 Andreas Menke
Andreas Menke- Center of Mental Health, Department of Psychiatry, Psychosomatics and Psychotherapy, University Hospital of Wuerzburg, Wuerzburg, Germany
Major depressive disorder (MDD) is a very common stress-related mental disorder that carries a huge burden for affected patients and the society. It is associated with a high mortality that derives from suicidality and the development of serious medical conditions such as heart diseases, diabetes, and stroke. Although a range of effective antidepressants are available, more than 50% of the patients do not respond to the first treatment they are prescribed and around 30% fail to respond even after several treatment attempts. The heterogeneous condition of MDD, the lack of biomarkers matching patients with the right treatments and the situation that almost all available drugs are only targeting the serotonin, norepinephrine, or dopamine signaling, without regulating other potentially dysregulated systems may explain the insufficient treatment status. The hypothalamic-pituitary-adrenal (HPA) axis is one of these other systems, there is numerous and robust evidence that it is implicated in MDD and other stress-related conditions, but up to date there is no specific drug targeting HPA axis components that is approved and no test that is routinely used in the clinical setting identifying patients for such a specific treatment. Is there still hope after these many years for a breakthrough of agents targeting the HPA axis? This review will cover tests detecting altered HPA axis function and the specific treatment options such as glucocorticoid receptor (GR) antagonists, corticotropin-releasing hormone 1 (CRH1) receptor antagonists, tryptophan 2,3-dioxygenase (TDO) inhibitors and FK506 binding protein 5 (FKBP5) receptor antagonists.
Introduction
With a life-time prevalence around 20% major depressive disorder (MDD) is a very common disorder. In Europe it is one of the three most disabling conditions, next to dementias and alcohol abuse (1) and the burden of disease is projected to climb (2). MDD is associated with a substantially increased mortality due to suicide and an increased risk for serious medical conditions such as heart diseases, diabetes, and stroke (3). Although a range of effective antidepressants are available, more than 50% of patients do not respond to the first antidepressant treatment they are prescribed (4) and around 30% fail to respond even after several treatment approaches (5). Of note, even remitted patients suffer from a functional impairment (6). These non-sufficient treatment options that are currently available are reflected by the high personal and societal burden with increased rates of sick leave and early retirement (1). The commonly used treatment options do not only struggle with high rates of partial or no response, but also with a delayed onset of treatment effects and uncomfortable or even threatening adverse side effects (3). Various factors may explain the current situation: MDD is a heterogeneous condition with poorly defined endophenotypes or subgroups, the currently available drugs have very similar treatment mechanisms and target almost only components of the serotonin, norepinephrine, or dopamine signaling, and there are no biomarkers to predict the response or side effects to specific interventions (7). Moreover, the diagnostic process and treatment choice are solely based on clinical experience and intuition. Fortunately, initiatives are under way to provide individualized treatment options for each patient: personalized medicine and precision medicine are employed to match individual patients with the most effective treatment options (7). Personalized and precision medicine are often used interchangeable, however, they describe two different concepts. Personalized treatment has been administered for the last decades, physicians considered sex, age, weight, co-medication together with renal, and liver functioning, comorbidities, core-symptoms (disturbances of sleep and appetite, psychotic vs. non-psychotic, agitated vs. non-agitated …) and patients preferences in the selection process of a suitable antidepressant. However, this personalization resembles a trial and error process and is highly dependent on the experience and the knowledge of the physician (8). The objective of precision medicine is to improve the selection of effective antidepressants with best possible response and minimal side effects using genetic markers or biomarkers derived from peripheral blood, imaging, neuropsychological tests, or behavioral measures (9–11). Given the high prevalence of MDD, another task of precision medicine will be the identification of individuals at risk and then to deliver specific interventions to avoid the full development of MDD (12).
Genetic and Environmental Factors Contributing to MDD
A meta-analysis with more than 21,000 individuals observed a heritability of MDD around 40%, common environmental factors had very small effects, but individual environmental factors showed a substantial contribution of around 60% (13). In fact, the development of MDD is crucially dependent on gene x environment interactions (14–17). Aversive environmental events such as sexual, physical, or emotional childhood trauma have been robustly associated with MDD (18). But still it is not understood how early aversive events interact with genetic and epigenetic factors to confer vulnerability to MDD and how to treat patients who have experienced early life adversity. Meanwhile there is growing evidence showing that childhood trauma substantially shapes biological systems that are responsible for a fight-or-flight response, such as the hypothalamic-pituitary-adrenal (HPA) axis. In fact, childhood trauma may lead to an increased sensitivity of the HPA axis and to heightened responses to subsequent stressors (19, 20). Thus, the HPA axis may be a suitable target for specific interventions.
