Mapping-by-Sequencing via MutMap Identifies a Mutation in ZmCLE7 Underlying Fasciation in a Newly Developed EMS Mutant Population in an Elite Tropical Maize Inbred

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
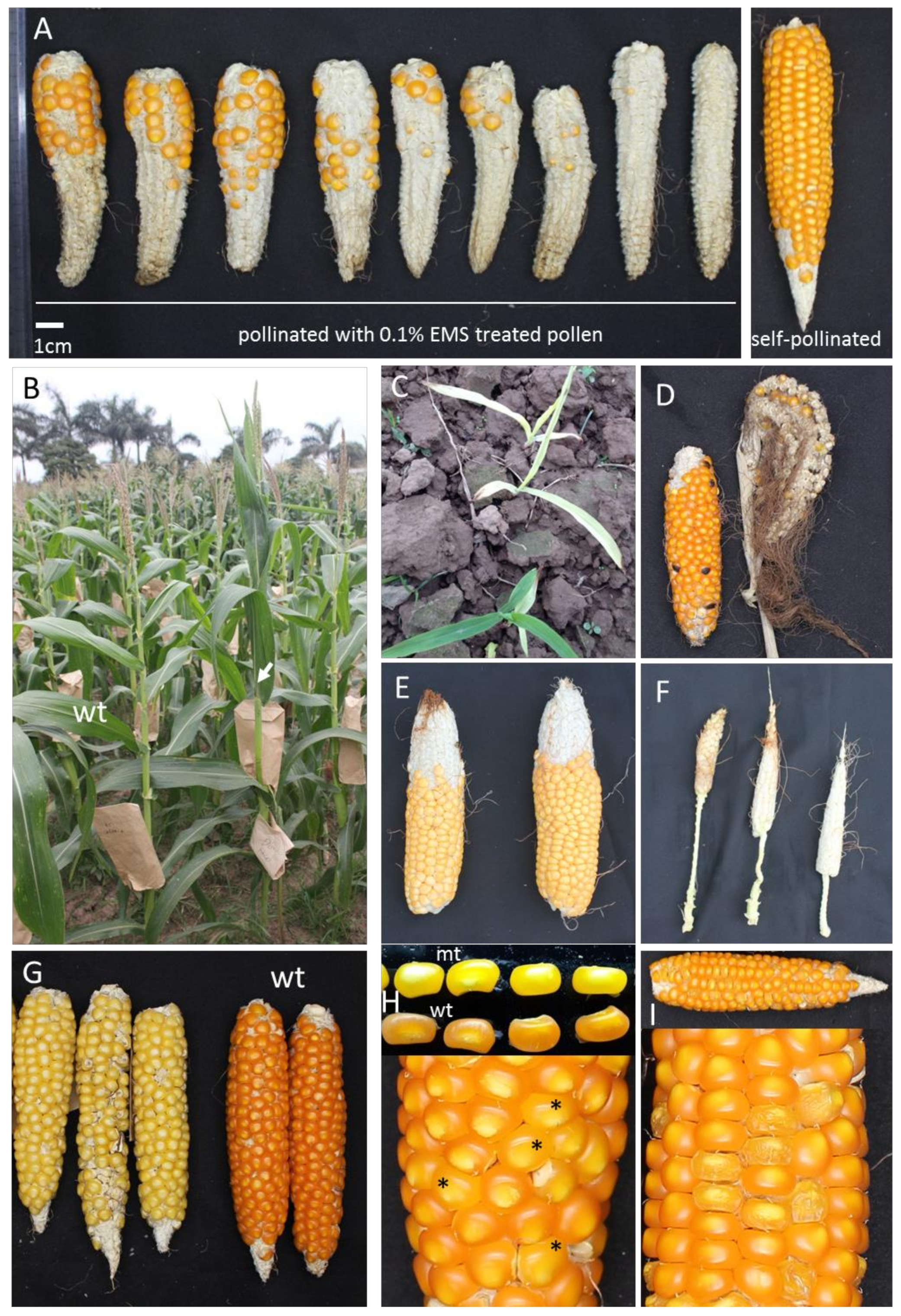
2.1. Mutagenesis, Phenotypic Screening and Plant Growth Conditions
2.2. Generating E-ML10
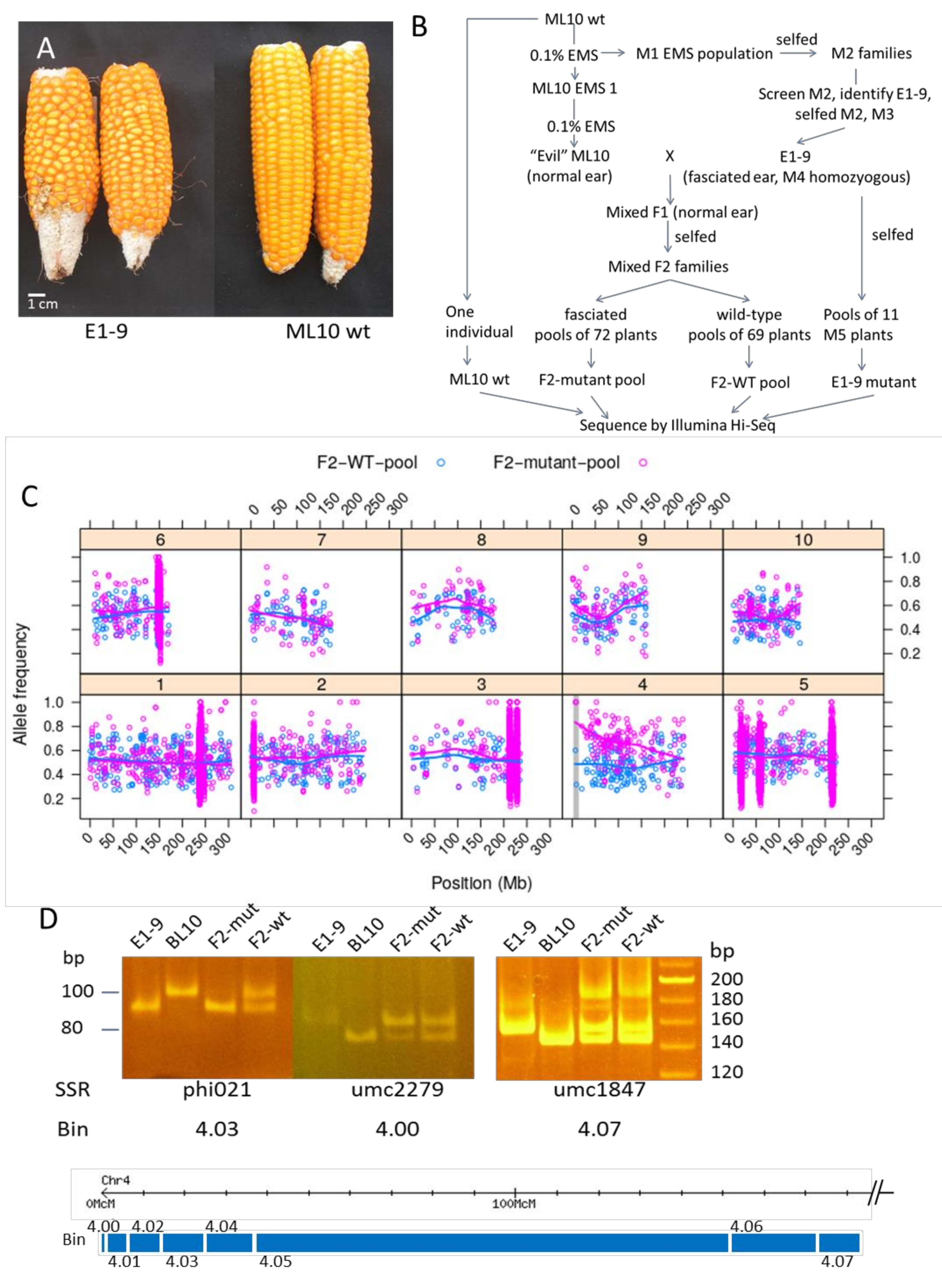
2.3. Mapping of E1-9
2.4. DNA Extraction
2.5. Sequencing and Bioinformatic Analysis
2.6. PCR Analysis
3. Results
3.1. Optimization of EMS Protocol for the ML10 Inbred and Development of the Mutant Population
