fanconi–Bickel syndrome: First report from China with novel mutations of SLC2A2 gene. J. Pediatr. Endocrinol. Metab. 2011, 24, 749–753. [Google Scholar] [CrossRef]
- Roy, M.; Bose, K.; Paul, D.K.; Anand, P. Hypophosphatemic rickets: Presenting features of Fanconi–Bickel syndrome. Case Rep. Pathol. 2011, 2011, 314696. [Google Scholar] [CrossRef] [Green Version]
- Karande, S.; Kumbhare, N.; Kulkarni, M. Fanconi–Bickel syndrome. Indian Pediatr. 2007, 44, 223–225. [Google Scholar]
- Riva, S.; Ghisalberti, C.; Parini, R.; Furlan, F.; Bettinelli, A.; Somaschini, M. The Fanconi–Bickel syndrome: A case of neonatal onset. J. Perinatol. 2004, 24, 322–323. [Google Scholar] [CrossRef] [Green Version]
- Ozer, E.A.; Aksu, N.; Uclar, E.; Erdogan, H.; Bakiler, A.R.; Tsuda, M.; Kitasawa, E.; Coker, M.; Ozer, E. No mutation in the SLC2A2 ( GLUT2) gene in a Turkish infant with Fanconi–Bickel syndrome. Pediatr. Nephrol. 2003, 18, 397–398. [Google Scholar] [CrossRef]
- Santer, R.; Groth, S.; Kinner, M.; Dombrowski, A.; Berry, G.T.; Brodehl, J.; Leonard, J.V.; Moses, S.; Norgren, S.; Skovby, F.; et al. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi–Bickel syndrome. Hum. Genet. 2002, 110, 21–29. [Google Scholar] [CrossRef]
- Muraoka, A.; Hashiramoto, M.; Clark, A.E.; Edwards, L.C.; Sakura, H.; Kadowaki, T.; Holman, G.D.; Kasuga, M. Analysis of the structural features of the C-terminus of GLUT1 that are required for transport catalytic activity. Biochem. J. 1995, 311, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Taha, D.; Al-Harbi, N.; Al-Sabban, E. Hyperglycemia and hypoinsulinemia in patients with Fanconi–Bickel syndrome. J. Pediatr. Endocrinol. Metab. 2008, 21, 581–586. [Google Scholar] [PubMed]
- Ganesh, R.; Arvindkumar, R.; Vasanthi, T. Infantile-onset diabetes mellitus: A 1-year follow-up study. Clin. Pediatr. 2009, 48, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Baker, L.; Kaplan, F.S.; Witzleben, C.L. Diabetes-like renal glomerular disease in Fanconi–Bickel syndrome. Pediatr. Nephrol. 1995, 9, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Garty, R.; Cooper, M.; Tabachnik, E. The Fanconi syndrome associated with hepatic glycogenosis and abnormal metabolism of galactose. J. Pediatr. 1974, 85, 821–823. [Google Scholar] [CrossRef]
- Guillam, M.T.; Hummler, E.; Schaerer, E.; Yeh, J.I.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Deriaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Lemelman, M.B.; Letourneau, L.; Greeley, S.A.W. Neonatal Diabetes Mellitus: An Update on Diagnosis and Management. Clin. Perinatol. 2018, 45, 41–59. [Google Scholar] [CrossRef]
- Saponaro, C.; Mühlemann, M.; Acosta-Montalvo, A.; Piron, A.; Gmyr, V.; Delalleau, N.; Moerman, E.; Thévenet, J.; Pasquetti, G.; Coddeville, A.; et al. Interindividual Heterogeneity of SGLT2 Expression and Function in Human Pancreatic Islets. Diabetes 2020, 69, 902–914. [Google Scholar] [CrossRef]
- Matsutani, A.; Koranyi, L.; Cox, N.; Permutt, M.A. Polymorphisms of GLUT2 and GLUT4 genes. Use in evaluation of genetic susceptibility to NIDDM in blacks. Diabetes 1990, 39, 1534–1542. [Google Scholar] [CrossRef]
- Lesage, S.; Zouali, H.; Vionnet, N.; Philippi, A.; Velho, G.; Serradas, P.; Passa, P.; Demenais, F.; Froguel, P. Genetic analyses of glucose transporter genes in French non-insulin-dependent diabetic families. Diabetes Metab. 1997, 23, 137–142. [Google Scholar]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet 2010, 42, 105–116. [Google Scholar] [CrossRef]
- Laukkanen, O.; Lindström, J.; Eriksson, J.; Valle, T.T.; Hämäläinen, H.; Ilanne-Parikka, P.; Keinänen-Kiukaanniemi, S.; Tuomilehto, J.; Uusitupa, M.; Laakso, M. Polymorphisms in the SLC2A2 (GLUT2) gene are associated with the conversion from impaired glucose tolerance to type 2 diabetes: The Finnish Diabetes Prevention Study. Diabetes 2005, 54, 2256–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemachandra, A.H.; Richard, M.C. Neonatal Hyperglycemia. Pediatrics Rev. 1999, 20, e16–e24. [Google Scholar] [CrossRef]
- Santer, R.; Hillebrand, G.; Steinmann, B.; Schaub, J. Intestinal glucose transport: Evidence for a membrane traffic-based pathway in humans. Gastroenterology 2003, 124, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Stümpel, F.; Burcelin, R.; Jungermann, K.; Thorens, B. Normal kinetics of intestinal glucose absorption in the absence of GLUT2: Evidence for a transport pathway requiring glucose phosphorylation and transfer into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 11330–11335. [Google Scholar] [CrossRef] [Green Version]