HPA Axis
Environmental stress activates the release of the monoamines serotonin, norepinephrine and dopamine from the amygdala, hippocampus, and other brain regions. Subsequently, the paraventricular nucleus (PVN) of the hypothalamus synthesizes corticotrophin-releasing hormone (CRH), that binds to corticotropin-releasing hormone 1 (CRH1) and CRH2 receptors in the anterior pituitary. Then ACTH is secreted in the circulation (see Figure 1). ACTH activates the production and release of glucocorticoids (GC) in the adrenal glands. To reinstate homeostasis negative feedback mechanisms are initiated: GCs bind to glucocorticoid receptors (GR) of the hippocampus, the PNV and the anterior pituitary gland and thus inhibit the further release of CRH (22). In MDD the sensitivity of the GR is impaired leading to a reduced negative feedback mechanism and subsequently to a central hypersecretion of CRH and an increased production of GCs (23, 24). The sensitivity of the GR is substantially regulated by FKBP5, encoding the FK 506 binding protein 51 or FKBP51, a co-chaperone of heat-shock protein 90 (hsp90) (25). When FKBP51 is bound to the GR complex, the affinity for glucocorticoid-binding is reduced and the GR is translocated into the nucleus less efficiently. FKBP5 mRNA and protein expression are induced by GR activation and provide an ultra-short negative feedback loop for GR sensitivity (25). Polymorphisms within FKBP5 have been shown to be associated with differential regulation of FKBP5 mRNA expression after activation of GR and differences in GR sensitivity (26, 27). FKBP5 has been implicated in several mental disorders and stress-related conditions such as major depression (26), bipolar disorder (28), childhood trauma and posttraumatic stress disorder (29), aggressive and suicidal behavior (30, 31). Above the cellular level these genetic variants in combination with epigenetic alterations were associated with structural and functional changes in several brain regions (32–34) and with impaired working memory and cardiac stress reactivity (35). Recently FKBP5 has been associated with metabolic function, diabetes and obesity (36–38) and pain (39, 40).
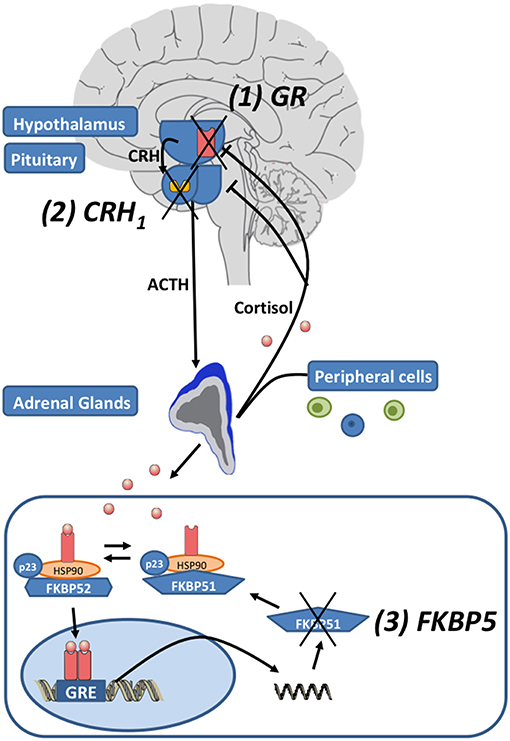
Figure 1. The hypothalamic-pituitary-adrenal (HPA) axis: Corticotropin-releasing hormone (CRH) is released by neurons in the paraventricular nucleus of the hypothalamus. Subsequently CRH1 receptors are activated and the secretion of adrenocorticotropic hormone (ACTH) from the pituitary is induced. ACTH induces the release of glucocorticoids (cortisol) by the adrenal glands. After the activation of the HPA axis, negative feedback loops are activated to reinstate homeostasis by cortisol activating glucocorticoid receptors (GR). The unliganded GR complex consists of the co-chaperones FKBP51 or FKBP52 (encoded by their respective genes FKBP5 and FKBP4), p23 (a co-chaperone molecule) and hsp90 dimer. When FKBP51 binds to the GR-complex via hsp90, the GR affinity for cortisol is reduced. When glucocorticoids bind to the GR, FKBP51 is exchanged against FKBP52 and the nuclear translocation of the ligand-bound GR is enabled. The GR directly binds to the DNA via glucocorticoid response elements (GREs) and induces FKBP5 mRNA expression and subsequently FKBP51 production, inducing an ultra-short negative feedback loop on GR sensitivity. Drugs regulating the function of the HPA axis target: (1) the GR, (2) the CRH1 receptors, and (3) FKBP5/FKBP51 (modified after Leistner and Menke (21); Copyright (2018), with permission from Elsevier).
Stress-induced cortisol excess was also observed to impact the kynurenine pathway by enhancing the hepatic activity of tryptophan 2,3-dioxygenase (TDO) (41–43). Aside indoleamine 2,3-dioxygenase (IDO) TDO is the first and rate-limiting enzyme that catalyzes the conversion of tryptophan into N-formyl-kynurenine (NFK) (44). Downstream several kynurenine pathways metabolites have been associated with the development of major depression as they exert neurotoxic effects, e.g., by activating N-methyl-D-aspartate (NMDA) receptors or enhancing free radical production (45–48).
