 Miguel A. Aon
Miguel A. Aon Niraj Bhatt
Niraj Bhatt Sonia C. Cortassa
Sonia C. Cortassa- Division of Cardiology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Current scientific debates center on the impact of lipids and mitochondrial function on diverse aspects of human health, nutrition and disease, among them the association of lipotoxicity with the onset of insulin resistance in skeletal muscle, and with heart dysfunction in obesity and diabetes. Mitochondria play a fundamental role in aging and in prevalent acute or chronic diseases. Lipids are main mitochondrial fuels however these molecules can also behave as uncouplers and inhibitors of oxidative phosphorylation. Knowledge about the functional composition of these contradictory effects and their impact on mitochondrial-cellular energetics/redox status is incomplete. Cells store fatty acids (FAs) as triacylglycerol and package them into cytoplasmic lipid droplets (LDs). New emerging data shows the LD as a highly dynamic storage pool of FAs that can be used for energy reserve. Lipid excess packaging into LDs can be seen as an adaptive response to fulfilling energy supply without hindering mitochondrial or cellular redox status and keeping low concentration of lipotoxic intermediates. Herein we review the mechanisms of action and utilization of lipids by mitochondria reported in liver, heart and skeletal muscle under relevant physiological situations, e.g., exercise. We report on perilipins, a family of proteins that associate with LDs in response to loading of cells with lipids. Evidence showing that in addition to physical contact, mitochondria and LDs exhibit metabolic interactions is presented and discussed. A hypothetical model of channeled lipid utilization by mitochondria is proposed. Direct delivery and channeled processing of lipids in mitochondria could represent a reliable and efficient way to maintain reactive oxygen species (ROS) within levels compatible with signaling while ensuring robust and reliable energy supply.
Discovery consists of seeing what everybody has seen and thinking what nobody has thought.
Albert Szent-Gyorgyi
Introduction
The role of lipids in human health, nutrition, and disease is taking center stage. Several circumstances including hotly debated issues concur to explain this unusual interest. Among them, pressing societal and biomedical issues concerning the epidemic proportions of obesity and related diseases in the United States and its increasing prevalence worldwide. Higher food consumption, decline in physical activity and a progressively aging population are among the social and behavioral roots of this phenomenon. Biologically, it adopts the form of a so-called “metabolic syndrome,” a set of comorbidities including upper body obesity, insulin resistance, dyslipidemia, and hypertension that increase the risk for developing type 2 diabetes, coronary artery disease, and stroke (Kok and Brindley, 2012; Schilling and Mann, 2012).
Functional impairments associated with increased circulating levels of lipids and their induced metabolic alterations in fatty acids (FAs) utilization and intracellular signaling, have been broadly termed lipotoxicity (Wende et al., 2012). Current scientific debates concern the association of lipotoxicity with the onset of insulin resistance in skeletal muscle, and with heart dysfunction in obese and diabetic patients.
Mitochondrial function is closely associated with the mounting attention on lipids. One obvious reason is that mitochondria are the main site of lipid degradation. However, the major driving force underlying this association is the fundamental role played by mitochondrial dysfunction in aging and acute or chronic disease conditions such as metabolic disorders (obesity, diabetes), cancer, inflammatory disorders, neurodegeneration, and cardiovascular disease (Akar et al., 2005; Aon et al., 2009; Bugger and Abel, 2010; Camara et al., 2011; Martinez-Outschoorn et al., 2012; Wallace, 2012; Helguera et al., 2013; Cortassa et al., 2014; Rossignol and Frye, 2014).
Cells protect themselves from lipotoxicity or death (Bernardi et al., 2002; Penzo et al., 2002) by either oxidizing FAs or sequestering them as triacylglycerol (TAG) within lipid droplets (LDs) (Greenberg et al., 2011; Walther and Farese, 2012) (Figure 1). The ability to store TAG in LDs is evolutionarily conserved and observed in yeast, plants, invertebrates, and vertebrates (Walther and Farese, 2012). LDs constitute a highly dynamic FA storage pool that can be used for energy reserve. Recent evidence shows that acute exercise can trigger changes in the dynamics of LD assembly, morphology, localization and mobilization in skeletal muscle, a process regulated by a broad genetic program affecting the spatial and metabolic interaction between mitochondria and LDs. In this process, the exercise-responsive transcriptional coactivator PGC-1α appears to play a key role in coordinating intramuscular LD programming with mitochondrial remodeling (Koves et al., 2013).
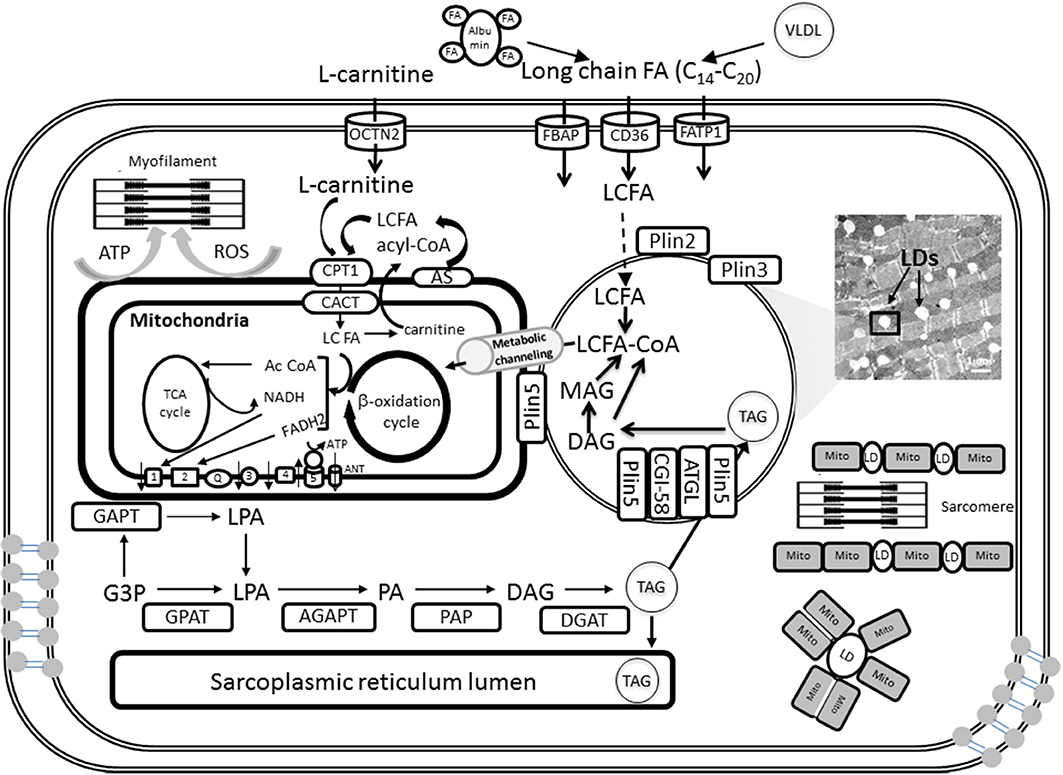
Figure 1. Triglyceride synthesis, storage in lipid droplets, and FA oxidation in cardiomyocyte mitochondria. A detailed explanation of the processes depicted in this figure will be found in sections Lipid Droplets and TAG Metabolism and Fatty Acids and Mitochondrial Function of the main text. LDs can be intercalated with mitochondria or surrounded by them as shown schematically at the right bottom. When mitochondria and LD interact in close contact the scheme suggests that FA degradation and activation occur at the contact sites between both organelles. FA precursors of β-oxidation will be subsequently metabolically channeled to the matrix, and likely β-oxidation, through known pathways (see section Metabolic Channeling of Lipid Utilization From Close Contacts Between Mitochondria and Lipid Droplets: A Hypothetical-Qualitative Model in the text for more details).
There is abundant anecdotal evidence describing close interaction between mitochondria and LD. Early observations indicated that mitochondria are often located near a supply of substrate, or at sites in the cell known to require the ATP generated by the mitochondrion (Lehninger, 1965). Occasional close associations between mitochondria and LDs were found in a variety of tissues such as myocardium, liver, pancreas, and brown adipose. As described by Ghadially (1997):
“… A single mitochondrion may appear close to, spread out over, or fused to the surface of a small LD, or several mitochondria may be seen surrounding a larger LD. In other instances, the LD may lie in a deep invagination of the mitochondrial envelope, and it is clear that in another plane of sectioning such a droplet could easily be mistaken for a lipid inclusion in the mitochondrion…, particularly if the invaginating membranes are not visualized.”
As early as 1958, Palade and Schidlowski suggested that these close associations were meaningful because they “bring the mitochondrial enzymes into close contact with the lipidic substrate” (Palade and Schidlowski, 1958, quoted by Ghadially, 1997). Although potential artifacts from sample preparation cannot be ruled out, and that pathologically altered mitochondria can have an influence, when describing lipidic inclusions in mitochondria, Ghadially (1997) wrote:
“… lipidic inclusions were noted in normal-looking mitochondria with well-formed cristae, where presumably the lipid has a physiological role.”
More recent experimental data puts on a more solid ground the idea that there are both physical and metabolic interactions between LD and mitochondria. These interactions appear to be modulated by relevant physiological situations such as fasting and exercise training. Available evidence also shows that proteins located in the LD surface closely interact with enzymes of the lypolytic cascade modulating FA acid efflux from the droplet.