- Michau, A.; Guillemain, G.; Grosfeld, A.; Vuillaumier-Barrot, S.; Grand, T.; Keck, M.; L’Hoste, S.; Chateau, D.; Serradas, P.; Teulon, J.; et al. Mutations in SLC2A2 gene reveal hGLUT2 function in pancreatic beta cell development. J. Biol. Chem. 2013, 288, 31080–31092. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Role of glucose transporter 2 (GLUT2) in pancreatic β cell glucose-stimulated insulin secretion. Glucose enters the β-cells facilitated by GLUT2, which is then phosphorylated by glucokinase (GK) and metabolized by glycolysis and tricarboxylic acid cycle (TCA), leading to increased generation of ATP. Glucagon like peptide-1 (GLP1), glucagon, and GIP (gastric inhibitory polypeptide) activation of G protein-coupled receptors (GPCRs) responsive adenylyl cyclases (AC), leading to increased formation of cyclic adenosine monophosphate (cAMP). cAMP activates protein kinase A (PKA) and epac2 (cAMP-regulated guanine nucleotide exchange factor) to potentiate glucose-stimulated secretion of insulin. Increased ratio of ATP/ADP due to increased glucose metabolism closes the ATP-gated K+ channel (KATP), causing membrane depolarization, and voltage-dependent calcium channels (VDCC) opening, leading to Ca2+ influx via VDCC, which activates insulin secretion. This action of Ca2+ is potentiated by PKA. For exocytosis of insulin from β-cells, Rab3a and Rab27a GTPases are important signaling mediators that become activated by this mechanism and contribute to the exocytosis of insulin from glucose-stimulated cells.
Figure 1.
Role of glucose transporter 2 (GLUT2) in pancreatic β cell glucose-stimulated insulin secretion. Glucose enters the β-cells facilitated by GLUT2, which is then phosphorylated by glucokinase (GK) and metabolized by glycolysis and tricarboxylic acid cycle (TCA), leading to increased generation of ATP. Glucagon like peptide-1 (GLP1), glucagon, and GIP (gastric inhibitory polypeptide) activation of G protein-coupled receptors (GPCRs) responsive adenylyl cyclases (AC), leading to increased formation of cyclic adenosine monophosphate (cAMP). cAMP activates protein kinase A (PKA) and epac2 (cAMP-regulated guanine nucleotide exchange factor) to potentiate glucose-stimulated secretion of insulin. Increased ratio of ATP/ADP due to increased glucose metabolism closes the ATP-gated K+ channel (KATP), causing membrane depolarization, and voltage-dependent calcium channels (VDCC) opening, leading to Ca2+ influx via VDCC, which activates insulin secretion. This action of Ca2+ is potentiated by PKA. For exocytosis of insulin from β-cells, Rab3a and Rab27a GTPases are important signaling mediators that become activated by this mechanism and contribute to the exocytosis of insulin from glucose-stimulated cells.

Figure 2.
Regulation of glycogen metabolism in liver by GLUT2. The insulin-independent GLUT2 facilitates glucose transport across the cell membranes. Sodium-glucose cotransporter (SGLT) transport activity can be regulated by protein kinases including PKA (protein kinase A) and PKC (protein kinase C). PKA activation positively regulates SGLT1 expression and activity. Increased glucose concentration causes conformational change and activation of glucokinase (GK), which phosphorylates glucose to glucose-6-phosphate (G6P) and serves as a substrate for glycogen synthesis or glycolysis. In hepatic glycogen metabolism, G6P serves as the central intermediate. During glycolysis, utilization of G6P provides energy in the form of pyruvate, NADH and ATP. Acetyl-CoA produced from pyruvate, enters the mitochondrial tricarboxylic acid cycle (TCA). During fasting, G6P serves as a substrate for synthesis of glucose during gluconeogenesis or glycogenolysis. Glycogen phosphorylase (GP) catalyze glycogenolysis, activated by AMP or PKA, and inhibited by insulin. GSK-3β (a serine/threonine kinase) is a downstream element of PI3K/AKT pathway, whose activity can be inhibited by protein kinase B (PKB)-mediated phosphorylation at Ser9 of GSK-3β. Glycogen synthase (GS) is phosphorylated and inactivated by GSK3. SGLT: sodium-glucose cotransporter; G: glucose. Green arrows (stimulation); Red arrows (inhibition); Green Sp (phosphorylated serine): activating phosphorylation; Red Sp: inhibiting phosphorylation. Dashed lines indicate many steps not listed in the gluconeogenesis and glycogenolysis pathways to glucose.
Figure 2.
Regulation of glycogen metabolism in liver by GLUT2. The insulin-independent GLUT2 facilitates glucose transport across the cell membranes. Sodium-glucose cotransporter (SGLT) transport activity can be regulated by protein kinases including PKA (protein kinase A) and PKC (protein kinase C). PKA activation positively regulates SGLT1 expression and activity. Increased glucose concentration causes conformational change and activation of glucokinase (GK), which phosphorylates glucose to glucose-6-phosphate (G6P) and serves as a substrate for glycogen synthesis or glycolysis. In hepatic glycogen metabolism, G6P serves as the central intermediate. During glycolysis, utilization of G6P provides energy in the form of pyruvate, NADH and ATP. Acetyl-CoA produced from pyruvate, enters the mitochondrial tricarboxylic acid cycle (TCA). During fasting, G6P serves as a substrate for synthesis of glucose during gluconeogenesis or glycogenolysis. Glycogen phosphorylase (GP) catalyze glycogenolysis, activated by AMP or PKA, and inhibited by insulin. GSK-3β (a serine/threonine kinase) is a downstream element of PI3K/AKT pathway, whose activity can be inhibited by protein kinase B (PKB)-mediated phosphorylation at Ser9 of GSK-3β. Glycogen synthase (GS) is phosphorylated and inactivated by GSK3. SGLT: sodium-glucose cotransporter; G: glucose. Green arrows (stimulation); Red arrows (inhibition); Green Sp (phosphorylated serine): activating phosphorylation; Red Sp: inhibiting phosphorylation. Dashed lines indicate many steps not listed in the gluconeogenesis and glycogenolysis pathways to glucose.