3.2. Development of a Mapping-by-Sequencing Method via Mutmap to Map the E1-9 Mutant
3.2.1. F2 Mapping Population Generated with a Heavily Mutagenized ML10 (E-ML10)
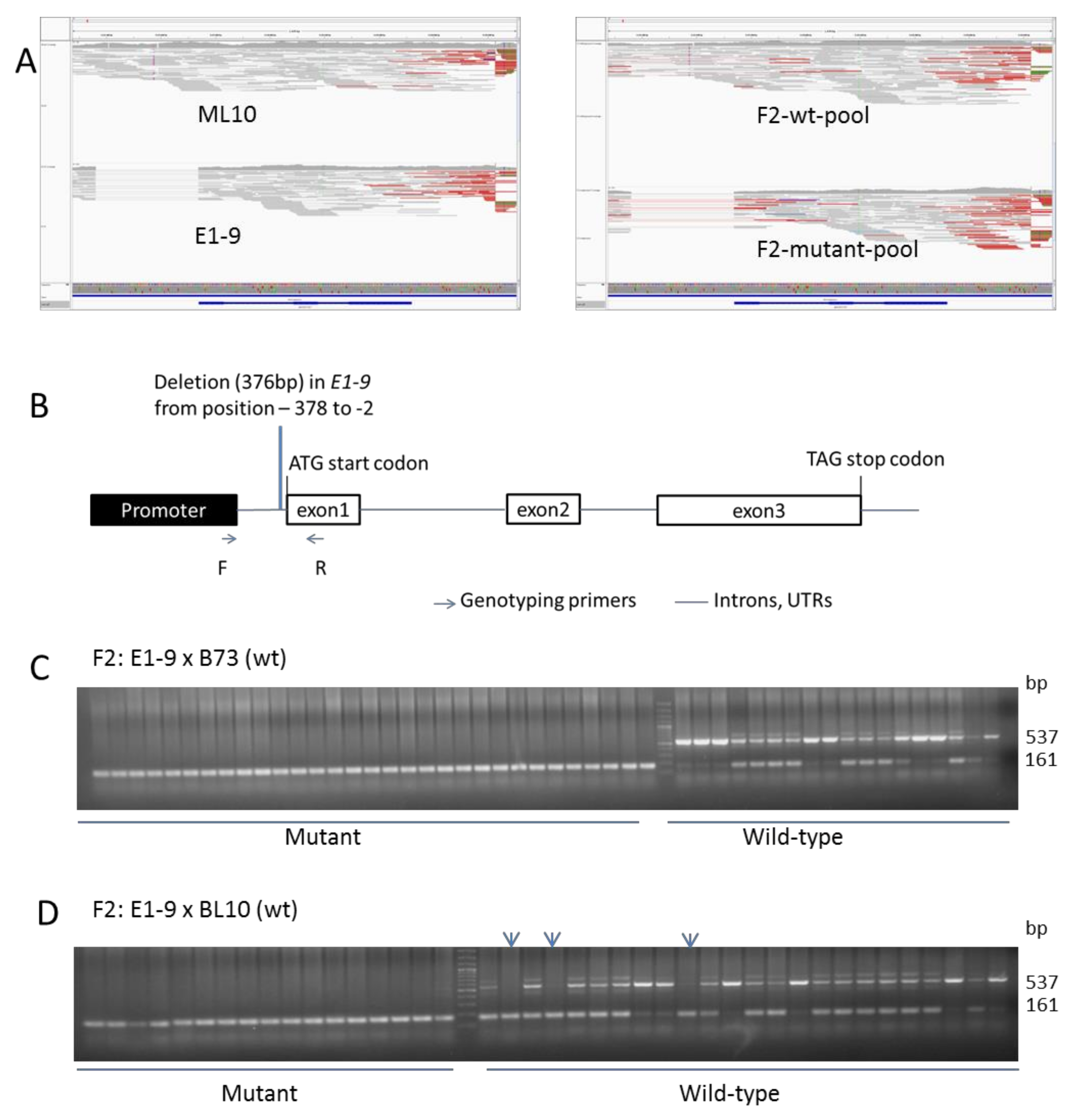
3.2.2. Sequencing and Bioinformatic Analysis
3.2.3. Linkage Confirmation by PCR Analysis and Identification of Causal Mutation
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307. [Google Scholar] [CrossRef] [Green Version]
- OECD; FAO. Agricultural Outlook 2018-2027; OECD-FAO Agricultural Outlook 2018-2027; OECD Publishing: Paris, France; Food and Agriculture, Organization of the United Nations: Rome, Italy, 2018; pp. 109–112. [Google Scholar]
- Rosegrant, M.W.; Msangi, S.; Ringler, C.; Sulser, T.B.; Zhu, T.; Cline, S.A. International Model for Policy Analysis of Agricultural Commodities and Trade (IMPACT): Model Description; International Food Policy Research Institute (IFPRI): Washington, DC, USA, 2012. [Google Scholar]
- Candela, H.; Hake, S. The art and design of genetic screens: Maize. Nat. Rev. Genet. 2008, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Neuffer, M. Mutagenesis. In The Maize Handbook; Springer: New York, NY, USA, 1994; pp. 212–219. [Google Scholar]
- maizegdb.org. Available online: https://www.maizegdb.org/data_center/phenotype (accessed on 9 February 2020).