Lipid Droplets and TAG Metabolism
TAG is the major form of energy storage that with sterol esters serve as reservoirs of membrane lipid components (Walther and Farese, 2009). In cardiomyocytes TAGs are synthesized by acyltransferases and phosphatases at the sarcoplasmic reticulum and mitochondrial membrane and then packaged into LDs (Walther and Farese, 2009; Singh and Cuervo, 2012; Kienesberger et al., 2013). TAG synthesis is initiated by glycerol-3-phosphate acyltransferases (GPAT) at the mitochondrial and sarcoplasmic reticulum membrane and then completed at the sarcoplasmic reticulum by sn-1-acyl-glycerol-3-phosphate acyltransferase (AGPAT), phosphatidic acid phosphatase (PAP), and sn-1,2-diacylglycerol acyltransferase (DGAT) reactions (Kienesberger et al., 2013) (Figure 1). Newly formed TAGs are packaged into cytoplasmic LDs. Thus, lipids are not stored as FAs but as TAGs (triglycerides) produced by a series of esterification reactions that combine three FA molecules with glycerol 3-phosphate; for example, the TAG for palmitate is tripalmitin.
LDs are considered dynamic cellular organelles rather than simple lipid storage depots that, relatively recently, have been implicated in many biological processes (Walther and Farese, 2009, 2012; Greenberg and Coleman, 2011; Singh and Cuervo, 2012). LDs size varies from a diameter of 0.1 μm in yeast to over 100 μm in a white adipocyte. LDs consist of a single protein-decorated phospholipid monolayer that delimits their hydrophobic core from the rest of the cell (Fujimoto and Parton, 2011). The hydrophobic core contains neutral lipids, most notably TAG and sterol esters. The adipose tissue LD has a core predominantly formed by TAG whereas in most cells cholesterol and TAG share the nuclear core of the LD (Singh and Cuervo, 2012). LDs are prominent in many types of mammalian cells, with adipocytes being the most highly specialized for lipid and energy storage. LDs interact with the endoplasmic reticulum and the mitochondria—the two organelles that have been proposed as sites of formation of the autophagosome limiting membrane (Fujimoto et al., 2008; Murphy et al., 2009; Singh and Cuervo, 2012). Such contact zones are also sites of active lipid synthesis enriched in Acyl CoA:diacylglycerol acyltransferase 2 (DGAT2), the major enzyme catalyzing TAG synthesis (Cases et al., 2001; Walther and Farese, 2009).
TAG stored in LDs is catabolized by the sequential hydrolysis of ester bonds between FAs and the glycerol backbone. TAG hydrolysis is a tightly regulated process that involves a complex interaction between lipases and regulatory proteins (Lass et al., 2011). TAG catabolism is performed by a cascade of lipolytic reactions that is initiated by adipose triglyceride lipase (ATGL) producing diacylglycerol (DAG). Hormone-sensitive lipase (HSL) and monoacylglycerol lipase (MGL) complete the lipolytic cascade by sequentially hydrolyzing DAG and monoacylglycerol (MAG), respectively, (Figure 1). MAG lipase (MGL) performs the final step in TAG catabolism by hydrolyzing MAGs to glycerol and FAs (Kienesberger et al., 2013). The rate of lipolysis can be dramatically stimulated by adrenergic hormones via activation of protein kinase A (PKA). PKA phosphorylates perilipin and HSL and causes a complex set of events leading to TAG hydrolysis.
The FAs released during TAG catabolism are mainly used for β-oxidation and subsequent ATP synthesis via OxPhos in mitochondria (Figure 1; see below: Fatty Acids and Mitochondrial Function). In oxidative tissues such as the heart, TAG-derived FAs are utilized as an energy source, but they also serve as signaling molecules as well as building blocks for membranes and complex lipids.
Hepatocytes, heart and skeletal myocytes, adrenocortical cells, enterocytes, and macrophages may all contain large amounts of LDs. Excessive LD accumulation is a hallmark of T2DM, obesity, atherosclerosis, hepatic steatosis, and other metabolic diseases. However, in certain organs like skeletal muscle, intramyocellular triacylglycerol (IMTG) accumulation is not strictly a pathologic phenomenon (see below: Mitochondria, Lipids and Insulin Resistance). Lipid content is elevated in red compared with white skeletal muscles and increases in response to habitual exercise in both oxidative and glycolytic fibers. The “athlete paradox” consists of IMTG accumulation observed in endurance-trained athletes that retain insulin sensitivity irrespective of the fact that in some cases IMTGs exceed those measured in sedentary obese or T2DM obese patients (Goodpaster et al., 2001; van Loon et al., 2003; Shaw et al., 2010; Egan and Zierath, 2013; Koves et al., 2013). As with aerobic exercise, both muscle glycogen and IMTG contribute to energy provision during resistance exercise (Koopman et al., 2006).
Mitochondria and Perilipins
The protein family of perilipins (Plin) is associated with LDs. As scaffolding proteins perilipins affect the spatial and metabolic interactions between LD and mitochondria (Figure 1). Development of tissue lipotoxicity and dysfunction is linked to alterations in LD biogenesis and regulation of TAG hydrolysis (Wang and Sztalryd, 2011). Since in response to lipid loading of cells perilipins associate with LDs the role of these proteins is under intense scrutiny.
The Plin protein family, or PAT for perilipin/ADRP/TIP47, is constituted by Plin1 to Plin5, and droplets may contain various combinations of them (Greenberg et al., 2011). Plin1 is the most abundant PAT protein in adipocytes and Plin2 in the liver, where it has been linked to hepatic steatosis. Whereas Plin1 and 4 are limited to adipose tissue, Plin2 and 3 are ubiquitous. Plin1 and 2 are always found in an LD-bound state whereas Plin3 to 5 can be either LD-bound or free in the cytoplasm.
Genetic manipulations aiming at ablating perilipins to infer about their physiological roles and impact on fat deposition have been performed. Plin1-null mice are lean and develop systemic insulin resistance as they grow older. Plin1-null adipocytes exhibited enhanced rates of constitutive (unstimulated) lipolysis and reduced catecholamine-stimulated lipolysis (Tansey et al., 2001). Together, these data suggested that Plin1 protein enhances catecholamine-stimulated lipolysis and, importantly, that a reduction in Plin1 protein expression is associated with increased constitutive lipolysis, which can promote systemic insulin resistance (Greenberg et al., 2011).
Plin5 is found primarily in oxidative tissues, e.g. skeletal and heart muscles, liver (Bickel et al., 2009). Plin5 knockout mice lacked detectable LDs in the heart and had significantly reduced myocardial TAG content, an effect that was rescued by lipase inhibition (Kuramoto et al., 2012). The excessive TAG catabolism exhibited by Plin5-deficient hearts was paralleled by increased FA oxidation (FAO) and enhanced ROS levels that led to an age-dependent decline in heart function. Thus, it was suggested that uncontrolled lipolysis and defective TAG storage impair cardiac function through chronic mitochondrial FA overload. Plin5 may regulate LD degradation and the flux of lipolysis-derived FAs to mitochondria for energy production (Figure 1) (Kienesberger et al., 2013). Plin5 overexpression in cardiac muscle produced a robust increase in LDs resulting in cardiac steatosis but without major consequences for heart function. This data indicated that Plin5 plays a critical role in droplet formation and stabilization via its regulatory role of lipolysis in vivo (Wang et al., 2013). Interestingly, mitochondria in heart tissue from the Plin5 overexpressor appeared to always be distributed in tight clusters around LDs exhibiting a significant increase in size without changes in number as revealed by morphometric analysis (Wang et al., 2013). In skeletal muscle, Plin5 overexpression increased IMCL content without hindering insulin mediated glucose uptake while promoting the expression of genes involved in mitochondrial FAO and fat catabolism (Bosma et al., 2013).
In liver, down-modulation of Plin2 promotes a reduction in hepatic steatosis and increases insulin sensitivity, but a reduction in both Plin2 and Plin3 causes insulin resistance (Greenberg et al., 2011). In the heart, Plin2 does not promote the interaction of mitochondria with LDs, but increased TAG accumulation associated with reduced presence of ATGL in LD and decreased lipolysis (Wang et al., 2011). As the first enzyme from the lipolytic cascade (Zimmermann et al., 2004), the constitutive activity of ATGL is predominantly responsible for basal levels of lipolysis (Greenberg et al., 2011). ATGL overexpression in a cardiomyocyte-specific manner decreased myocardial TAG and lipotoxic intermediates accumulation in type 1 diabetic mice (Pulinilkunnil et al., 2013). This resulted in decreased reliance on FAO, and preserved content of respiratory complexes as well as cardiac function during early stages of diabetes.
Overall, the reported data indicate that reduced expression of perilipins may promote both lipolysis and fat oxidation, resulting in reduced intracellular TAG and adipose mass. On the other hand, excessive lypolysis and defective lipid storage may promote insulin resistance and impaired cardiac function through chronic mitochondrial FA overload. Consequently, lipid storage and utilization appears to be a tightly regulated cellular process.
Fatty Acids and Mitochondrial Function
Preservation of the intracellular redox environment (RE) is crucial for vital functions such as division, differentiation, contractile work and survival amongst others (Schafer and Buettner, 2001; Aon et al., 2007, 2009; Brown et al., 2010; Fisher-Wellman and Neufer, 2012; Jeong et al., 2012; Lloyd et al., 2012; Muoio and Neufer, 2012; Aggarwal and Makielski, 2013). Mitochondria are main drivers of the intracellular RE (Aon et al., 2010, 2012; Stanley et al., 2011; Tocchetti et al., 2012; Fisher-Wellman et al., 2013; Kembro et al., 2013) and together with peroxisomes constitute the main subcellular compartments where lipid degradation occurs. Yet, the impact of lipids on mitochondrial redox status and ROS emission, and their links to energetics are not fully elucidated.