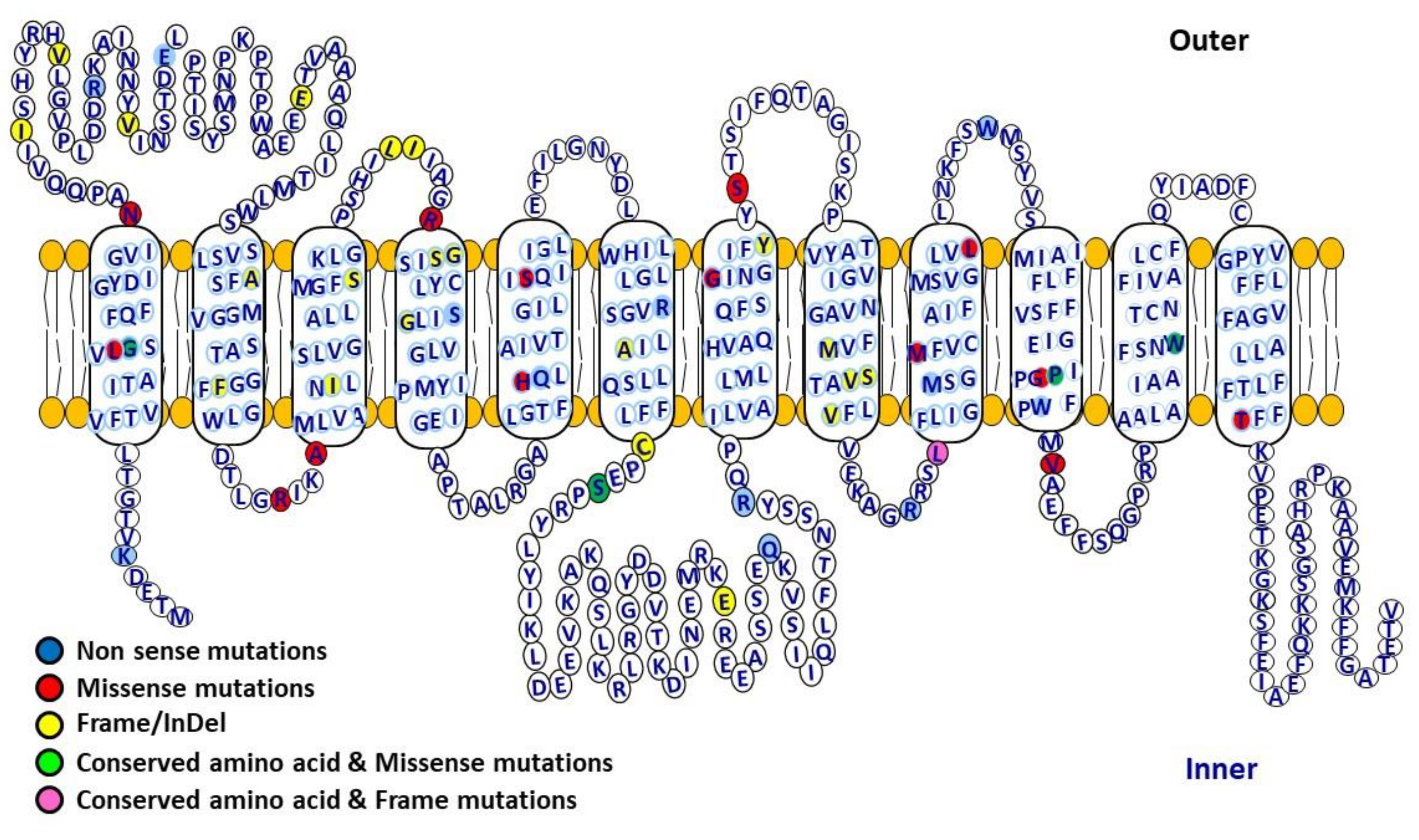
Figure 3.
Human glucose transporter 2 (GLUT2) structural topology including location of variants and conserved amino acids. GLUT2 is a glucose transporter containing 12 transmembrane domains (524 amino acids) connected with extracellular and intracellular loops, facilitating the movement of glucose across the cell membranes. It has high capacity but low affinity (high km) for glucose, and functions as a glucose sensor. GLUT2 mediates bidirectional glucose transport, and is mostly expressed in pancreatic β-cells, liver, small intestine, brain and renal tubular cells.
Figure 3.
Human glucose transporter 2 (GLUT2) structural topology including location of variants and conserved amino acids. GLUT2 is a glucose transporter containing 12 transmembrane domains (524 amino acids) connected with extracellular and intracellular loops, facilitating the movement of glucose across the cell membranes. It has high capacity but low affinity (high km) for glucose, and functions as a glucose sensor. GLUT2 mediates bidirectional glucose transport, and is mostly expressed in pancreatic β-cells, liver, small intestine, brain and renal tubular cells.