Tests Detecting the Function of the HPA Axis
Different tests have been developed to measure the function of the HPA axis (21). The dexamethasone suppression test [DST, (49)] identifies an impaired suppression of dexamethasone on cortisol, as observed in depressed patients (50–52). However, the DST has not reached clinical relevance as a diagnostic tool because of its low sensitivity, which ranges between 20 and 50% (52–54). To increase the sensitivity and the specificity the DST was combined with the CRH stimulation test, the dexamethasone-corticotropin-releasing hormone [dex-CRH, (55, 56)] test, which actually led to an improved sensitivity in detecting alterations of the HPA axis with a successful classification of up to 80% of depressed patients (55, 56). These findings could be replicated in several studies (57–59), but others observed negative results when analyzing case-control differences (60–62). Interestingly, in addition to its ability to identify depressed patients, several studies observed that the dex-CRH test may allow a stratification of depressed patients and predict treatment outcome and disease course (21). Studies reported an increased cortisol response to the dex-CRH test in patients after remission at risk of relapse (59, 63, 64), in subjects with violent suicide attempts and suicide completion (65) and in melancholic patients compared to non-melancholic depressed patients (66). Contrary, a reduced cortisol response in the dex-CRH test was found in depressed patients with suicidal behavior (67) and women with chronic social stressors (68). An early normalization of the dex-CRH test results has been associated with response to antidepressant medication (59). However, we observed previously, that the readouts of the dex-CRH test are substantially dependent on the plasma concentrations of dexamethasone, thus several factors that influence the plasma concentration do also impact the readout of the test (69). Recently we reported on the potential use of the dexamethasone-induced gene expression changes as an additional indicator for alterations of the HPA axis and as a potential biomarker in depression (70) and other stress-related mental disorders such as job-related exhaustion (71). For this test, before and 3 h after a GR activation by dexamethasone cortisol, ACTH, blood count, and gene expression signatures are measured to detect GR sensitivity alterations (21). Of note, this test was not dependent on dexamethasone plasma concentrations (69). Applying this test we observed an increased GR sensitivity in patients with anxious depression compared with non-anxious depression, an enhanced leukocyte reactivity in patients with childhood trauma (72) and an increased GR sensitivity in healthy women compared to healthy men (73). In a broader, stimulated expression quantitative trait locus (eQTL) approach we combined these gene expression signatures after GR-activation with genome-wide single nucleotide polymorphism (SNP) data and found that common genetic variants that modulate the transcriptional response to GR-activation mediate the risk for MDD as well as other mental disorders (74).
Specific Targets of the HPA Axis
GR Antagonists
Based on numerous findings of a HPA axis hyperactivity in patients with psychotic depression open-label and double-blind trials with the GR antagonist (and also progesterone antagonist) mifepristone were conducted (75) (Figure 1). The studies using dosages between 300 and 1,200 mg /d showed mixed results, with both positive studies and failed trials (75). A combined analysis of similarly designed double-blind phase 2 or 3 studies assessing the efficacy and safety of 7 day mifepristone treatment revealed a meaningful efficacy (p < 0.004) for mifepristone in reducing psychotic symptoms, adverse events were similar in mifepristone and placebo treated patients (76). Interestingly, high mifepristone plasma concentrations were associated with the strongest response, followed by changes in cortisol and ACTH (76). There is also accumulating evidence that mifepristone ameliorates cognitive deficits in major depression and bipolar disorder (77). Thus, for depressed patients with psychotic features a GR antagonist such as mifepristone may be an individualized treatment option.
CRH1 Receptor Antagonists
In preclinical models central administration of CRH produces behavioral effects that closely resemble the symptoms of depression in humans (78, 79). These effects are attenuated by central administration of a specific CRH receptor antagonist (79, 80). Moreover, also clinical studies provided evidence of a CRH hyperactivity in depression and anxiety (79). A clinical trial using the CRH1 receptor antagonist R121919 in the treatment of major depression revealed significant reductions in the Hamilton Depression Rating Scale (HAMD) over the 30 day treatment period (81). The stress-elicited secretion of cortisol was reduced, however, it did not impair the CRH-induced release of ACTH and cortisol and thus the stress hormone system responsivity to CRH remained unchanged (81). However, the study did not include design components such as blinding, randomization or a placebo control and R121919 was withdrawn due to liver enzyme elevations. A further trial using another CRH1 receptor antagonist, CP-316,311 did not observe a significant difference between patients treated with CP-316,311 and placebo (82). Other trials using CRH1 receptor antagonists in patients with major depression, social and generalized anxiety disorder and suicidal ideation could also not reveal beneficial effects (83). In a trial with anxious, alcohol-dependent women the CRH1 receptor antagonist Verucerfont (also GSK561679) produced a dampening of the HPA axis response to social stressors and attenuated amygdala response to negative affective stimuli, while alcohol craving was unaffected (84). Recently a double-blind, randomized and placebo-controlled trial investigated the efficacy of the same CRH1 receptor antagonist in women suffering from Posttraumatic stress disorder (85, 86). The trial did not observe a significant improvement of PTSD symptoms in patients treated with GSK561679 compared to placebo overall (86). However, subjects with a moderate or severe history of childhood abuse and a certain CRH1 receptor SNP genotype did only response to GSK561679, not to placebo (86). Nevertheless, the authors concluded that CRH1 receptor antagonists as a class are ineffective as monotherapy for stress-related mental disorders (86) and the question arose whether it is time to call it quits for the CRH1 receptor antagonists (83, 87). CRH is a key regulator of the stress response and controls endocrine activity by direct modulation of the HPA axis. As stated above, numerous preclinical and clinical data support the involvement of CRH and CRH1 receptors in stress-related mental disorder (88). However, some of the tested agents did only show meaningful effects in some of the preclinical stress tests, moreover, preclinical data not always translate to clinical trials without complications (83, 87). For CRH1 receptor antagonists, the traditional clinical trial design is probably not suitable. Instead, patients with an overactivity of the CRH—CRH1 receptor signaling should be identified by reliable biological measures in terms of precision medicine, that is already well-established in other medical fields, such as oncology (87), and then included in a respective trial. Thus, CRH1 receptor antagonists are still promising agents for stress-related mental disorders, but probably only in those patients who are subject to a significant CRH signaling dysfunction.
TDO Inhibitors
TDO inhibition by directly targeting the kynurenine production is supposed to decrease neurotoxic metabolites and thus ameliorate depressive symptoms (44). TDO inhibition is a mechanism shared by the largest number of antidepressants, e.g., citalopram effectively decreases TDO activity (44, 89). Interestingly, agents inhibiting glucocorticoids such as RU486 showed antidepressive properties by inhibiting TDO activity (90, 91). Additionally, co-treatment with allopurinol, also a TDO inhibitor, improved chronic stress induced depressive-like behavior (92). Recently, the agent NSC 36398, a flavonoid compound, was observed to be a first selective TDO inhibitor (93).