FAs are main metabolic fuels in heart and skeletal muscle, and β-oxidation represents their main degradation pathway. The rate of β-oxidation is led by demand since an increase in work rate and ATP utilization leads to faster oxidative phosphorylation (OxPhos) and tricarboxylic acid (TCA) cycle activity. In turn, the decrease in NADH and acetyl-CoA (AcCoA) levels leads to an increase of the β-oxidation flux (Neely et al., 1969; Oram et al., 1973; Eaton et al., 1996a; Eaton, 2002; Lopaschuk et al., 2010).
Lipids are supplied in the form of albumin-bound FAs secreted from adipose tissue or by catabolism of very low density lipoprotein (VLDL) complex by coronary vascular endothelial lipoprotein lipases (Figure 1). Long chain FA (LCFA) transport requires carrier proteins in the sarcolemma (FATP1, fatty acid transporter protein 1; FABP, plasma membrane-associated fatty acid-binding protein; LCFAT, long-chain fatty acid transporter; OCTN2, plasma membrane sodium-dependent carnitine transporter; FAT/CD36, fatty acid translocase CD36) and the mitochondria (CPT1, carnitine palmitoyltransferase 1; CACT, carnitine:acylcarnitine translocase).
Upon entry into the cell, LCFA first gets activated by forming thioesters with coenzyme A (CoA), LCFA-CoA, and is either oxidized in the mitochondria via β-oxidation or forms TAG by esterification (Figure 1). Subsequently TAGs can be stored in the form of LD. Long-chain FAs are activated on the mitochondrial outer membrane by the long-chain acyl-CoA synthetase but the mitochondrial inner membrane is not permeable to these acyl-CoAs. CPT1 catalyzes the conversion of long-chain acyl CoA to long-chain acylcarnitine, which is subsequently shuttled into the mitochondria (Lopaschuk et al., 2010). Control at the level of CPT1 activity appears to be important in heart and skeletal muscle β-oxidation flux (Awan and Saggerson, 1993; Lopaschuk et al., 1994; Zammit, 1999; Eaton, 2002).
After its formation by CPT1, the long-chain acylcarnitine is translocated across the inner mitochondrial membrane by CACT that involves the exchange of carnitine for acylcarnitine. CACT has extremely high activity in most cell types with active β-oxidation (Ramsay and Tubbs, 1976; Noel et al., 1985; Eaton, 2002). CACT is a critical step in the translocation of FA moieties into the mitochondria, as evidenced by the development of cardiomyopathies and irregular heartbeats in individuals with CACT deficiencies (Lopaschuk et al., 1994, 2010).
In the matrix, acylcarnitine is converted back to acyl CoA and catabolized via β-oxidation. The β-oxidation of activated FAs occurs within the mitochondrial matrix and is catalyzed by the sequential action of four enzyme families (acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl- CoA thiolase), with acyl-CoA dehydrogenase exhibiting different substrate specificity for short-, medium-, long- and very long-chain acyl-CoAs (Kunau et al., 1995; Eaton et al., 1996a; Kerner and Hoppel, 2000). The end product of each cycle of β-oxidation is AcCoA, shortening the LCFA by 2 carbons. Ac CoA then enters the TCA cycle for complete oxidation rendering reducing equivalents in the form of the electron donors NADH and FADH2 leading to ATP synthesis via OxPhos in the respiratory chain (Figure 1). Ultimately, ATP is utilized by the contractile machinery to transduce chemical energy into mechanical work. ROS may also affect contractile performance via signaling or redox modification of sensitive cysteines from, e.g., myosin heavy chain (Canton et al., 2011; Steinberg, 2013).
Besides their metabolic role in the provision of energy, long-chain free FAs exert diverse effects on cellular membranes and on the catalytic activities of many enzymes (Loskovich et al., 2005). FAs play the dual role of uncouplers and inhibitors of mitochondrial respiration (Wojtczak and Schonfeld, 1993) through a protonophoric effect on the inner membrane, and an inhibitory action on the electron transfer chain (Schonfeld and Reiser, 2006; Schonfeld and Wojtczak, 2007, 2008). Additionally, FAs have the potential to drastically alter mitochondrial membranes permeability through opening of the permeability transition pore (Scorrano et al., 2001; Bernardi et al., 2002; Penzo et al., 2002, 2004). Excluded from these effects are the acyl-CoAs that do not exert protonophoric activity and do not uncouple OxPhos because they are unable to cross the inner mitochondrial membrane (Wojtczak, 1976).
Free FAs can act as specific complex I-directed inhibitors (Loskovich et al., 2005; Schonfeld and Wojtczak, 2008), and long-chain acyl-CoAs are known inhibitors of ANT (Pande and Blanchaer, 1971; Lerner et al., 1972; Wojtczak, 1976). The inhibition is of a competitive character (Duszynski and Wojtczak, 1975) and strongly depends on the carbon chain length of the fatty acyl moiety (Morel et al., 1974). Further evidence that FAs, in their anionic form, can be substrates for transport by ANT was given by their inhibitory effect on ATP and ADP exchanges (Wojtczak and Zaluska, 1967; Schonfeld et al., 1996; Klingenberg, 2008). According to the FA cycling model (Skulachev, 1991) undissociated FA molecules can undergo a spontaneous flip-flop from the outer to the inner leaflet of the inner mitochondrial membrane where they release protons because of the alkaline milieu of the matrix. Then, in the form of anions, they are transported back to the external leaflet by ANT; one proton is transferred from the external space to the matrix compartment per molecule of the FA per cycle. In this manner, FAs can lead to energy dissipation through a selective protonophoric action mediated by coupling of transmembrane movement of the fatty acyl anion (via the ANT, uncoupling proteins, UCPs, and/or other inner membrane carriers). These events result in dissipative proton cycling that decreases the proton motive force thereby affecting respiration, ATP synthesis, and ion homeostasis.
Palmitoyl CoA inhibits the ANT independently from β-oxidation, according to more recent evidence obtained in isolated mitochondria from rat liver (Ciapaite et al., 2005) and guinea pig heart (Aon and Cortassa, unpublished) respiring on G/M. In the case of liver mitochondria it was shown that the ANT inhibition induced changes in intra- and extra-mitochondrial ATP concentrations and ΔΨm. This interference with the ANT carrier increased ΔΨm and the reduction level of coenzyme Q (Bakker et al., 2000) both expected to promote the formation of ROS. Studies further showed that the PCoA-elicited concentration-dependent H2O2 formation can be explained by its effect on ΔΨm that in the presence of 5 μM PCoA showed a 13 mV increase (Ciapaite et al., 2006). The specific action of PCoA on the ANT in the liver (Ciapaite et al., 2006), is in contrast with an apparent multi target effect in the heart (Aon and Cortassa, unpublished). These differences may be given by intrinsic functional differences due to species (rat, guinea pig) or organ specificity, e.g., liver and heart mitochondria. Differences may also be linked to the presence of distinct FA transporters (FATPs or SLC27As) or FA binding proteins (FABPs).
Mitochondria, Lipids, and Insulin Resistance
The shift from intermediate values of RE, corresponding to ROS levels compatible with signaling functions (Aon et al., 2010; Cortassa et al., 2014), toward either more reducing or oxidizing conditions is a topic of great potential importance and interest with implications for insulin signaling. Indeed, the association between lipotoxicity and the onset of insulin resistance in skeletal muscle is a hotly debated subject (Muoio and Neufer, 2012). One side posits that it is due to dysfunctional mitochondria with intrinsic deficiencies in OxPhos and deficits in fat oxidation. These impairments impinge on insulin signaling by diverting FAs away from oxidation and toward production of DAGs, ceramide and other toxic lipid species (Lowell and Shulman, 2005; Roden, 2005). The other side of the debate notes that this idea is incompatible with the principles of bioenergetics because mitochondrial respiration is governed by energy demand; intracellular lipids will accumulate whenever FAs supply exceeds the energy needs of the cell. Consequently, they suggest that the etiology of muscle insulin resistance is grounded on the fundamental principles that govern cellular and mitochondrial bioenergetics and the redox stress that is placed on the respiratory system when energy supply persistently outpaces energy demand (Muoio and Neufer, 2012). In agreement with this idea other authors have emphasized that the matching between increased FA availability and oxidative capacity distinguishes the increase in IMTG following endurance training from obesity/diabetic conditions. Chronic exercise training can elicit high oxidative capacity conferred by higher mitochondrial content but not mitochondrial function. Under these conditions, lipid infusion in endurance-trained athletes is able to reduce insulin sensitivity only by 29% as compared to 63% in untrained subjects (Phielix et al., 2012).
Whereas in exercise training IMTG reflects an increased reliance on fats as substrate, in obesity/diabetes will imply accumulation of lipid metabolites [long chain fatty acyl-CoA (LCFA-CoA), DAG, and ceramide] that are responsible for the impairment in insulin action rather than the IMTG pool contained in LDs (Schrauwen et al., 2010; Fisher-Wellman and Neufer, 2012). Apparently, increased concentrations of intramuscular LCFA-CoA and DAG activate PKC, which appears to induce impairments in insulin signaling via serine phosphorylation of the insulin receptor substrate-1. In a model of diet-induced obesity, accumulation of acylcarnitines, as products of incomplete β-oxidation, was shown in skeletal muscle (Koves et al., 2008). These findings led to the idea of a mitochondria-derived signal that couples incomplete β-oxidation with insulin resistance. Chronic elevations of incomplete oxidation intermediates of FAs and branched-chain amino acids (Newgard, 2012) might foster a mitochondrial microenvironment that is conducive to higher H2O2 release from mitochondria with potential to modulate insulin signaling (Fisher-Wellman and Neufer, 2012; Muoio and Neufer, 2012).