- Till, B.J.; Reynolds, S.H.; Weil, C.; Springer, N.; Burtner, C.; Young, K.; Bowers, E.; Codomo, C.A.; Enns, L.C.; Odden, A.R.; et al. Discovery of induced point mutations in maize genes by TILLING. BMC Plant. Biol. 2004, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Liu, J.; Ren, W.; Yang, Q.; Chai, Z.; Chen, R.; Wang, L.; Zhao, J.; Lang, Z.; Wang, H.; et al. Gene-Indexed mutations in maize. Mol. Plant. 2018, 11, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuermann, M.C.; Rosso, M.G.; Mascher, M.; Brandt, R.; Tschiersch, H.; Altschmied, L.; Altmann, T. Combining next-generation sequencing and progeny testing for rapid identification of induced recessive and dominant mutations in maize M2 individuals. Plant J. 2019, 100, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settles, A.M. Transposon tagging and reverse genetics. In Molecular Genetic Approaches to Maize Improvement; Springer: Berlin/Heidelberg, Germany, 2009; pp. 143–159. [Google Scholar]
- Raizada, M.N.; Nan, G.L.; Walbot, V. Somatic and germinal mobility of the RescueMu transposon in transgenic maize. Plant Cell 2001, 13, 1587–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, B.P.; Liu, H.; Vollbrecht, E.; Senior, L.; Rabinowicz, P.D.; Roh, D.; Pan, X.; Stein, L.; Freeling, M.; Alexander, D.; et al. Maize-targeted mutagenesis: A knockout resource for maize. Proc. Natl. Acad. Sci. USA 2003, 100, 11541–11546. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.R.; Settles, A.M.; Suzuki, M.; Tan, B.C.; Latshaw, S.; Porch, T.; Robin, K.; Baier, J.; Avigne, W.; Lai, J.; et al. Steady-state transposon mutagenesis in inbred maize. Plant J. 2005, 44, 52–61. [Google Scholar] [CrossRef]
- Settles, A.M.; Holding, D.R.; Tan, B.C.; Latshaw, S.P.; Liu, J.; Suzuki, M.; Li, L.; O’Brien, B.A.; Fajardo, D.S.; Wroclawska, E. Sequence-indexed mutations in maize using the UniformMu transposon-tagging population. BMC Genom. 2007, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Williams-Carrier, R.; Stiffler, N.; Belcher, S.; Kroeger, T.; Stern, D.B.; Monde, R.A.; Coalter, R.; Barkan, A. Use of Illumina sequencing to identify transposon insertions underlying mutant phenotypes in high-copy Mutator lines of maize. Plant J. 2010, 63, 167–177. [Google Scholar] [CrossRef]
- Liang, L.; Zhou, L.; Tang, Y.; Li, N.; Song, T.; Shao, W.; Zhang, Z.; Cai, P.; Feng, F.; Ma, Y.; et al. A Sequence-Indexed mutator insertional library for maize functional genomics study. Plant Physiol. 2019, 181, 1404–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Li, A.; Morton, K.; Avoles-Kianian, P.; Kianian, S.F.; Zhang, C.; Holding, D. A Population of deletion mutants and an integrated mapping and exome-seq pipeline for gene discovery in maize. G3 (Bethesda) 2016, 6, 2385–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Wang, H.; Liu, S.; Li, Z.; Yang, X.; Yan, J.; Li, J.; Tran, L.S.; Qin, F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 2015, 6, 8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Nyaga, C.; Gowda, M.; Beyene, Y.; Muriithi, W.T.; Makumbi, D.; Olsen, M.S.; Suresh, L.M.; Bright, J.M.; Das, B.; Prasanna, B.M. Genome-wide analyses and prediction of resistance to MLN in large tropical maize germplasm. Genes (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Chen, J.; Mu, C.; Makumbi, D.; Xu, Y.; Mahuku, G. Combined linkage and association mapping reveal QTL for host plant resistance to common rust (Puccinia sorghi) in tropical maize. BMC Plant Biol. 2018, 18, 310. [Google Scholar] [CrossRef]