FKBP5 Antagonists
As described above FKBP5, respectively, FK506 binding protein 51/FKBP51 regulates the responsiveness of the GR and the HPA axis and is also implicated in important gene x environment interactions underlying stress-related mental disorders (25, 94), making it a promising drug target. In fact, several research groups have consistently observed protective effects of FKBP5 knock-out or knock-down on stress-coping behavior and stress endocrinology in preclinical models of depression and anxiety (95). The prototypic FKBP ligand FK506 and rapamycin showed the principal druggability. In addition, FKBP51 is highly suited for X-ray cocrystallography, which facilitates the rational drug design (96). Sulfonamide analogs have been found that possess FKBP51 binding properties (97). However, drug discovery has been hampered by the inability that all known ligands cannot differentiate FKBP51 and the opposing homolog FKBP52 (98, 99). Recently with SAFit1 and SAFit2 two promising potent and highly selective inhibitors of FKBP51 were discovered, that achieved selectivity by an induced-fit mechanism and improved neuroendocrine feedback and stress-coping behavior in mice (100, 101). Of note, co-application of SAFit2 with the selective serotonin reuptake inhibitor escitalopram, a common antidepressant, lowered the efficacy of escitalopram in anxiety-related tests but improved stress coping behavior in a mouse model (102). FKBP5 antagonists may be promising new treatment options for patients suffering from stress-related mental disorders and who have an altered functioning of FKBP5/GR/HPA axis signaling.
Conclusion
Despite the very strong preclinical and clinical data of a dysregulation of the HPA axis in stress-related mental disorders, such as major depression, no drug has been approved that targets specific components of the HPA axis. In addition, no test is routinely used in the clinical setting to identify patients with a measureable HPA axis dysfunction. In fact, there is evidence that not all depressed patients display alterations of the HPA axis, and therefore not all of them would benefit of a very specific treatment, targeting only HPA axis components. This has become abundantly clear with the failing CRH1 receptor antagonists for major depression and posttraumatic stress disorder. However, even in the failed trials, there were initial hints that subgroups of patients carrying certain genetic risk variants or having a history of childhood trauma would indeed benefit from these very specific treatment options (86). Therefore, precision medicine has to be employed to match the specific antidepressant agent to the specific underlying biological alteration and the individual patient. Biological markers derived from tests detecting the HPA axis function, GR sensitivity, or FKBP5 dysregulation are necessary to identify suitable patients for these specific agents. In addition, clinical variables such as psychotic symptoms or history of childhood trauma combined with certain genetic risk variants may further improve the accuracy of such a test. Still in its infancy, the dexamethasone-induced gene expression test may become a promising tool to assess the GR sensitivity and FKBP5 function (21), because it combines neuroendocrine results with molecular genetic patterns of a GR challenged gene expression integrating genetic risk polymorphisms and additional clinical data (21). Thus, the future algorithms defining the treatment of major depression or other stress-related mental disorders will incorporate tests that stratify patients groups and match individual patients with highly specific agents, for example that target HPA axis components such as the GR, CRH1 receptors or FKBP5. GR antagonists, especially mifepristone, have provided very promising results for the treatment of psychotic depression so far and therefore could gain more relevance (76). CRH1 receptor antagonists have experienced a setback, but after employing suitable tests to find susceptible patients this development could be reversed. FKBP5, representing a molecular hub modulating many cellular pathways, is a novel and very promising candidate to target component of the stress hormone system and to ameliorate stress-related mental disorders and other sequelae of stress.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This publication was funded by the German Research Foundation (DFG) and the University of Wuerzburg in the funding programme Open Access Publishing.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. (2011) 21:655–79. doi: 10.1016/j.euroneuro.2011.07.018
2. Kessler RC, Bromet EJ. The epidemiology of depression across cultures. Annu Rev Public Health. (2013) 34:119–38. doi: 10.1146/annurev-publhealth-031912-114409
3. Otte C, Gold SM, Penninx BW, Pariante CM, Etkin A, Fava M, et al. Major depressive disorder. Nat Rev Dis Primers. (2016) 2:16065. doi: 10.1038/nrdp.2016.65
4. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selective serotonin reuptake inhibitor in major depressive disorder: a systematic review. J Clin Psychiatry. (2006) 67:1836–55. doi: 10.4088/JCP.v67n1203
5. Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. (2006) 354:1231–42. doi: 10.1056/NEJMoa052963
6. Ormel J, Oldehinkel AJ, Nolen WA, Vollebergh W. Psychosocial disability before, during, and after a major depressive episode: a 3-wave population-based study of state, scar, and trait effects. Arch Gen Psychiatry. (2004) 61:387–92. doi: 10.1001/archpsyc.61.4.387
7. Menke A. Precision pharmacotherapy: psychiatry's future direction in preventing, diagnosing, and treating mental disorders. Pharmacogenom Pers Med. (2018) 11:211–22. doi: 10.2147/PGPM.S146110
8. Perlis RH. Abandoning personalization to get to precision in the pharmacotherapy of depression. World Psychiatry. (2016) 15:228–35. doi: 10.1002/wps.20345
9. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. (2015) 372:793–5. doi: 10.1056/NEJMp1500523
10. Jameson JL, Longo DL. Precision medicine–personalized, problematic, and promising. N Engl J Med. (2015) 372:2229–34. doi: 10.1056/NEJMsb1503104