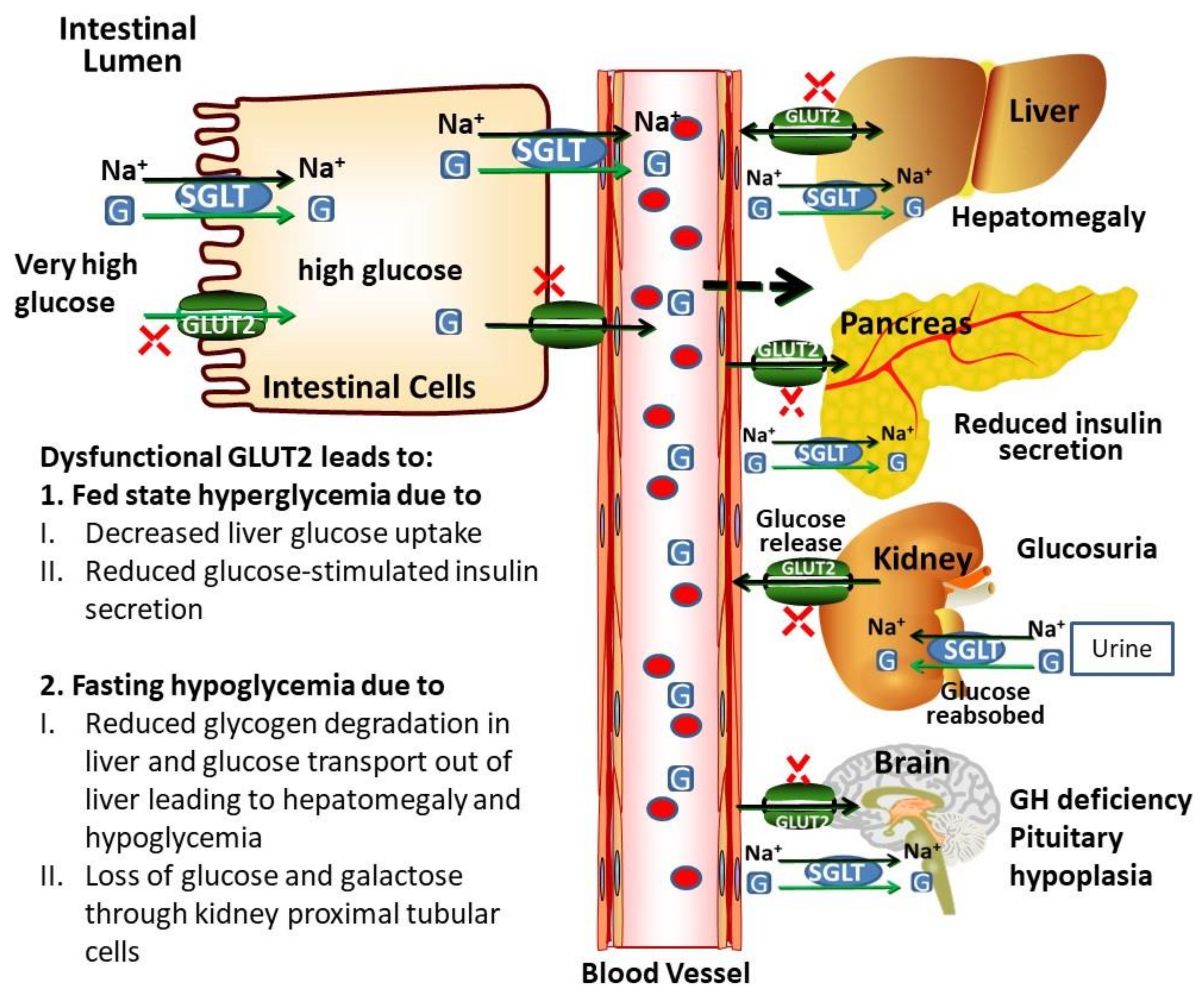
Figure 4.
GLUT2-mediated glucose transport across cell membranes and communication to different organs. GLUT2 is a low affinity and high capacity glucose transporter and facilitates the transport of glucose in intestinal cells. SGLTs are high-affinity and low capacity transporters, capable of transporting glucose against a concentration gradient. Ghrelin increases the GLUT2 expression by modulating the GLUT2 transcription via GHS-R1 (growth hormone secretagogue receptor 1a) and PLC/PKC pathway. Ghrelin also stimulates translocation of GLUT2 to the surfaces of intestinal cell from intracellular vesicles, leading to increased glucose absorption. GLUT2 is highly expressed in the liver, pancreas, brain and kidney cells. Activation of nervous signals, induces the first phase of insulin release from pancreatic β-cells induced by increased glucose transport to pancreatic β-cells. These signals also induce other physiological processes. In the liver, GLUT2 maintains the glucose homeostasis during fasting and fed state by regulating the expression of glucose-sensitive genes. GLUT2 is more highly expressed in rodent β-cells than in human β-cells. In the brain, glucose-sensing cells expressing GLUT2, regulate glucose by parasympathetic and sympathetic systems. GLUT2-dependent glucose-sensing cells also regulate leptin sensitivity and regulate the expressions of uncoupling protein 1 (UCP1) and thermogenesis. Homozygous GLUT2 mutation leads to a dysfunctional and reduced expression of GLUT2, which causes FBS. Dysfunctional GLUT2 causes fasting hypoglycemia, postprandial hyperglycemia, glucose and galactose intolerance, hepatomegaly, glucosuria, reduced GSIS (glucose-stimulated insulin secretion), rickets, GH (growth hormone) deficiency and poor, growth. (G; glucose).