- Zuo, W.; Chao, Q.; Zhang, N.; Ye, J.; Tan, G.; Li, B.; Xing, Y.; Zhang, B.; Liu, H.; Fengler, K.A.; et al. A maize wall-associated kinase confers quantitative resistance to head smut. Nat. Genet. 2015, 47, 151–157. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide association studies in maize: Praise and stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.; Xiao, Y.; Conklin, P.A.; Govindarajulu, R.; Kelly, J.A.; Scanlon, M.J.; Whipple, C.J.; Bartlett, M. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3: Genes Genomes Genet. 2018, 8, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Addo-Quaye, C.; Buescher, E.; Best, N.; Chaikam, V.; Baxter, I.; Dilkes, B.P. Forward genetics by sequencing EMS variation-Induced inbred lines. G3 (Bethesda) 2017, 7, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap+: Genetic mapping and mutant identification without crossing in rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef] [PubMed]
- Gerpacio, R.V. Impact of Public-and Private-Sector Maize Breeding Research in Asia, 1966–1997/98; CIMMYT: El Batán, Mexico, 2001; pp. 105–120. [Google Scholar]
- Clarke, J.D. Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harbor Protocols 2009, 2009, pdb. prot5177. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef]
- Sarkar, D. Lattice: Multivariate Data Visualization with R; Springer: New York, NY, USA, 2008. [Google Scholar]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Schnable, J.C. RNA-Seq based analysis of population structure within the maize inbred B73. PLoS ONE 2016, 11, e0157942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommert, P.; Je, B.I.; Goldshmidt, A.; Jackson, D. The maize Gα gene COMPACT PLANT2 functions in CLAVATA signalling to control shoot meristem size. Nature 2013, 502, 555–558. [Google Scholar] [CrossRef]
- Taguchi-Shiobara, F.; Yuan, Z.; Hake, S.; Jackson, D. The fasciated ear2 gene encodes a leucine-rich repeat receptor-like protein that regulates shoot meristem proliferation in maize. Genes Dev. 2001, 15, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Bommert, P.; Nardmann, J.; Vollbrecht, E.; Running, M.; Jackson, D.; Hake, S.; Werr, W. Thick tassel dwarf1 encodes a putative maize ortholog of the Arabidopsis CLAVATA1 leucine-rich repeat receptor-like kinase. Development 2005, 132, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Xu, F.; Liu, L.; Char, S.N.; Ding, Y.; Je, B.I.; Schmelz, E.; Yang, B.; Jackson, D. The maize heterotrimeric G protein β subunit controls shoot meristem development and immune responses. Proc. Natl. Acad. Sci. USA 2020, 117, 1799–1805. [Google Scholar] [CrossRef]
- Je, B.I.; Gruel, J.; Lee, Y.K.; Bommert, P.; Arevalo, E.D.; Eveland, A.L.; Wu, Q.; Goldshmidt, A.; Meeley, R.; Bartlett, M. Signaling from maize organ primordia via FASCIATED EAR3 regulates stem cell proliferation and yield traits. Nat. Genet. 2016, 48, 785. [Google Scholar] [CrossRef] [Green Version]
- Pautler, M.; Eveland, A.L.; LaRue, T.; Yang, F.; Weeks, R.; Je, B.I.; Meeley, R.; Komatsu, M.; Vollbrecht, E.; Sakai, H. FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. Plant Cell 2015, 27, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Je, B.I.; Xu, F.; Wu, Q.; Liu, L.; Meeley, R.; Gallagher, J.P.; Corcilius, L.; Payne, R.J.; Bartlett, M.E.; Jackson, D. The CLAVATA receptor FASCIATED EAR2 responds to distinct CLE peptides by signaling through two downstream effectors. Elife 2018, 7, e35673. [Google Scholar] [CrossRef]