11. Holsboer F. How can we realize the promise of personalized antidepressant medicines? Nat Rev Neurosci. (2008) 9:638–46. doi: 10.1038/nrn2453
12. van Zoonen K, Buntrock C, Ebert DD, Smit F, Reynolds CF III, Beekman AT, et al. Preventing the onset of major depressive disorder: a meta-analytic review of psychological interventions. Int J Epidemiol. (2014) 43:318–29. doi: 10.1093/ije/dyt175
13. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. (2000) 157:1552–62. doi: 10.1176/appi.ajp.157.10.1552
14. Klengel T, Binder EB. Epigenetics of stress-related psychiatric disorders and gene x environment interactions. Neuron. (2015) 86:1343–57. doi: 10.1016/j.neuron.2015.05.036
15. Binder EB. Dissecting the molecular mechanisms of gene x environment interactions: implications for diagnosis and treatment of stress-related psychiatric disorders. Eur J Psychotraumatol. (2017) 8(Suppl. 5):1412745. doi: 10.1080/20008198.2017.1412745
16. Kendler KS, Davis CG, Kessler RC. The familial aggregation of common psychiatric and substance use disorders in the National Comorbidity Survey: a family history study. Br J Psychiatry. (1997) 170:541–8. doi: 10.1192/bjp.170.6.541
17. Kessler RC, Davis CG, Kendler KS. Childhood adversity and adult psychiatric disorder in the US National Comorbidity Survey. Psychol Med. (1997) 27:1101–19. doi: 10.1017/S0033291797005588
18. Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. (2001) 49:1023–39. doi: 10.1016/S0006-3223(01)01157-X
19. Danese A, Lewis SJ. Psychoneuroimmunology of early-life stress: the hidden wounds of childhood trauma? Neuropsychopharmacology. (2017) 42:99–114. doi: 10.1038/npp.2016.198
20. Kuhlman KR, Chiang JJ, Horn S, Bower JE. Developmental psychoneuroendocrine and psychoneuroimmune pathways from childhood adversity to disease. Neurosci Biobehav Rev. (2017) 80:166–84. doi: 10.1016/j.neubiorev.2017.05.020
21. Leistner C, Menke A. How to measure glucocorticoid receptor's sensitivity in patients with stress-related psychiatric disorders. Psychoneuroendocrinology. (2018) 91:235–60. doi: 10.1016/j.psyneuen.2018.01.023
22. de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. (2005) 6:463–75. doi: 10.1038/nrn1683
23. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. (2000) 23:477–501. doi: 10.1016/S0893-133X(00)00159-7
24. Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. (2001) 49:391–404. doi: 10.1016/S0006-3223(00)01088-X
25. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. (2009) 34(Suppl. 1):S186–95. doi: 10.1016/j.psyneuen.2009.05.021
26. Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. (2004) 36:1319–25. doi: 10.1038/ng1479
27. Menke A, Klengel T, Rubel J, Bruckl T, Pfister H, Lucae S, et al. Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav. (2013) 12:289–96. doi: 10.1111/gbb.12026
28. Willour VL, Chen H, Toolan J, Belmonte P, Cutler DJ, Goes FS, et al. Family-based association of FKBP5 in bipolar disorder. Mol Psychiatry. (2008) 14: 261–268. doi: 10.1038/sj.mp.4002141
29. Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. (2008) 299:1291–305. doi: 10.1001/jama.299.11.1291
30. Roy A, Gorodetsky E, Yuan Q, Goldman D, Enoch MA. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology. (2010) 35:1674–83. doi: 10.1038/npp.2009.236
31. Bevilacqua L, Carli V, Sarchiapone M, George DK, Goldman D, Roy A, et al. Interaction between FKBP5 and childhood trauma and risk of aggressive behavior. Arch Gen Psychiatry. (2012) 69:62–70. doi: 10.1001/archgenpsychiatry.2011.152
32. Grabe HJ, Wittfeld K, Van der Auwera S, Janowitz D, Hegenscheid K, Habes M, et al. Effect of the interaction between childhood abuse and rs1360780 of the FKBP5 gene on gray matter volume in a general population sample. Hum Brain Mapp. (2016) 37:1602–13. doi: 10.1002/hbm.23123
33. Han KM, Won E, Sim Y, Kang J, Han C, Kim YK, et al. Influence of FKBP5 polymorphism and DNA methylation on structural changes of the brain in major depressive disorder. Sci Rep. (2017) 7:42621. doi: 10.1038/srep42621
34. Tozzi L, Farrell C, Booij L, Doolin K, Nemoda Z, Szyf M, et al. Epigenetic changes of FKBP5 as a link connecting genetic and environmental risk factors with structural and functional brain changes in major depression. Neuropsychopharmacology. (2018) 43:1138–45. doi: 10.1038/npp.2017.290
35. Lovallo WR, Enoch MA, Acheson A, Cohoon AJ, Sorocco KH, Hodgkinson CA, et al. Early-life adversity interacts with FKBP5 genotypes: altered working memory and cardiac stress reactivity in the oklahoma family health patterns project. Neuropsychopharmacology. (2016) 41:1724–32. doi: 10.1038/npp.2015.347
36. Balsevich G, Hausl AS, Meyer CW, Karamihalev S, Feng X, Pohlmann ML, et al. Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nat Commun. (2017) 8:1725. doi: 10.1038/s41467-017-01783-y
37. Ortiz R, Joseph JJ, Lee R, Wand GS, Golden SH. Type 2 diabetes and cardiometabolic risk may be associated with increase in DNA methylation of FKBP5. Clin Epigenetics. (2018) 10:82. doi: 10.1186/s13148-018-0513-0
38. Lee RS, Mahon PB, Zandi PP, McCaul ME, Yang X, Bali U, et al. DNA methylation and sex-specific expression of FKBP5 as correlates of one-month bedtime cortisol levels in healthy individuals. Psychoneuroendocrinology. (2018) 97:164–73. doi: 10.1016/j.psyneuen.2018.07.003
39. Bortsov AV, Smith JE, Diatchenko L, Soward AC, Ulirsch JC, Rossi C, et al. Polymorphisms in the glucocorticoid receptor co-chaperone FKBP5 predict persistent musculoskeletal pain after traumatic stress exposure. Pain. (2013) 154:1419–26. doi: 10.1016/j.pain.2013.04.037
40. Linnstaedt SD, Riker KD, Rueckeis CA, Kutchko KM, Lackey L, McCarthy KR, et al. A functional riboSNitch in the 3' untranslated region of FKBP5 alters MicroRNA-320a binding efficiency and mediates vulnerability to chronic post-traumatic pain. J Neurosci. (2018) 38:8407–20. doi: 10.1523/JNEUROSCI.3458-17.2018
41. Badawy AA. Tryptophan: the key to boosting brain serotonin synthesis in depressive illness. J Psychopharmacol. (2013) 27:878–93. doi: 10.1177/0269881113499209
42. Knox WE. Two mechanisms which increase in vivo the liver tryptophan peroxidase activity: specific enzyme adaptation and stimulation of the pituitary adrenal system. Br J Exp Pathol. (1951) 32:462–9.