Figure 4.
GLUT2-mediated glucose transport across cell membranes and communication to different organs. GLUT2 is a low affinity and high capacity glucose transporter and facilitates the transport of glucose in intestinal cells. SGLTs are high-affinity and low capacity transporters, capable of transporting glucose against a concentration gradient. Ghrelin increases the GLUT2 expression by modulating the GLUT2 transcription via GHS-R1 (growth hormone secretagogue receptor 1a) and PLC/PKC pathway. Ghrelin also stimulates translocation of GLUT2 to the surfaces of intestinal cell from intracellular vesicles, leading to increased glucose absorption. GLUT2 is highly expressed in the liver, pancreas, brain and kidney cells. Activation of nervous signals, induces the first phase of insulin release from pancreatic β-cells induced by increased glucose transport to pancreatic β-cells. These signals also induce other physiological processes. In the liver, GLUT2 maintains the glucose homeostasis during fasting and fed state by regulating the expression of glucose-sensitive genes. GLUT2 is more highly expressed in rodent β-cells than in human β-cells. In the brain, glucose-sensing cells expressing GLUT2, regulate glucose by parasympathetic and sympathetic systems. GLUT2-dependent glucose-sensing cells also regulate leptin sensitivity and regulate the expressions of uncoupling protein 1 (UCP1) and thermogenesis. Homozygous GLUT2 mutation leads to a dysfunctional and reduced expression of GLUT2, which causes FBS. Dysfunctional GLUT2 causes fasting hypoglycemia, postprandial hyperglycemia, glucose and galactose intolerance, hepatomegaly, glucosuria, reduced GSIS (glucose-stimulated insulin secretion), rickets, GH (growth hormone) deficiency and poor, growth. (G; glucose).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Fanconi–Bickel Syndrome (FBS) cases reported from 1987 to 2020.
| Patient Number | Reference | Sex | Origin | Mutation | Amino Acid Change |
|---|---|---|---|---|---|
| 1 | [9] | F | Pakistani | c.497-2A > T | p.(Gly166_Ser169del)] |
| 2 | [9] | M | Pakistani | c.497-2A > T | p.(Gly166_Ser169del)] |
| 3 | [54] | F | Turkish | IVS8g.24401-24406del6 | NR |
| 4 | [55] | F | India | c.952G>A | p.Gly318Arg |
| 5 | [55] | M | India | c.952G>A | p.Gly318Arg |
| 6 | [55] | F | India | c.952G>A | p.Gly318Arg |
| 7 | [56] | M | Kuwait | c.474A>C | p. Arg158Ser |
| 8 | [57] | M | Turkish | c.108+5G>A | NR |
| 9 | [58] | M | Turkish | c.1069delGinsAATAA | p. Val357AsnfsTer37 |
| 10 | [58] | F | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 11 | [58] | NR | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 12 | [58] | M | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 13 | [58] | NR | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 14 | [58] | M | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 15 | [58] | M | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 16 | [58] | M | Turkish | c.575A>G | p. His192Arg |
| 17 | [59] | M | Iran | C.685_701del GCCATCCTTCAGTCTCT ins CAGAAAA | p.A229 QfsX19 |
| 18 | [60] | M | NR | NR | E85fsX177 and G189D |
| 19 | [61] | F | Indian | c.1246G>A | p. Gly416Ser |
| 20 | [60] | M | NR | NR | NR |
| 21 | [62] | M | Kashmir | NR | NR |
| 22 | [63] | F | Indian | NR | NR |
| 23 | [64] | M | Indian? | c.16-1G>A or IVS 1-1G>A | NR |
| 24 | [65] | F | African American | c.670 | Cys224Del |
| 25 | [66] | F | Palestinian | c.901C>T | p. Arg301Ter |
| 26 | [66] | M | Palestinian | c.901C>T | p. Arg301Ter |
| 27 | [67] | F | Pakistani | c.339delC | p. Phe114LeufsTer16 |
| 28 | [67] | M | Pakistani | c.339delC | p. Phe114LeufsTer16 |