The debate about the role of mitochondrial and lipid metabolism at the origin of insulin resistance is highly relevant for the diabetic heart because of its heavy dependence on fats for function (Holloway et al., 2009, 2011). The debate centered on the mitochondrial load-oxidative potential in skeletal muscle, is also relevant for the heart where function is led by energy demand. In fact, lipid accumulation in the heart is largely seen as a mismatch between supply and demand, i.e., lipids amass when supply outpaces demand.
A fundamentally important question still heavily debated is whether or not a shift in substrate preference toward fat oxidation lowers disease risk (Muoio and Neufer, 2012). FAs and glucose are the two major fuels driving heart contraction. In type 2 diabetes and obesity FAO is increased (Lopaschuk, 2002; Carley and Severson, 2005) but our knowledge about the combined effects of hyperglycemia, a hallmark of diabetes, and high FA availability, on metabolism, redox/ROS balance and their impact on heart function is incomplete. Although the healthy heart is flexible regarding fuel selection, in the metabolically challenged diabetic heart by high levels of glucose and fat, the factors contributing to dysfunction and which are beneficial as energy source or redox donors are still unclear. Existing compelling evidence indicates that substrate-driven redox status plays a critical role in cardiac contractile performance in type 2 diabetes where cellular/mitochondrial redox and energetics are altered (see below: Mitochondrial, Cellular and Organ Mechanisms for Managing Lipid Affluence) (Anderson et al., 2009a; Tocchetti et al., 2012). Overall, there is no disputing that lipid oxidation confers a metabolic advantage during starvation and exercise, but the role of fuel selection per se in defending against metabolic disease needs further investigation.
Mitochondrial, Cellular, and Organ Mechanisms for Managing Lipid Affluence
As important fuels of cellular function it is very well known how FAs are degraded by mitochondria. Yet, the mechanisms by which mitochondria manage lipid excess are largely unknown. The role of β-oxidation per se as an underlying cause of obesity-associated glucose intolerance remains a topic of active research and debate (Fisher-Wellman and Neufer, 2012; Muoio and Neufer, 2012). Furthermore, mitochondria play a central role in the development of diabetes and obesity complications (Bugger and Abel, 2010; Sivitz and Yorek, 2010) and their energetic/redox dysfunction is directly involved in the redox imbalance exhibited by the heart (Tocchetti et al., 2012; Frasier et al., 2013) and skeletal muscle (Anderson et al., 2009a).
Mitochondria and lipid oxidation play a predominant role as drivers of the intracellular RE. FAs are a major source of cellular ATP which, in the heart, is synthesized up to two thirds via reducing equivalents (e.g., 24 NADH, 8 FADH2 for palmitate) derived from β-oxidation in mitochondria. The higher energetic budget provided by the saturated FA palmitate (three times higher than from glucose when ATP/mol substrate is considered) in the form of reducing power provides electrons to antioxidant systems and the mitochondria respiratory/energetic machinery. In agreement with the prominent role of lipids on the intracellular redox status, it was shown that Palm determined a transition from oxidized-to-reduced cellular redox status in cardiomyocytes from type-2 diabetic (db/db) hearts abating ROS levels drastically (Tocchetti et al., 2012). This effect was coupled to a marked GSH rise both in wild type and db/db myocytes. As a consequence of its favorable effect on cellular redox balance, Palm significantly improved isoproterenol-induced contractile reserve in db/db cardiomyocytes (Tocchetti et al., 2012).
Keeping a proper cellular/mitochondrial RE is vital for optimal excitation-contraction (EC) coupling as well as energy supply in the heart (Burgoyne et al., 2012; Christians and Benjamin, 2012; Nickel et al., 2013, 2014). Intracellular redox balance affects Ca2+ handling by interfering with a wide range of proteins implicated in EC coupling (Fauconnier et al., 2007) including the SR Ca2+ release channels [the ryanodine receptors], the SR Ca2+ pumps, and the sarcolemmal Na+/Ca2+ exchanger (Zima and Blatter, 2006; Dedkova and Blatter, 2008). Also unknown is whether the mechanisms utilized by mitochondria to deal with lipid excess differ between organs. Important examples are the skeletal and cardiac muscles where β-oxidation predominates due to their lack of de novo lipogenesis (Eaton, 2002). Certainly, the organ's functional specificity plays a role. As a matter of fact, skeletal muscle is the largest glycogen storage organ (~4-fold the capacity of the liver) thus critical for glycemic control as the predominant (~80%) site of glucose disposal under insulin-stimulated conditions (DeFronzo et al., 1981; Egan and Zierath, 2013). On the other hand, the heart carries out its pump function transducing the chemical energy stored in FAs and glucose into mechanical and electrical energy. At rest, the heart cycles about 6 kg of ATP every day while beating about 100,000 times (Neubauer, 2007). Mitochondria provide the bulk of the ATP needed for cardiac muscle contraction (about two thirds) and sarcolemmal and sarcoplasmic ion transport (one third), responsible for the Ca2+ transients and electrical activity in cardiac cells (Solaini and Harris, 2005; Cortassa et al., 2009; Nickel et al., 2013).
The far higher amounts of O2 processed by the heart on a specific basis with respect to, e.g., brain and skeletal muscle (Rolfe and Brown, 1997), and its continuous activity, make this organ susceptible to oxidative damage (Burgoyne et al., 2012; Christians and Benjamin, 2012). As a matter of fact, myocardial function and the ability of the heart to tolerate stress decline with age (Lakatta and Sollott, 2002). Although the mechanisms contributing to age-related alterations in myocardial function are not fully understood, mitochondrial dysfunction, oxidative stress and the accumulation of oxidant-induced damage are major factors (Fannin et al., 1999; Suh et al., 2003; Judge et al., 2005).
Defects in mitochondrial FA β-oxidation lead to several well-known metabolic disorders, such as Reye syndrome, cardiomyopathy and sudden infant death syndrome (Roe and Ding, 2001; Yang et al., 2001). The maintenance of high levels of mitochondrial β-oxidation could reduce the excessive fat accumulation and storage leading to human obesity. Lipid overload involving TAG accumulation in non-adipose tissues characterizes disorders such as hyperlipidemia and lipodystrophies, heart dysfunction, liver disease, in both humans and in animal models of obesity and diabetes.
It is becoming increasingly clear that adequate regulation of TAG metabolism in different organs is critical for both energy metabolism and function. Liver and heart respond to the massive influx of lipids from blood by up regulating LD biogenesis, as a mechanism of defense against the toxicity of FAs, which upon esterification get converted into TAG and stored into LD (Lass et al., 2011). Failure to do so in the liver originates pathogenic conditions such as steatosis and steatohepatitis (Greenberg et al., 2011). The lipid excess situation is also relevant for heart function in T2DM where FAs are preferred fuels (Lopaschuk et al., 2010). However, under acute, non-chronic, conditions FAs can exhibit advantageous actions, especially in the heart under diabetic conditions (Tocchetti et al., 2012). Cellular TAG accumulation in LDs may be beneficial rather than detrimental because it diverts FAs from pathways leading to cytotoxicity thus serving as a buffer against lipotoxicity (Listenberger et al., 2003).
From the examples and arguments above, it is clear that lipids have a considerable impact on many cellular processes, including mitochondria. This impact influences the functional outcome of several organs such as the liver, skeletal and cardiac muscles. Deregulation of lipid metabolism produces overload that is at the origin or as an aggravating consequence of many diseases. Consequently, the fundamental as well as practical importance of unraveling the mechanisms by which mitochondria handle lipids excess cannot be overstated. First, at the most basic level, we do not know enough about lipids action on mitochondrial energetic and redox functions. Lipids can act both as uncouplers and inhibitors of OxPhos (Wojtczak and Schonfeld, 1993; Bernardi et al., 2002), and the consequences of these contradictory effects on mitochondrial energetic, redox and signaling functions are just starting to be unraveled (Schonfeld and Wojtczak, 2008). Second, besides being the main site of lipid degradation, mitochondria may be actively modulating the balance between lipid storage and utilization.
In the following sections we explore some of the new emerging mechanisms of lipid storage and utilization by mitochondria at the organelle, cellular and organ level in different physiological settings.
Close Contact Mitochondria-Lipid Droplet
Regular exercise and physical activity are considered cornerstones in the prevention, management, and treatment of numerous chronic conditions, including hypertension, coronary heart disease, obesity, T2DM, and age-related muscle wasting (sarcopenia) (Haskell et al., 2007; Colberg et al., 2010; Egan and Zierath, 2013).
Exercise training enhances mitochondrial biogenesis and performance in skeletal muscle (Irrcher et al., 2003), but not in the heart (Li et al., 2011). Whether the same is true in T2DM hearts is unclear. In electron micrographs LDs can be easily detected in type 2 diabetic (db/db) (Boudina et al., 2007) or ob/ob (Ge et al., 2012) but not in WT mice hearts. In cells LDs can be readily visualized using the fluorescent FA analog (dodecanoic acid) BODIPY that labels neutral lipids in cytoplasmic droplets (Walther and Farese, 2012).