43. Salter M, Pogson CI. The role of tryptophan 2,3-dioxygenase in the hormonal control of tryptophan metabolism in isolated rat liver cells. Effects of glucocorticoids and experimental diabetes. Biochem J. (1985) 229:499–504. doi: 10.1042/bj2290499
44. Qin Y, Wang N, Zhang X, Han X, Zhai X, Lu Y. IDO and TDO as a potential therapeutic target in different types of depression. Metab Brain Dis. (2018) 33:1787–800. doi: 10.1007/s11011-018-0290-7
45. Guillemin GJ. Quinolinic acid, the inescapable neurotoxin. FEBS J. (2012) 279:1356–65. doi: 10.1111/j.1742-4658.2012.08485.x
46. Maes M, Galecki P, Verkerk R, Rief W. Somatization, but not depression, is characterized by disorders in the tryptophan catabolite (TRYCAT) pathway, indicating increased indoleamine 2,3-dioxygenase and lowered kynurenine aminotransferase activity. Neuro Endocrinol Lett. (2011) 32:264–73.
47. Savitz J, Drevets WC, Wurfel BE, Ford BN, Bellgowan PS, Victor TA, et al. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav Immun. (2015) 46:55–9. doi: 10.1016/j.bbi.2015.02.007
48. Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci. (2013) 34:136–43. doi: 10.1016/j.tips.2012.09.006
49. Carroll BJ, Martin FI, Davies B. Resistance to suppression by dexamethasone of plasma 11-O.H.C.S. levels in severe depressive illness. Br Med J. (1968) 3:285–7. doi: 10.1136/bmj.3.5613.285
50. Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry. (1981) 38:15–22. doi: 10.1001/archpsyc.1981.01780260017001
51. Nelson JC, Davis JM. DST studies in psychotic depression: a meta-analysis. Am J Psychiatry. (1997) 154:1497–503. doi: 10.1176/ajp.154.11.1497
52. Nierenberg AA, Feinstein AR. How to evaluate a diagnostic marker test. Lessons from the rise and fall of dexamethasone suppression test. JAMA. (1988) 259:1699–702. doi: 10.1001/jama.1988.03720110061036
53. Holsboer F, Bender W, Benkert O, Klein HE, Schmauss M. Diagnostic value of dexamethasone suppression test in depression. Lancet. (1980) 2:706. doi: 10.1016/S0140-6736(80)92755-5
54. Arana GW, Baldessarini RJ, Ornsteen M. The dexamethasone suppression test for diagnosis and prognosis in psychiatry. Commentary and review. Arch Gen Psychiatry. (1985) 42:1193–204. doi: 10.1001/archpsyc.1985.01790350067012
55. Bardeleben U, Holsboer F. Cortisol response to a combined dexamethasone-human corticotrophin-releasing hormone challenge in patients with depression. J Neuroendocrinol. (1989) 1:485–8. doi: 10.1111/j.1365-2826.1989.tb00150.x
56. Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res. (1994) 28:341–56. doi: 10.1016/0022-3956(94)90017-5
57. Modell S, Yassouridis A, Huber J, Holsboer F. Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology. (1997) 65:216–22. doi: 10.1159/000127275
58. Kunugi H, Ida I, Owashi T, Kimura M, Inoue Y, Nakagawa S, et al. Assessment of the Dexamethasone/CRH test as a state-dependent marker for Hypothalamic-Pituitary-Adrenal (HPA) axis abnormalities in major depressive episode: a multicenter study. Neuropsychopharmacology. (2006) 31:212–20. doi: 10.1038/sj.npp.1300868
59. Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression - a potential biomarker? Biol Psychiatry. (2007) 62:47–54. doi: 10.1016/j.biopsych.2006.07.039
60. Oshima A, Yamashita S, Owashi T, Murata T, Tadokoro C, Miyaoka H, et al. The differential ACTH responses to combined dexamethasone/CRH administration in major depressive and dysthymic disorders. J Psychiatr Res. (2000) 34:325–8. doi: 10.1016/S0022-3956(00)00021-2
61. Gervasoni N, Bertschy G, Osiek C, Perret G, Denis R, Golaz J, et al. Cortisol responses to combined dexamethasone/CRH test in outpatients with a major depressive episode. J Psychiatr Res. (2004) 38:553–7. doi: 10.1016/j.jpsychires.2004.04.008
62. Carpenter LL, Ross NS, Tyrka AR, Anderson GM, Kelly M, Price LH. Dex/CRH test cortisol response in outpatients with major depression and matched healthy controls. Psychoneuroendocrinology. (2009) 34:1208–13. doi: 10.1016/j.psyneuen.2009.03.009