| 29 | [68] | NR | Chinese | c.380C>A and c.970dupT | p.Ala127Asp and p.324TyrfsX392 |
| 30 | [68] | NR | Chinese | c.1068+5G>C | IVS8+5G>C |
| 31 | [68] | NR | Chinese | c.1194T>A | p.Tyr398X |
| 32 | [69] | F | Iranian | c.1061_1066del6 | p.V355_S356del2 |
| 33 | [69] | M | Iranian | c.1061_1066del6 | p.V355_S356del2 |
| 34 | [70] | M | Algerian | IVS 3+2T>C/IVS 3+2T>C | NR |
| 35 | [10] | M | Indian | c.1330T>C | p. W444R |
| 36 | [71] | F | Iranian | c. 685_701del GCCATCCTTCAGTCTCTins CAGAAAA | P.A229QFsX19 |
| 37 | [72] | F | Indian | c.56T>C | p. Leu19Pro |
| 38 | [73] | F | Indian | NR | NR |
| 39 | [74] | F | Chinese | c.609T>A | p.Ser203Arg |
| 40 | [74] | M | Oman | c.1127T>G | p.Met376Arg |
| 41 | [74,75] | F | Iran | c.963+1G>A | NR |
| 42 | [74] | M | Sudanese | c.157C>T | p.Arg53X |
| 43 | [74] | M | Saudi Arabia | c.339del | p.Phe114LeufsX16 |
| 44 | [76] | F | Egyptian | c.1250C>T | p. P417L |
| 45 | [76] | M | Egyptian | c.253_254del GA | p. Glu85fs |
| 46 | [77] | M | Egyptian | c.776-1G>C | NR |
| 47 | [78] | F | Caucasian | c.1439C>G and c.1469delA | T480R and L490SfsX24 |
| 48 | [79] | M | Turkish | c.835_836delGA | p.E279KfsX6 |
| 49 | [80] | F | NR | c.1213C>T | NR |
| 50 | [81] | M | Turkish | 783del17 | NR |
| 51 | [81] | F | Turkish | c.818C>G | NR |
| 52 | [81] | F | Turkish | IVS5+1 G>T | NR |
| 53 | [82] | M | Italian | 425_7/delTAA | NR |
| 54 | [83] | F | Korean | c.322A>T | K5X |
| 55 | [84] | F | Japanese | c.96T>G | N32K |
| 56 | [85] | M | Japanese | c.1093 C>T and c.1642 T>C | p. Arg365Ter and p. Trp444Arg |
| 57 | [86] | F | Japanese | nt 1580T>A | V423E |
| 58 | [86] | M | Japanese | IVS2–2A>G | NR |
| 59 | [86] | M | Japanese | c.1171C>T and c.1478T>C | Q287X and L389P |
| 60 | [87] | M | Japanese | c.1159G>A | W420X |
| 61 | [87] | F | Japanese | NR | NR |
| 62 | [88] | M | Saudi-Arabian | c.1250 C>T | Pro417Leu |
| 63 | NR | Arabian | c.1250C>T | NR | |
| 64 | NR | Arabian | c.1250 C>T | NR | |
| 65 | NR | Arabian | c.1250 C>T | NR | |
| 66 | NR | Arabian | c.1250 C>T | NR | |
| 67 | NR | Arabian | c.1250 C>T | NR | |
| 68 | [6,89] | M | Swiss | c.1251C>T or 1213 C>T | R301X |
| 69 | [6] | F | NR | ΔT446-449 | |
| 70 | [6] | M | NR | ΔT446-449 | |
| 71 | [6] | M | NR | c.1405C>T | R365X |
| 72 | [90] | M | Japanese | c.1171C>T and c.1478 T>C | NR |
| 73 | [91] | M | Caucasian | NR | NR |
| 74 | [91] | M | Caucasian | NR | NR |
| 75 | [4] | NR | Japanese | c.1571G>A | NR |
| 76 | [2] | F | Arabian | c.1562C>T | NR |
| 77 | M | Arabian | c.1562C>T | NR | |
| 78 | M | Arabian | c.1562C>T | NR | |
| 79 | NR | Arabian | c.1562C>T | NR | |
| 80 | NR | Arabian | c.1562C>T | NR | |
| 81 | NR | Arabian | c.1562C>T | NR | |
| 82 | NR | Arabian | c.1562C>T | NR | |
| 83 | NR | Arabian | c.1562C>T | NR | |
| 84 | NR | Arabian | c.1562C>T | NR | |
| 85 | [92] | M | Iran | IVS8+1 G>T | NR |
| 86 | [93] | M | Caucasian | c.457_462delCTTATA and c.1250C>G | p.153_4delLI and p.P417R |
| 87 | [93] | F | Caucasian | c.457_462delCTTATA and c.1250C>G | p.153_4delLI and p.P417R |
| 88 | [45] | M | Dominican Republic | IVS 4-2A>G | p. Gln166AspfsTer4 |
| 89 | [45] | M | Dominican Republic | IVS 4-2A>G | p. Gln166AspfsTer4 |
| 90 | [45] | F | Israeli | c.372A>C | p. Arg124Ser |
| 91 | [45] | M | Israeli | c.372A>C | p. Arg124Ser |
| 92 | [45] | F | Israeli | c.372A>C | p. Arg124Ser |
| 93 | [94] | F | Chinese Han | c.682C>T and c.1185 G>A | p. Arg228X and p. Trp395X |
| 94 | [94] | M | Chinese Han/Yao | c.196G>T and c.1117delA | p. Glu66X and p. Met373X |
| 95 | [95] | M | NR | NR | NR |
| 96 | [80] | F | NR | c.1213C>T | NR |