The occurrence of close contact between mitochondria and LD in the heart is remarkable because of its dependence on mitochondrial energetics preferentially fueled by FAs. More noteworthy though is the fact that these close contacts occur in the T2DM heart, where the dependence on fat fueling is even more prominent (Lopaschuk, 2002; Bugger and Abel, 2010). Interestingly, Plin5 overexpression in heart tissue rendered tight mitochondrial clusters around LDs with mitochondria significantly larger but not higher in number (Wang et al., 2013). The same authors proposed that Plin5 could play a regulatory role in the FA flux from LDs to mitochondria under conditions of increased cellular FA influx (Wang and Sztalryd, 2011). These data also suggest that Plin5 with its role of favoring LD accumulation may act to keep the intracellular levels of FA metabolites (e.g., DAG, ceramide) below lipotoxic amounts (see below: Metabolic Channeling of Lipid Utilization From Close Contacts Between Mitochondria and Lipid Droplets: A Hypothetical-Qualitative Model).
In skeletal muscle IMTG accumulates and is actively utilized during exercise (Shaw et al., 2010; Egan and Zierath, 2013; Koves et al., 2013). Endurance exercise training increases mitochondrial content (by size not numbers) for men and women but healthy active women have higher IMTG accumulation compared with men due to greater number rather than size of LDs (Tarnopolsky et al., 2007). Interestingly, this study also reported an increase in the physical contact between mitochondria and IMTGs following endurance exercise training. Rates of whole body fat oxidation and IMTG utilization are determined by factors such as diet, intensity and duration of exercise, and fitness. During acute exercise, the contribution of various metabolic pathways to energy provision is determined by the relative intensity and absolute power output of the exercise bout (Egan and Zierath, 2013). The rate of ATP demand and energy expenditure is determined by the absolute power output whereas the relative exercise intensity influences the relative contributions of carbohydrate oxidation and lipid sources, and circulating (extramuscular) and intramuscular fuel stores, to energy provision. As exercise intensity increases, muscle utilization of circulating free FAs slightly declines, whereas utilization of circulating glucose increases progressively up to near-maximal intensities (van Loon et al., 2001).
IMTG breakdown occurs primarily via HSL and ATGL (Watt and Spriet, 2010). Although IMTGs constitute only a small fraction (~1–2%) of whole-body lipid stores they represent an important fuel source during prolonged (>90 min) but moderate intensity exercise. IMTGs can provide ~25% of total energy however their contribution decreases at either higher or lower intensities of exercise (Romijn et al., 1993; van Loon et al., 2001). Maximal rates of fat oxidation occur at moderate exercise intensities (~ 60% VO2 max) (Shaw et al., 2010; Egan and Zierath, 2013). At low-to-moderate exercise intensity, the primary substrates fueling skeletal muscle are glucose, derived from hepatic glycogenolysis (or gluconeogenesis) or oral ingestion, and free FAs released by adipose tissue lipolysis. Prolonged exercise (>60 min) at a fixed intensity increases the energy contribution from lipid oxidation (Egan and Zierath, 2013). IMTG stores can be reduced by ~60% following exercise, predominantly in type I muscle fibers (van Loon et al., 2003; Stellingwerff et al., 2007; Shaw et al., 2010; Egan and Zierath, 2013).
Lipophagy, i.e., the turnover of LDs by autophagy, may occur due to random sequestration of cytosolic material by “in bulk” autophagy. However, when lipophagy is activated in response to a lipid challenge or prolonged starvation, a switch toward the preferential sequestration of LD seems to happen, supporting some level of selectivity in this process (Singh et al., 2009). We suggest that this may also be the case for close contacts mitochondria-LD, and that energy demand may be a main elicitor of the interaction between these two organelles. Consonant with this idea, it has been proposed that LDs assembly in skeletal muscle under exercise training would improve the management of high FA influx enabling a more precisely regulated trafficking of substrate to and from IMTG thus contributing to optimal mitochondrial performance and metabolic flexibility (Koves et al., 2013).
Lipotoxicity and LD Accumulation Dynamics
In pathologic states lipotoxicity may occur over time, despite TAG accumulation, when either the cellular capacity for TAG storage is exceeded or when triglyceride pools are hydrolyzed, resulting in increased cellular free FA levels. Thus, the duration and extent of lipid overload may determine if a cell is protected or damaged. Whether mitochondrial energy/redox status can alter the balance LD formation and utilization in the short-term is a question that has not been hitherto addressed.
Studies performed with non-invasive spectroscopic techniques have shown elevated IMCL triglyceride content in the left ventricle (i.e., LV steatosis) of obese and T2DM patients (McGavock et al., 2007; Rijzewijk et al., 2008) but its association with early diastolic dysfunction leading to subsequent systolic dysfunction remains controversial (Anderson et al., 2009b; Lopaschuk et al., 2010). Again, lipids through accumulation of triglycerides are at the center of the controversy. In skeletal (Liu et al., 2007) and cardiac (Ussher et al., 2009) muscle, IMCL accumulation as a result of diet-induced obesity is not at all pathogenic, but may even be protective against obesity-associated maladies.
Previous reports have linked ROS-mediated mitochondrial dysfunction to DAG and ceramide, two main products of lipid degradation (Coen and Goodpaster, 2012). Lipid channeling to mitochondria may represent a mechanism by which concentration build-up of these intermediaries is avoided, especially under high energy demand. Based on these premises, we suggest that temporary lipid storage in LDs does not necessarily represent pathophysiological behavior. On the contrary, it may embody an adaptive response, at least in the short-term thus representing an adaptive strategy of lipids utilization ensuring energy supply without affecting neither mitochondrial nor cellular redox status.
Redox Optimized ROS Balance and Mitochondrial Redox and Energetics
Lipid metabolites can damage the respiratory chain leading to impaired energetic transition in mitochondria through their dual effect as uncouplers and inhibitors (Wojtczak and Schonfeld, 1993). Impairment of the key state 4→3 energetic transition can occur via inhibition of ANT or ATPsynthase thereby producing a continuous release of ROS irrespective of ADP addition (Tocchetti et al., 2012).
Mitochondria are a main source of ROS but can also be their target. The RE is a major driving force of the crucial energy-redox link of mitochondrial function (Cortassa et al., 2014). The mitochondrial RE depends on the intrinsic redox potential and instantaneous reducing capacity of this organelle as well as its response to the cytoplasmic redox status (Aon et al., 2010; Kembro et al., 2013). In this context, Redox-Optimized ROS Balance (R-ORB) provides a useful conceptual framework to rationalize many results described in the present review. One of the main R-ORB postulates is that ROS efflux from mitochondria will attain a minimum at intermediate values of RE, when VO2 reaches a maximum following ADP stimulation (Figure 2) (Cortassa et al., 2014). Under state 3 respiration, glutathione and thioredoxin systems are essential for minimizing ROS release from mitochondria (Aon et al., 2010, 2012; Stanley et al., 2011; Kudin et al., 2012; Cortassa et al., 2014). In excess, lipid precursors of β-oxidation can promote mitochondrial uncoupling and oxidized redox status (Aon and Cortassa, unpublished). In more oxidized RE, away from the optimum (intermediate) RE compatible with minimal ROS, antioxidant systems become overwhelmed leading to pathological ROS overflow (Aon et al., 2010; Cortassa et al., 2014).
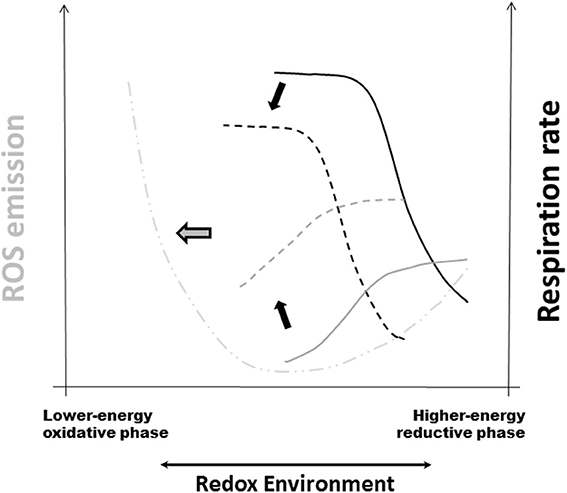
Figure 2. Redox-Optimized ROS Balance and the effect of oxidative stress on mitochondrial respiration, H2O2 emission, and the RE. R-ORB postulates that ROS levels (as the net result of production and scavenging) depend on the intra-cellular and -mitochondrial redox environment (RE). It also proposes that there is a minimum level of ROS emission when mitochondria maximize their energetic output. Under high energy demand, and despite large respiratory rates, ROS emission levels will be kept to a minimum by ROS scavenging systems (Stanley et al., 2011; Aon et al., 2012). Oxidative stress can happen at either extreme of RE, either highly reduced or highly oxidized, but governed by different mechanisms (Aon et al., 2010; Kembro et al., 2014). The plot displays schematically the summary of the response of respiration (black traces) and ROS emission in stressed mitochondria (gray traces) plus further addition of the uncoupler FCCP (dashed-dotted line). Continuous lines correspond to the absence of stress whereas dashed lines belong to mitochondria under stressed conditions (Cortassa et al., 2014). Black arrows indicate the direction of change in VO2 and ROS elicited by stress. Notice the shift toward more oxidized RE in the curves corresponding to stressful conditions. The thick gray arrow pointing to the left denotes pathological conditions arising, e.g., from chronic diseases, where severe stress will affect both energetic (e.g., ΔΨm, ADP consumption) and redox [e.g., NAD(P)H, GSH, Trx] functions thus increased mitochondrial ROS emission and higher cytoplasmic ROS levels. Reprinted from Cortassa et al. (2014).