63. Zobel AW, Nickel T, Sonntag A, Uhr M, Holsboer F, Ising M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression. a prospective study. J Psychiatr Res. (2001) 35:83–94. doi: 10.1016/S0022-3956(01)00013-9
64. Appelhof BC, Huyser J, Verweij M, Brouwer JP, van Dyck R, Fliers E, et al. Glucocorticoids and relapse of major depression (dexamethasone/corticotropin-releasing hormone test in relation to relapse of major depression). Biol Psychiatry. (2006) 59:696–701. doi: 10.1016/j.biopsych.2005.09.008
65. Coryell W, Schlesser M. The dexamethasone suppression test and suicide prediction. Am J Psychiatry. (2001) 158:748–53. doi: 10.1176/appi.ajp.158.5.748
66. Kaestner F, Hettich M, Peters M, Sibrowski W, Hetzel G, Ponath G, et al. Different activation patterns of proinflammatory cytokines in melancholic and non-melancholic major depression are associated with HPA axis activity. J Affect Disord. (2005) 87:305–11. doi: 10.1016/j.jad.2005.03.012
67. Pfennig A, Kunzel HE, Kern N, Ising M, Majer M, Fuchs B, et al. Hypothalamus-pituitary-adrenal system regulation and suicidal behavior in depression. Biol Psychiatry. (2005) 57:336–42. doi: 10.1016/j.biopsych.2004.11.017
68. Rydmark I, Wahlberg K, Ghatan PH, Modell S, Nygren A, Ingvar M, et al. Neuroendocrine, cognitive and structural imaging characteristics of women on longterm sickleave with job stress-induced depression. Biol Psychiatry. (2006) 60:867–73. doi: 10.1016/j.biopsych.2006.04.029
69. Menke A, Arloth J, Best J, Namendorf C, Gerlach T, Czamara D, et al. Time-dependent effects of dexamethasone plasma concentrations on glucocorticoid receptor challenge tests. Psychoneuroendocrinology. (2016) 69:161–71. doi: 10.1016/j.psyneuen.2016.04.003
70. Menke A, Arloth J, Putz B, Weber P, Klengel T, Mehta D, et al. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology. (2012) 37:1455–64. doi: 10.1038/npp.2011.331
71. Menke A, Arloth J, Gerber M, Rex-Haffner M, Uhr M, Holsboer F, et al. Dexamethasone stimulated gene expression in peripheral blood indicates glucocorticoid-receptor hypersensitivity in job-related exhaustion. Psychoneuroendocrinology. (2014) 44:35–46. doi: 10.1016/j.psyneuen.2014.02.013
72. Menke A, Lehrieder D, Fietz J, Leistner C, Wurst C, Stonawski S, et al. Childhood trauma dependent anxious depression sensitizes HPA axis function. Psychoneuroendocrinology. (2018) 98:22–9. doi: 10.1016/j.psyneuen.2018.07.025
73. Rampp C, Eichelkraut A, Best J, Czamara D, Rex-Haffner M, Uhr M, et al. Sex-related differential response to dexamethasone in endocrine and immune measures in depressed in-patients and healthy controls. J Psychiatr Res. (2018) 98:107–15. doi: 10.1016/j.jpsychires.2017.12.020
74. Arloth J, Bogdan R, Weber P, Frishman G, Menke A, Wagner KV, et al. Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron. (2015) 86:1189–202. doi: 10.1016/j.neuron.2015.05.034
75. Schatzberg AF. Anna-Monika Award Lecture, DGPPN Kongress, 2013: the role of the hypothalamic-pituitary-adrenal (HPA) axis in the pathogenesis of psychotic major depression. World J Biol Psychiatry. (2015) 16:2–11. doi: 10.3109/15622975.2014.916414
76. Block TS, Kushner H, Kalin N, Nelson C, Belanoff J, Schatzberg A. Combined analysis of mifepristone for psychotic depression: plasma levels associated with clinical response. Biol Psychiatry. (2018) 84:46–54. doi: 10.1016/j.biopsych.2018.01.008
77. Soria V, Gonzalez-Rodriguez A, Huerta-Ramos E, Usall J, Cobo J, Bioque M, et al. Targeting hypothalamic-pituitary-adrenal axis hormones and sex steroids for improving cognition in major mood disorders and schizophrenia: a systematic review and narrative synthesis. Psychoneuroendocrinology. (2018) 93:8–19. doi: 10.1016/j.psyneuen.2018.04.012
78. Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. (1991) 43:425–73.