- Rodriguez-Leal, D.; Xu, C.; Kwon, C.-T.; Soyars, C.; Demesa-Arevalo, E.; Man, J.; Liu, L.; Lemmon, Z.H.; Jones, D.S.; Van Eck, J. Evolution of buffering in a genetic circuit controlling plant stem cell proliferation. Nat. Genet. 2019, 51, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Henry, I.M.; Nagalakshmi, U.; Lieberman, M.C.; Ngo, K.J.; Krasileva, K.V.; Vasquez-Gross, H.; Akhunova, A.; Akhunov, E.; Dubcovsky, J.; Tai, T.H.; et al. Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 2014, 26, 1382–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjes, C.E.; Rocheford, T.R.; Bai, L.; Brutnell, T.P.; Kandianis, C.B.; Sowinski, S.G.; Stapleton, A.E.; Vallabhaneni, R.; Williams, M.; Wurtzel, E.T.; et al. Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science 2008, 319, 330–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Kandianis, C.B.; Harjes, C.E.; Bai, L.; Kim, E.-H.; Yang, X.; Skinner, D.J.; Fu, Z.; Mitchell, S.; Li, Q. Rare genetic variation at Zea mays crtRB1 increases β-carotene in maize grain. Nat. Genet. 2010, 42, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Season | Sowing Date | Pollen Shedding Date | Estimated Time from Sowing to Shedding |
|---|---|---|---|
| Spring | Jan 11 (±10 d) | Apr 5 (±10 d) | 85 days |
| Autumn | Aug 8 to Sep 8 | Oct 12 to Nov 12 | 65 days |
| Phenotype | Number of M2 Families | Frequency (%) |
|---|---|---|
| Defective kernel | 33 | 2.78 |
| Small kernel | 7 | 0.59 |
| Dwarf plants | 4 | 0.34 |
| Kernel color | 4 | 0.34 |
| Narrow leaf | 4 | 0.34 |
| Albino | 3 | 0.25 |
| Purple stalk/leaf | 3 | 0.25 |
| Fasciated ear | 2 | 0.17 |
| Chlorotic lesion leaf | 2 | 0.17 |
| Liguleless/upright leaf | 2 | 0.17 |
| Wrinkled kernel | 3 | 0.25 |
| Tassel branch angle | 2 | 0.17 |
| Tassel branch number | 2 | 0.17 |
| Tassel seed | 2 | 0.17 |
| Branched ear (ramosa) | 1 | 0.08 |
| Anther color | 1 | 0.08 |
| Broad leaf | 1 | 0.08 |
| Early senescence | 1 | 0.08 |
| Sample | F2 Mutant Pool | F2 Wild-Type Pool | E1-9 | ML10 |
|---|---|---|---|---|
| Number of reads | 470,223,828 | 414,380,508 | 347,522,864 | 370,346,096 |
| Number of mapped reads | 462,796,855 | 407,731,780 | 344,923,846 | 361,795,129 |
| % of mapped reads | 98.42% | 98.40% | 99.25% | 97.69% |
| Properly paired reads | 405,367,374 | 359,813,078 | 305,113,228 | 320,163,016 |
| % of properly paired reads | 86.21% | 86.83% | 87.80% | 86.45% |
| Coverage (x) | 26 | 23 | 19 | 21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, Q.H.; Bui, N.H.; Kappel, C.; Dau, N.T.N.; Nguyen, L.T.; Tran, T.T.; Khanh, T.D.; Trung, K.H.; Lenhard, M.; Vi, S.L. Mapping-by-Sequencing via MutMap Identifies a Mutation in ZmCLE7 Underlying Fasciation in a Newly Developed EMS Mutant Population in an Elite Tropical Maize Inbred. Genes 2020, 11, 281. https://doi.org/10.3390/genes11030281
Tran QH, Bui NH, Kappel C, Dau NTN, Nguyen LT, Tran TT, Khanh TD, Trung KH, Lenhard M, Vi SL. Mapping-by-Sequencing via MutMap Identifies a Mutation in ZmCLE7 Underlying Fasciation in a Newly Developed EMS Mutant Population in an Elite Tropical Maize Inbred. Genes. 2020; 11(3):281. https://doi.org/10.3390/genes11030281
Chicago/Turabian StyleTran, Quan Hong, Ngoc Hong Bui, Christian Kappel, Nga Thi Ngoc Dau, Loan Thi Nguyen, Thuy Thi Tran, Tran Dang Khanh, Khuat Huu Trung, Michael Lenhard, and Son Lang Vi. 2020. "Mapping-by-Sequencing via MutMap Identifies a Mutation in ZmCLE7 Underlying Fasciation in a Newly Developed EMS Mutant Population in an Elite Tropical Maize Inbred" Genes 11, no. 3: 281. https://doi.org/10.3390/genes11030281