| 97 | [96] | F | Indian | NR | NR |
| 98 | [97] | M | Arab | c.1213C>T | p.Phe405>Leu |
| 99 | [98] | F | Turkish | NR | NR |
| 100 | [99] | F | German | NR | NR |
| 101 | M | German | 627 delAGTTGGTGins GT | NR | |
| 102 | M | Turkish-Assyrian | 793–4 ins C | NR | |
| 103 | F | Turkish-Assyrian | 793–4 ins C | NR | |
| 104 | M | German | NR | NR | |
| 105 | M | German | NR | NR | |
| 106 | M | Italian (?) | c.1213C>T | NR | |
| 107 | F | Italian (?) | c.889C>T | NR | |
| 108 | M | English | 1363 del G and 1405C>T | NR | |
| 109 | M | English | 1364 del G and 1405C>T | NR | |
| 110 | M | Caucasian | 1264 G>A and 469C>T | NR | |
| 111 | M | Turkish | 449 del T | NR | |
| 112 | F | Turkish | 450 del T | NR | |
| 113 | M | Turkish | c.1405C>T | NR | |
| 114 | M | Caucasian | c.1405C>T and 1008 ins A | NR | |
| 115 | M | Arabian | c.1213C>T | NR | |
| 116 | M | Polish (?) | NR | NR | |
| 117 | F | Polish (?) | c.469C>T | NR | |
| 118 | F | Polish (?) | NR | NR | |
| 119 | M | Algerian | IVS 6 +1 G>C | NR | |
| 120 | M | Moroccan | 1288–9TC>AA | NR | |
| 121 | F | NR | c.1562C>T and IVS 8 + | NR | |
| 122 | 1 G>A | NR | |||
| 123 | M | Algerian | IVS 5 +5 G>C | NR | |
| 124 | M | Algerian | IVS 5 +5 G>C | NR | |
| 125 | F | French (?) | 1573 ins GT and 1751C>G | NR | |
| 126 | M | French (?) | 1574 ins GT and 1751C>G | NR | |
| 127 | F | Italian | 1264G>A | NR | |
| 128 | F | Italian | 371G>A and 1751C>G | NR | |
| 129 | M | Turkish | 1562C>T | NR | |
| 130 | F | NR | IVS 6 +1 g>a | NR | |
| 131 | M | NR | NR | NR | |
| 132 | M | NR | NR | NR | |
| 133 | F | French-Canadian | 494 ins 26 and 1751C>G | NR | |
| 134 | F | Eskimo | 1415–6 del TC | NR | |
| 135 | M | Arabian | 1213C>T | NR | |
| 136 | F | NR | IVS 8 +1 G>A | NR | |
| 137 | F | NR | NR | NR | |
| 138 | F | Turkish-Assyrian | 793–4 ins C | NR | |
| 139 | F | Greek | 712–3 del CT | NR | |
| 140 | M | Algerian | IVS 3 +2 T>C | NR | |
| 141 | M | NR | 1092C>A | NR | |
| 142 | M | Turkish | 738 del 17 | NR | |
| 143 | F | Turkish | IVS 5 +1 G>T | NR | |
| 144 | [100] | M | Japanese | c.1405C>T and c.1642T>C | NR |
NR: not reported.
Table 2.
Summary of reported seventy different SLC2A2 mutations (Missense, Nonsense, fs/indel, Intronic, and Compound Heterozygous Mutations).
Table 2.
Summary of reported seventy different SLC2A2 mutations (Missense, Nonsense, fs/indel, Intronic, and Compound Heterozygous Mutations).
| Missense Mutations | Nonsense Mutations | fs/indel Mutations | Intronic Mutations | Compound Heterozygous Mutations |
|---|---|---|---|---|
| G20E | K5X | I39 | c.497-2A>T | p.Ala127Asp and p.324TyrfsX392 |
| N32K | R53X | L153_I154 | c.108+5G>A | p. Arg365Ter and p. Trp444Arg |
| R158S | E66X | C239 | IVS 3+2T>C | Q287X and L389P |
| S203R | S169X | V355_S356 | c.963+1G>A | Gly20Glu and T480R |
| S242R | Q193X | V45 | c.776-1G>C | T480R and L490SfsX24 |
| G318R | R228X | V60 | IVS5+1 G>T | p.153_4delLI and p.P417R |
| S326K | Q287X | A105 | IVS 2 - 2 A>G | E85fsX177 and G189D |
| M376R | R301X | I133 | IVS4+1G>A | p. Arg228X and p. Trp395X |
| L389P | R365X | S145 | IVS 8+1 G>T | p. Glu66X and p. Met373X |
| G416S | W420X | S161 | IVS 5-1 G>A | |
| P417L | M350 | c.16-1G>A or IVS 1-1G>A | ||
| P417R | L368 | IVS8g.24401-24406del6 | ||
| V423E | W420 | c.1068+5 G>C | ||
| W444R | Glu85fs | |||
| T480R | E279KfsX6 | |||
| His192Arg | Val357AsnfsTer37 | |||
| Arg124Ser | Gly162ArgfsTer17 | |||
| Leu19Pro ** | A229QFsX19 | |||
| p.Phe405>Leu | Phe114LeufsX16 | |||
| Gln166AspfsTer4 | ||||
| Cys224Del |
Table 3.
All of the SLC2A2 gene mutations where dysglycaemia associated with FBS has been reported.