79. Holsboer F. The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J Psychiatr Res. (1999) 33:181–214. doi: 10.1016/S0022-3956(98)90056-5
80. Berridge CW, Dunn AJ. A corticotropin-releasing factor antagonist reverses the stress-induced changes of exploratory behavior in mice. Horm Behav. (1987) 21:393–401. doi: 10.1016/0018-506X(87)90023-7
81. Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M, et al. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. (2000) 34:171–81. doi: 10.1016/S0022-3956(00)00016-9
82. Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry. (2008) 165:617–20. doi: 10.1176/appi.ajp.2008.07071199
83. Spierling SR, Zorrilla EP. Don't stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology. (2017) 234:1467–81. doi: 10.1007/s00213-017-4556-2
84. Schwandt ML, Cortes CR, Kwako LE, George DT, Momenan R, Sinha R, et al. The CRF1 antagonist verucerfont in anxious alcohol-dependent women: translation of neuroendocrine, but not of anti-craving effects. Neuropsychopharmacology. (2016) 41:2818–29. doi: 10.1038/npp.2016.61
85. Dunlop BW, Rothbaum BO, Binder EB, Duncan E, Harvey PD, Jovanovic T, et al. Evaluation of a corticotropin releasing hormone type 1 receptor antagonist in women with posttraumatic stress disorder: study protocol for a randomized controlled trial. Trials. (2014) 15:240. doi: 10.1186/1745-6215-15-240
86. Dunlop BW, Binder EB, Iosifescu D, Mathew SJ, Neylan TC, Pape JC, et al. Corticotropin-releasing factor receptor 1 antagonism is ineffective for women with posttraumatic stress disorder. Biol Psychiatry. (2017) 82:866–74. doi: 10.1016/j.biopsych.2017.06.024
87. Murrough JW, Charney DS. Corticotropin-releasing factor type 1 receptor antagonists for stress-related disorders: time to call it quits? Biol Psychiatry. (2017) 82:858–60. doi: 10.1016/j.biopsych.2017.10.012
88. Lloyd RB, Nemeroff CB. The role of corticotropin-releasing hormone in the pathophysiology of depression: therapeutic implications. Curr Top Med Chem. (2011) 11:609–17. doi: 10.2174/1568026611109060609
89. Ara I, Bano S. Citalopram decreases tryptophan 2,3-dioxygenase activity and brain 5-HT turnover in swim stressed rats. Pharmacol Rep. (2012) 64:558–66. doi: 10.1016/S1734-1140(12)70851-4
90. Gjerde H, Morland J, Olsen H. The antiglucocorticoid RU486 inhibits the ethanol-induced increase of tryptophan oxygenase. J Steroid Biochem. (1985) 23:1091–2. doi: 10.1016/0022-4731(85)90074-3
91. Wolkowitz OM, Reus VI. Treatment of depression with antiglucocorticoid drugs. Psychosom Med. (1999) 61:698–711. doi: 10.1097/00006842-199909000-00011
92. Gibney SM, Fagan EM, Waldron AM, O'Byrne J, Connor TJ, Harkin A. Inhibition of stress-induced hepatic tryptophan 2,3-dioxygenase exhibits antidepressant activity in an animal model of depressive behaviour. Int J Neuropsychopharmacol. (2014) 17:917–28. doi: 10.1017/S1461145713001673
93. Pantouris G, Mowat CG. Antitumour agents as inhibitors of tryptophan 2,3-dioxygenase. Biochem Biophys Res Commun. (2014) 443:28–31. doi: 10.1016/j.bbrc.2013.11.037
94. Matosin N, Halldorsdottir T, Binder EB. Understanding the molecular mechanisms underpinning gene by environment interactions in psychiatric disorders: the FKBP5 model. Biol Psychiatry. (2018) 83:821–30. doi: 10.1016/j.biopsych.2018.01.021
95. Schmidt MV, Paez-Pereda M, Holsboer F, Hausch F. The prospect of FKBP51 as a drug target. ChemMedChem. (2012) 7:1351–9. doi: 10.1002/cmdc.201200137
96. Gopalakrishnan R, Kozany C, Gaali S, Kress C, Hoogeland B, Bracher A, et al. Evaluation of synthetic FK506 analogues as ligands for the FK506-binding proteins 51 and 52. J Med Chem. (2012) 55:4114–22. doi: 10.1021/jm201746x
97. Gopalakrishnan R, Kozany C, Wang Y, Schneider S, Hoogeland B, Bracher A, et al. Exploration of pipecolate sulfonamides as binders of the FK506-binding proteins 51 and 52. J Med Chem. (2012) 55:4123–31. doi: 10.1021/jm201747c
98. Gaali S, Gopalakrishnan R, Wang Y, Kozany C, Hausch F. The chemical biology of immunophilin ligands. Curr Med Chem. (2011) 18:5355–79. doi: 10.2174/092986711798194342
99. Blackburn EA, Walkinshaw MD. Targeting FKBP isoforms with small-molecule ligands. Curr Opin Pharmacol. (2011) 11:365–71. doi: 10.1016/j.coph.2011.04.007
100. Gaali S, Kirschner A, Cuboni S, Hartmann J, Kozany C, Balsevich G, et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol. (2015) 11:33–7. doi: 10.1038/nchembio.1699
101. Kolos JM, Voll AM, Bauder M, Hausch F. FKBP Ligands—where we are and where to go? Front Pharmacol. (2018) 9:1425. doi: 10.3389/fphar.2018.01425
Keywords: precision medicine, personalized medicine, biomarker, depression, HPA axis, glucocorticoid receptor, CRH1, FKBP5
Citation: Menke A (2019) Is the HPA Axis as Target for Depression Outdated, or Is There a New Hope? Front. Psychiatry 10:101. doi: 10.3389/fpsyt.2019.00101
Received: 15 October 2018; Accepted: 11 February 2019;
Published: 28 February 2019.
Edited by:
Christian Sander, Leipzig University, GermanyReviewed by:
Casimiro Cabrera Abreu, Queens University, CanadaAndrea Norcini Pala, Columbia University, United States
Copyright © 2019 Menke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Menke, Menke_A@ukw.de