Mitochondria function in more oxidative environments in chronic diseases (Tocchetti et al., 2012). Thus, it becomes fundamental to understand how oxidative stress influences the dependence of ROS emission on respiration (Cortassa et al., 2014). When oxidant challenged, mitochondria displayed H2O2 emission levels 2-fold higher than controls, and exhibited lower respiration (Figure 2). Oxidative stress shifted redox balance toward the more oxidized range where the sensitivity of the ROS efflux to the RE decreases more drastically in state 4 than in state 3 respiration. A 50% decrease in reduced glutathione (GSH) was mainly responsible for the shift of the RE to a more oxidized state (Cortassa et al., 2014).
Metabolic Channeling of Lipid Utilization from Close Contacts Between Mitochondria and Lipid Droplets: A Hypothetical-Qualitative Model
Recent evidence supports physical and metabolic interactions between LDs and mitochondria mediated by the scaffolding protein Plin 5 (Wang and Sztalryd, 2011; Wang et al., 2011; Koves et al., 2013). Wang and collaborators observed that Plin5-overexpressing cells show decreased LD hydrolysis and palmitate β-oxidation when compared with controls. Instead, palmitate increasingly incorporated into TAGs under basal conditions whereas in protein kinase A-stimulated state LD hydrolysis inhibition was removed and FAs released for β-oxidation. These results suggested that Plin5 regulates LD hydrolysis and controls local FA flux to protect mitochondria against excessive exposure to FA (Wang and Sztalryd, 2011). All these observations are in agreement with the relatively recent realization that the LD proteome is highly dynamic and more complex than previously thought. The LD proteome contains key components of the fat mobilization system and proteins that suggest LD interactions with a variety of cell organelles, including the mitochondria (Beller et al., 2010).
Based on the premise of metabolic links extending beyond physical contact between mitochondria and LDs, we propose a model of metabolic channeling for lipid utilization by mitochondria. According to our model, metabolic channeling represents a way mitochondria can manage lipid affluence in an energetically and redox-controlled fashion. Qualitatively, the lipid utilization channeling model postulates that after TAG degradation, lipids are directly delivered for activation, transport and β-oxidation from the LD to the mitochondrion at the contact site (Figure 1). The model also proposes that β-oxidation may also happen metabolically channeled through the enzymatic components of the lipid degradation pathway organized as a multienzyme complex (Eaton, 2002).
From a structural standpoint, the model is based on direct and close contact between LDs and mitochondria involving their recruitment and surrounding of the LD. The model also postulates membrane fusion-mediated reorganization of intra-mitochondrial membrane and molecular components (Walther and Farese, 2009) as well as lipids segregation within the droplet (Fujimoto and Parton, 2011).
Biochemically, the pathway of long-chain FAO to AcCoA is one of the longest unbranched pathways in metabolism, containing 27 intermediates between palmitoyl-CoA and AcCoA (Eaton, 2002). That the enzymes of β-oxidation may be organized into a multienzyme complex was suggested long ago. In these biomolecular assemblies, sequential catalytic reactions proceed via transfer of the intermediates between individual component enzymes, precluding their diffusion into the bulk aqueous medium, thus “metabolically channeled” (Welch, 1977; Sumegi et al., 1991).
An earlier proposal of metabolic channeling in β-oxidation was based on the detection of low concentrations of intermediates (Garland et al., 1965) and the observation that β-oxidation intermediates that accumulate behaved more like products than intermediates (Stewart et al., 1973; Stanley and Tubbs, 1974, 1975; Eaton et al., 1994, 1996a,b, 1999). This led to the “leaky hosepipe” model for the control of β-oxidation flux (Stewart et al., 1973; Stanley and Tubbs, 1974, 1975) in which channeling of a small, quickly turning-over pool of intermediates was implied (see Eaton, 2002 for a review).
Some aspects of the structural basis for a channeling mechanism in β-oxidation have been described (Ishikawa et al., 2004). Evidence in support of a multifunctional FAO complex within mitochondria, physically associated with respiratory chain supercomplexes that favor metabolic channeling, has been recently reported (Wang et al., 2010). Functionally, the direct delivery of lipids at contact sites, and their channeled processing will avoid elevation of their concentration, thus ruling out the potential inhibitory as well as uncoupling action of FAs (Wojtczak and Schonfeld, 1993). The latter will ensure a reliable and efficient energy supply.
Concluding Remarks
Mitochondria, cells and organs have developed mechanisms that allow managing heavy influx of FAs within functionally reliable limits. The LD as a dynamic storage of FAs can also be seen as a protective mechanism employed by cells to avoid excessive intracellular concentration of FAs thus hindering their potential deleterious effects on mitochondrial function. The tight and reciprocal regulation of lipid storage and utilization is evidenced by genetic manipulation of perilipins indicating that their reduced expression leads to increased lipid oxidation and reduced accumulation of intracellular fat and adipose mass. On the other hand, however, excessive lipolysis and defective lipid storage promotes insulin resistance through mitochondrial FA overload and ROS overflow.
Preservation of the intracellular RE is crucial for vital functions. Mitochondria play a decisive role as the organelle that specifically handles the highest amounts of oxygen processed by the organism thus prone not only to be the source but also the target of oxidative stress. Mitochondrial function needs to sustain energy supply reliably while releasing ROS levels compatible with signaling. However, lipids can derail both of these critical functions. Consequently, the hypothetical lipid utilization channeling model we are proposing herein satisfies the fundamentals of cellular and mitochondrial energetics and redox. In principle, diversion of excess lipids to LDs can be an effective cytoplasmic mechanism for “sequestering” FAs thereby helping to keep low concentration of lipotoxic intermediates resulting from lipid oxidation. Functionally, direct delivery and channeled processing of lipids in mitochondria could represent a reliable and efficient way to ensure energy supply and redox control. Such a mechanism would avoid exceeding the lipid storage capacity thus becoming crucial for skeletal muscle or heart subjected to high workload, and therefore, heavy influx of FAs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health grants R01-HL091923 (Miguel A. Aon) and R21HL106054 (Sonia C. Cortassa).
References
Aggarwal, N. T., and Makielski, J. C. (2013). Redox control of cardiac excitability. Antioxid. Redox Signal. 18, 432–468. doi: 10.1089/ars.2011.4234
Akar, F. G., Aon, M. A., Tomaselli, G. F., and O'Rourke, B. (2005). The mitochondrial origin of postischemic arrhythmias. J. Clin. Invest. 115, 3527–3535. doi: 10.1172/JCI25371
Anderson, E. J., Kypson, A. P., Rodriguez, E., Anderson, C. A., Lehr, E. J., and Neufer, P. D. (2009b). Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898. doi: 10.1016/j.jacc.2009.07.031
Anderson, E. J., Lustig, M. E., Boyle, K. E., Woodlief, T. L., Kane, D. A., Lin, C. T., et al. (2009a). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581. doi: 10.1172/JCI37048
Aon, M. A., Cortassa, S., Akar, F. G., Brown, D. A., Zhou, L., and O'Rourke, B. (2009). From mitochondrial dynamics to arrhythmias. Int. J. Biochem. Cell Biol. 41, 1940–1948. doi: 10.1016/j.biocel.2009.02.016
Aon, M. A., Cortassa, S., Maack, C., and O'Rourke, B. (2007). Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 282, 21889–21900. doi: 10.1074/jbc.M702841200
Aon, M. A., Cortassa, S., and O'Rourke, B. (2010). Redox-optimized ROS balance: a unifying hypothesis. Biochim. Biophys. Acta 1797, 865–877. doi: 10.1016/j.bbabio.2010.02.016
Aon, M. A., Stanley, B. A., Sivakumaran, V., Kembro, J. M., O'Rourke, B., Paolocci, N., et al. (2012). Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J. Gen. Physiol. 139, 479–491. doi: 10.1085/jgp.201210772
Awan, M. M., and Saggerson, E. D. (1993). Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation. Biochem. J. 295(pt 1), 61–66.