| Type of Dysglycaemia | Mutation | Amino Acid Change | Reference |
|---|---|---|---|
| Transient neonatal diabetes | c.952G>A | p.Gly318Arg | [55] |
| c.609 T>A | p.Ser203Arg | [74] | |
| c.1127 T>G | p.Met376Arg | [74] | |
| c.963+1G>A | NR | [74] | |
| c.157C>T | p.Arg53X | [74] | |
| c.339del | p.Phe114LeufsX16 | [74] | |
| 322 A>T | K5X | [83] | |
| Glucose intolerance/diabetes mellitus | c.482_483insC | p. Gly162ArgfsTer17 | [58] |
| c.575A>G | p. His192Arg | [58] | |
| c.56 T>C | p. Leu19Pro ** | [72] | |
| Gestational diabetes | c.1439C>G and c.1469delA | T480R and L490SfsX24 | [78] |
| NR | valine 197 to isoleucine | [4] | |
| Other: 2 patients [101], and 1 patient [102] | |||
| Fasting hypoglycemia | c.108+5G>A | NR | [57] |
| 783del17 | NR | [81] | |
| 818C>G | NR | [81] | |
| IVS5+1 G>T | NR | [81] | |
| c.1580T>A | V423E | [86] | |
| IVS 2 - 2 A>G | NR | [86] | |
| c.952G>A | p.Gly318Arg | [55] | |
| NR | E85fsX177 and G189D | [60] | |
| NR | NR | [62] | |
| NR | NR | [91] | |
| NR | NR | [103] | |
| c.1246 G>A | p. Gly416Ser | [61] | |
| c.339delC | p. Phe114LeufsTer16 | [67] | |
| c.339delC | p. Phe114LeufsTer16 | [67] | |
| Post-prandial hyperglycemia | c.1061_1066del6 | p.V355_S356del2 | [69] |
| IVS8g.24401-24406del6 | NR | [54] | |
| NR | NR | [73] | |
| NR | NR | [96] | |
| Fasting hypoglycemia and postprandial hyperglycemia | c.1069delGinsAATAA | p. Val357AsnfsTer37 | [58] |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.901C > T | p. Arg301Ter | [66] | |
| c.16-1G>A or IVS 1-1G>A | [64] | ||
| c.380C>A and c.970dupT | p.Ala127Asp and p.324TyrfsX392 | [68] | |
| c.1068+5 G>C | IVS8+5G>C | [68] | |
| c.1194T>A | p.Tyr398X | [68] | |
| c.1250C>T | p. P417L | [76] | |
| IVS 3+2t>c/IVS 3+2t>c) | NR | [70] | |
| c. 685_70l del GCCATCCTTCAGTCTCTins CAGAAAA | P.A229QFsX19 | [71] | |
| c.253_254delGA | p. Glu85fs | [77] | |
| c.776-1G>C | NR | [77] | |
| c.835_836delGA | p.E279KfsX6 | [79] | |
| C1213T | NR | [80] | |
| 96T>G | N32K | [84] | |
| c.1171C>T and c.1478T>C | Q287X and L389P | [86] | |
| c.1213C>T | R301X | [89] | |
| ΔT446-449 | [6] | ||
| ΔT446-449 | [6] | ||
| C1405T | R365X | [6] | |
| c.1213 C>T | p.Phe405>Leu | [97] | |
| C.685_701del GCCATCCTTCAGTCTCT ins CAGAAAA | p.A229 QfsX19 | [59] | |
| c.670 | Cys224Del | [65] | |
| c.682C>T and c.1185 G>A | p. Arg228X and p. Trp395X | [94] | |
| c.196G>T and c.1117delA | p. Glu66X and p. Met373X | [94] | |
| c.1330 T>C | p. W444R | [10] |
NR: not reported.
Table 4.
Summary of different types of GLUT2 mutations associated with Fanconi–Bickel Syndrome and the Patient Birth Weights.
Table 4.
Summary of different types of GLUT2 mutations associated with Fanconi–Bickel Syndrome and the Patient Birth Weights.
| Mutation | Birth Weight (kg) | References | |
|---|---|---|---|
| DNA | Protein | ||
| Missense | |||
| c.609T>A | p.Ser203Arg | 1.85 | [74] |
| c.1127T>G | p.Met376Arg | 2.5 | [74] |
| c.952G>A | p.Gly318>Arg | 2.4 | [55] |
| c.952G>A | p.Gly318>Arg | 2.8 | [55] |
| c.952G>A | p.Gly318>Arg | 2.3 | [55] |
| c.1213C>T | p.Phe405>Leu | 3.23 | [97] |
| Non-sense | |||
| c.901C>T | p.Arg301X | 2.8 | [66] |
| c.901C>T | p.Arg301X | 2.2 | [66] |
| fs/indel | |||
| c.322A>T | p.Lys5>X | 2 | [83] |
| c.339delC | p.Phe114LeufsX16 | 2.5 | [74] |
| c. 685_701 del GCCATCCTTCAGTCTCT ins CAGAAAA | P.A229QFsX19 | 2.6 | [71] |
| c.1069delGinsAATAA | p. Val357AsnfsTer37 | 3 | [58] |
| C.685_701del GCCATCCTTCAGTCTCT ins CAGAAAA | p.A229 QfsX19 | 2.6 | [59] |
| c.783del17 | 2.6 | [81] | |
| c.670 | Cys224Del | 2.09 | [65] |
| Intronic | |||
| c.963+1G>A | NR | 2 | [74] |
| c.16-1G>A or IVS 1- 1G>A | NR | 2.5 | [64] |
| IVS8 g.24401-24406del6 | NR | 2.6 | [54] |
| c.963+1 G>A | NR | 2.05 | [75] |
| (IVS2+5G>A[c.108+5G>A]) | NR | 3.25 | [57] |
| Compound Heterozygous | |||
| c.457_462delCTTATA in Exon 4 and c.1250C>G in Exon 10 | (p.153_4delLI) and (p.Pro417Arg) | 3.773.97 | [93] |
| E85fsX177 and G189D | 3.0 | [60] | |
| Undefined | |||
| 2.8 | [62] | ||
| 2.5 | [63] | ||
| 2.9 | [73] | ||
| 2.1 | [96] | ||
| 2.5 | [95] | ||
| 2.8 | [104] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharari, S.; Abou-Alloul, M.; Hussain, K.; Ahmad Khan, F. Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. Int. J. Mol. Sci. 2020, 21, 6286. https://doi.org/10.3390/ijms21176286
AMA Style
Sharari S, Abou-Alloul M, Hussain K, Ahmad Khan F. Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. International Journal of Molecular Sciences. 2020; 21(17):6286. https://doi.org/10.3390/ijms21176286
Chicago/Turabian StyleSharari, Sanaa, Mohamad Abou-Alloul, Khalid Hussain, and Faiyaz Ahmad Khan. 2020. "Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia" International Journal of Molecular Sciences 21, no. 17: 6286. https://doi.org/10.3390/ijms21176286
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.