Bakker, S. J., IJzerman, R. G., Teerlink, T., Westerhoff, H. V., Gans, R. O., and Heine, R. J. (2000). Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis 148, 17–21. doi: 10.1016/S0021-9150(99)00329-9
Beller, M., Thiel, K., Thul, P. J., and Jackle, H. (2010). Lipid droplets: a dynamic organelle moves into focus. FEBS Lett. 584, 2176–2182. doi: 10.1016/j.febslet.2010.03.022
Bernardi, P., Penzo, D., and Wojtczak, L. (2002). Mitochondrial energy dissipation by fatty acids. Mechanisms and implications for cell death. Vitam. Horm. 65, 97–126. doi: 10.1016/S0083-6729(02)65061-7
Bickel, P. E., Tansey, J. T., and Welte, M. A. (2009). PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 1791, 419–440. doi: 10.1016/j.bbalip.2009.04.002
Bosma, M., Sparks, L. M., Hooiveld, G. J., Jorgensen, J. A., Houten, S. M., Schrauwen, P., et al. (2013). Overexpression of PLIN5 in skeletal muscle promotes oxidative gene expression and intramyocellular lipid content without compromising insulin sensitivity. Biochim. Biophys. Acta 1831, 844–852. doi: 10.1016/j.bbalip.2013.01.007
Boudina, S., Sena, S., Theobald, H., Sheng, X., Wright, J. J., Hu, X. X., et al. (2007). Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56, 2457–2466. doi: 10.2337/db07-0481
Brown, D. A., Aon, M. A., Frasier, C. R., Sloan, R. C., Maloney, A. H., Anderson, E. J., et al. (2010). Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J. Mol. Cell. Cardiol. 48, 673–679. doi: 10.1016/j.yjmcc.2009.11.011
Bugger, H., and Abel, E. D. (2010). Mitochondria in the diabetic heart. Cardiovasc. Res. 88, 229–240. doi: 10.1093/cvr/cvq239
Burgoyne, J. R., Mongue-Din, H., Eaton, P., and Shah, A. M. (2012). Redox signaling in cardiac physiology and pathology. Circ. Res. 111, 1091–1106. doi: 10.1161/CIRCRESAHA.111.255216
Camara, A. K., Bienengraeber, M., and Stowe, D. F. (2011). Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2:13. doi: 10.3389/fphys.2011.00013
Canton, M., Menazza, S., Sheeran, F. L., Polverino de Laureto, P., Di Lisa, F., and Pepe, S. (2011). Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 57, 300–309. doi: 10.1016/j.jacc.2010.06.058
Carley, A. N., and Severson, D. L. (2005). Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim. Biophys. Acta 1734, 112–126. doi: 10.1016/j.bbalip.2005.03.005
Cases, S., Stone, S. J., Zhou, P., Yen, E., Tow, B., Lardizabal, K. D., et al. (2001). Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 276, 38870–38876. doi: 10.1074/jbc.M106219200
Christians, E. S., and Benjamin, I. J. (2012). Proteostasis and REDOX state in the heart. Am. J. Physiol. Heart Circ. Physiol. 302, H24–H37. doi: 10.1152/ajpheart.00903.2011
Ciapaite, J., Bakker, S. J., Diamant, M., van Eikenhorst, G., Heine, R. J., Westerhoff, H. V., et al. (2006). Metabolic control of mitochondrial properties by adenine nucleotide translocator determines palmitoyl-CoA effects. Implications for a mechanism linking obesity and type 2 diabetes. FEBS J. 273, 5288–5302. doi: 10.1111/j.1742-4658.2006.05523.x
Ciapaite, J., Van Eikenhorst, G., Bakker, S. J., Diamant, M., Heine, R. J., Wagner, M. J., et al. (2005). Modular kinetic analysis of the adenine nucleotide translocator-mediated effects of palmitoyl-CoA on the oxidative phosphorylation in isolated rat liver mitochondria. Diabetes 54, 944–951. doi: 10.2337/diabetes.54.4.944
Coen, P. M., and Goodpaster, B. H. (2012). Role of intramyocelluar lipids in human health. Trends Endocrinol. Metab. 23, 391–398. doi: 10.1016/j.tem.2012.05.009
Colberg, S. R., Albright, A. L., Blissmer, B. J., Braun, B., Chasan-Taber, L., Fernhall, B., et al. (2010). Exercise and type 2 diabetes: American College of Sports Medicine and the American Diabetes Association: joint position statement. Exercise and type 2 diabetes. Med. Sci. Sports Exerc. 42, 2282–2303. doi: 10.1249/MSS.0b013e3181eeb61c
Cortassa, S., O'Rourke, B., and Aon, M. A. (2014). Redox-Optimized ROS Balance and the relationship between mitochondrial respiration and ROS. Biochim. Biophys. Acta 1837, 287–295. doi: 10.1016/j.bbabio.2013.11.007
Cortassa, S., O'Rourke, B., Winslow, R. L., and Aon, M. A. (2009). Control and regulation of mitochondrial energetics in an integrated model of cardiomyocyte function. Biophys. J. 96, 2466–2478. doi: 10.1016/j.bpj.2008.12.3893
Dedkova, E. N., and Blatter, L. A. (2008). Mitochondrial Ca2+ and the heart. Cell Calcium 44, 77–91. doi: 10.1016/j.ceca.2007.11.002
DeFronzo, R. A., Jacot, E., Jequier, E., Maeder, E., Wahren, J., and Felber, J. P. (1981). The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30, 1000–1007. doi: 10.2337/diab.30.12.1000
Duszynski, J., and Wojtczak, L. (1975). Effect of metal cations on the inhibition of adenine nucleotide translocation by acyl-CoA. FEBS Lett. 50, 74–78. doi: 10.1016/0014-5793(75)81044-1
Eaton, S. (2002). Control of mitochondrial beta-oxidation flux. Prog. Lipid Res. 41, 197–239. doi: 10.1016/S0163-7827(01)00024-8
Eaton, S., Bartlett, K., and Pourfarzam, M. (1996a). Mammalian mitochondrial beta-oxidation. Biochem. J. 320(pt 2), 345–357.
Eaton, S., Bartlett, K., and Pourfarzam, M. (1999). Intermediates of myocardial mitochondrial beta-oxidation: possible channelling of NADH and of CoA esters. Biochim. Biophys. Acta 1437, 402–408. doi: 10.1016/S1388-1981(99)00027-X
Eaton, S., Pourfarzam, M., and Bartlett, K. (1996b). The effect of respiratory chain impairment of beta-oxidation in rat heart mitochondria. Biochem. J. 319(pt 2), 633–640.
Eaton, S., Turnbull, D. M., and Bartlett, K. (1994). Redox control of beta-oxidation in rat liver mitochondria. Eur. J. Biochem. 220, 671–681. doi: 10.1111/j.1432-1033.1994.tb18668.x
Egan, B., and Zierath, J. R. (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184. doi: 10.1016/j.cmet.2012.12.012
Fannin, S. W., Lesnefsky, E. J., Slabe, T. J., Hassan, M. O., and Hoppel, C. L. (1999). Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 372, 399–407. doi: 10.1006/abbi.1999.1508
Fauconnier, J., Andersson, D. C., Zhang, S. J., Lanner, J. T., Wibom, R., Katz, A., et al. (2007). Effects of palmitate on Ca(2+) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes 56, 1136–1142. doi: 10.2337/db06-0739
Fisher-Wellman, K. H., Mattox, T. A., Thayne, K., Katunga, L. A., La Favor, J. D., Neufer, P. D., et al. (2013). Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. 591, 3471–3486. doi: 10.1113/jphysiol.2013.254193
Fisher-Wellman, K. H., and Neufer, P. D. (2012). Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 23, 142–153. doi: 10.1016/j.tem.2011.12.008
Frasier, C. R., Moukdar, F., Patel, H. D., Sloan, R. C., Stewart, L. M., Alleman, R. J., et al. (2013). Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc. Res. 98, 47–55. doi: 10.1093/cvr/cvt009
Fujimoto, T., Ohsaki, Y., Cheng, J., Suzuki, M., and Shinohara, Y. (2008). Lipid droplets: a classic organelle with new outfits. Histochem. Cell Biol. 130, 263–279. doi: 10.1007/s00418-008-0449-0
Fujimoto, T., and Parton, R. G. (2011). Not just fat: the structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol. 3, 1–17. doi: 10.1101/cshperspect.a004838
Garland, P. B., Shepherd, D., and Yates, D. W. (1965). Steady-state concentrations of coenzyme A, acetyl-coenzyme A and long-chain fatty acyl-coenzyme A in rat-liver mitochondria oxidizing palmitate. Biochem. J. 97, 587–594.
Ge, F., Hu, C., Hyodo, E., Arai, K., Zhou, S., Lobdell, H. 4th., et al. (2012). Cardiomyocyte triglyceride accumulation and reduced ventricular function in mice with obesity reflect increased long chain fatty acid uptake and de novo fatty acid synthesis. J. Obes. 2012:205648. doi: 10.1155/2012/205648
Ghadially, F. N. (1997). Ultrastructural Pathology of the Cell and Matrix. Boston, MA: Butterworth-Heinemann.
Goodpaster, B. H., He, J., Watkins, S., and Kelley, D. E. (2001). Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 86, 5755–5761. doi: 10.1210/jcem.86.12.8075
Greenberg, A. S., and Coleman, R. A. (2011). Expanding roles for lipid droplets. Trends Endocrinol. Metab. 22, 195–196. doi: 10.1016/j.tem.2011.04.002
Greenberg, A. S., Coleman, R. A., Kraemer, F. B., McManaman, J. L., Obin, M. S., Puri, V., et al. (2011). The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Invest. 121, 2102–2110. doi: 10.1172/JCI46069
Haskell, W. L., Lee, I. M., Pate, R. R., Powell, K. E., Blair, S. N., Franklin, B. A., et al. (2007). Physical activity and public health: updated recommendation for adults from the American College of Sports Medicine and the American Heart Association. Med. Sci. Sports Exerc. 39, 1423–1434. doi: 10.1249/mss.0b013e3180616b27
Helguera, P., Seiglie, J., Rodriguez, J., Hanna, M., Helguera, G., and Busciglio, J. (2013). Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 17, 132–140. doi: 10.1016/j.cmet.2012.12.005
Holloway, G. P., Benton, C. R., Mullen, K. L., Yoshida, Y., Snook, L. A., Han, X. X., et al. (2009). In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am. J. Physiol. Endocrinol. Metab. 296, E738–E747. doi: 10.1152/ajpendo.90896.2008
Holloway, G. P., Snook, L. A., Harris, R. J., Glatz, J. F., Luiken, J. J., and Bonen, A. (2011). In obese Zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J. Physiol. 589, 169–180. doi: 10.1113/jphysiol.2010.198663
Irrcher, I., Adhihetty, P. J., Joseph, A. M., Ljubicic, V., and Hood, D. A. (2003). Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sports Med. 33, 783–793. doi: 10.2165/00007256-200333110-00001
Ishikawa, M., Tsuchiya, D., Oyama, T., Tsunaka, Y., and Morikawa, K. (2004). Structural basis for channelling mechanism of a fatty acid beta-oxidation multienzyme complex. EMBO J. 23, 2745–2754. doi: 10.1038/sj.emboj.7600298
Jeong, E. M., Liu, M., Sturdy, M., Gao, G., Varghese, S. T., Sovari, A. A., et al. (2012). Metabolic stress, reactive oxygen species, and arrhythmia. J. Mol. Cell. Cardiol. 52, 454–463. doi: 10.1016/j.yjmcc.2011.09.018
Judge, S., Jang, Y. M., Smith, A., Hagen, T., and Leeuwenburgh, C. (2005). Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 19, 419–421. doi: 10.1096/fj.04-2622fje
Kembro, J. M., Aon, M. A., Winslow, R. L., O’Rourke, B., and Cortassa, S. (2013). Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys. J. 104, 332–343. doi: 10.1016/j.bpj.2012.11.3808
Kembro, J. M., Cortassa, S., and Aon, M. A. (2014). “Mitochondrial reactive oxygen species and arrhythmias,” in Systems Biology of Free Radicals and Antioxidants, ed I. Laher (Berlin-Heidelberg: Springer-Verlag), 1047–1076.
Kerner, J., and Hoppel, C. (2000). Fatty acid import into mitochondria. Biochim. Biophys. Acta 1486, 1–17. doi: 10.1016/S1388-1981(00)00044-5
Kienesberger, P. C., Pulinilkunnil, T., Nagendran, J., and Dyck, J. R. (2013). Myocardial triacylglycerol metabolism. J. Mol. Cell. Cardiol. 55, 101–110. doi: 10.1016/j.yjmcc.2012.06.018
Klingenberg, M. (2008). The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 1778, 1978–2021. doi: 10.1016/j.bbamem.2008.04.011
Kok, B. P., and Brindley, D. N. (2012). Myocardial fatty acid metabolism and lipotoxicity in the setting of insulin resistance. Heart Fail. Clin. 8, 643–661. doi: 10.1016/j.hfc.2012.06.008
Koopman, R., Manders, R. J., Jonkers, R. A., Hul, G. B., Kuipers, H., and van Loon, L. J. (2006). Intramyocellular lipid and glycogen content are reduced following resistance exercise in untrained healthy males. Eur. J. Appl. Physiol. 96, 525–534. doi: 10.1007/s00421-005-0118-0
Koves, T. R., Sparks, L. M., Kovalik, J. P., Mosedale, M., Arumugam, R., DeBalsi, K. L., et al. (2013). PPARgamma coactivator-1alpha contributes to exercise-induced regulation of intramuscular lipid droplet programming in mice and humans. J. Lipid Res. 54, 522–534. doi: 10.1194/jlr.P028910
Koves, T. R., Ussher, J. R., Noland, R. C., Slentz, D., Mosedale, M., Ilkayeva, O., et al. (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56. doi: 10.1016/j.cmet.2007.10.013
Kudin, A. P., Augustynek, B., Lehmann, A. K., Kovacs, R., and Kunz, W. S. (2012). The contribution of thioredoxin-2 reductase and glutathione peroxidase to H(2)O(2) detoxification of rat brain mitochondria. Biochim. Biophys. Acta 1817, 1901–1906. doi: 10.1016/j.bbabio.2012.02.023
Kunau, W. H., Dommes, V., and Schulz, H. (1995). beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog. Lipid Res. 34, 267–342. doi: 10.1016/0163-7827(95)00011-9
Kuramoto, K., Okamura, T., Yamaguchi, T., Nakamura, T. Y., Wakabayashi, S., Morinaga, H., et al. (2012). Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 287, 23852–23863. doi: 10.1074/jbc.M111.328708
Lakatta, E. G., and Sollott, S. J. (2002). Perspectives on mammalian cardiovascular aging: humans to molecules. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 132, 699–721. doi: 10.1016/S1095-6433(02)00124-1
Lass, A., Zimmermann, R., Oberer, M., and Zechner, R. (2011). Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 50, 14–27. doi: 10.1016/j.plipres.2010.10.004
Lehninger, A. L. (1965). The Mitochondrion. Molecular Basis of Structure and Function. New York, NY: W.A. Benjamin.
Lerner, E., Shug, A. L., Elson, C., and Shrago, E. (1972). Reversible inhibition of adenine nucleotide translocation by long chain fatty acyl coenzyme A esters in liver mitochondria of diabetic and hibernating animals. J. Biol. Chem. 247, 1513–1519.
Li, L., Muhlfeld, C., Niemann, B., Pan, R., Li, R., Hilfiker-Kleiner, D., et al. (2011). Mitochondrial biogenesis and PGC-1alpha deacetylation by chronic treadmill exercise: differential response in cardiac and skeletal muscle. Basic Res. Cardiol. 106, 1221–1234. doi: 10.1007/s00395-011-0213-9
Listenberger, L. L., Han, X., Lewis, S. E., Cases, S., Farese, R. V. Jr., Ory, D. S., et al. (2003). Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U.S.A. 100, 3077–3082. doi: 10.1073/pnas.0630588100
Liu, L., Zhang, Y., Chen, N., Shi, X., Tsang, B., and Yu, Y. H. (2007). Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J. Clin. Invest. 117, 1679–1689. doi: 10.1172/JCI30565
Lloyd, D., Cortassa, S., O'Rourke, B., and Aon, M. A. (2012). What yeast and cardiomyocytes share: ultradian oscillatory redox mechanisms of cellular coherence and survival. Integr. Biol. (Camb.) 4, 65–74. doi: 10.1039/c1ib00124h
Lopaschuk, G. D. (2002). Metabolic abnormalities in the diabetic heart. Heart Fail. Rev. 7, 149–159. doi: 10.1023/A:1015328625394
Lopaschuk, G. D., Belke, D. D., Gamble, J., Itoi, T., and Schonekess, B. O. (1994). Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim. Biophys. Acta 1213, 263–276. doi: 10.1016/0005-2760(94)00082-4
Lopaschuk, G. D., Ussher, J. R., Folmes, C. D., Jaswal, J. S., and Stanley, W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. doi: 10.1152/physrev.00015.2009
Loskovich, M. V., Grivennikova, V. G., Cecchini, G., and Vinogradov, A. D. (2005). Inhibitory effect of palmitate on the mitochondrial NADH:ubiquinone oxidoreductase (complex I) as related to the active-de-active enzyme transition. Biochem. J. 387, 677–683. doi: 10.1042/BJ20041703
Lowell, B. B., and Shulman, G. I. (2005). Mitochondrial dysfunction and type 2 diabetes. Science 307, 384–387. doi: 10.1126/science.1104343
Martinez-Outschoorn, U. E., Sotgia, F., and Lisanti, M. P. (2012). Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 15, 4–5. doi: 10.1016/j.cmet.2011.12.011
McGavock, J. M., Lingvay, I., Zib, I., Tillery, T., Salas, N., Unger, R., et al. (2007). Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation 116, 1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614
Morel, F., Lauquin, G., Lunardi, J., Duszynski, J., and Vignais, P. V. (1974). An appraisal of the functional significance of the inhibitory effect of long chain acyl-CoAs on mitochondrial transports. FEBS Lett. 39, 133–138. doi: 10.1016/0014-5793(74)80035-9
Muoio, D. M., and Neufer, P. D. (2012). Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 15, 595–605. doi: 10.1016/j.cmet.2012.04.010
Murphy, S., Martin, S., and Parton, R. G. (2009). Lipid droplet-organelle interactions; sharing the fats. Biochim. Biophys. Acta 1791, 441–447. doi: 10.1016/j.bbalip.2008.07.004
Neely, J. R., Bowman, R. H., and Morgan, H. E. (1969). Effects of ventricular pressure development and palmitate on glucose transport. Am. J. Physiol. 216, 804–811.
Neubauer, S. (2007). The failing heart–an engine out of fuel. N.Engl. J. Med. 356, 1140–1151. doi: 10.1056/NEJMra063052
Newgard, C. B. (2012). Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 15, 606–614. doi: 10.1016/j.cmet.2012.01.024
Nickel, A., Kohlhaas, M., and Maack, C. (2014). Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 73, 26–33. doi: 10.1016/j.yjmcc.2014.03.011
Nickel, A., Loffler, J., and Maack, C. (2013). Myocardial energetics in heart failure. Basic Res. Cardiol. 108:358. doi: 10.1007/s00395-013-0358-9
Noel, H., Goswami, T., and Pande, S. V. (1985). Solubilization and reconstitution of rat liver mitochondrial carnitine acylcarnitine translocase. Biochemistry 24, 4504–4509. doi: 10.1021/bi00338a003
Oram, J. F., Bennetch, S. L., and Neely, J. R. (1973). Regulation of fatty acid utilization in isolated perfused rat hearts. J. Biol. Chem. 248, 5299–5309.
Palade, G. E., and Schidlowski, G. (1958). Functional association of mitochondria and lipid inclusions. Anat. Rec. 130:352.
Pande, S. V., and Blanchaer, M. C. (1971). Reversible inhibition of mitochondrial adenosine diphosphate phosphorylation by long chain acyl coenzyme A esters. J. Biol. Chem. 246, 402–411.
Penzo, D., Petronilli, V., Angelin, A., Cusan, C., Colonna, R., Scorrano, L., et al. (2004). Arachidonic acid released by phospholipase A2 activation triggers Ca2+-dependent apoptosis through the mitochondrial pathway. J. Biol. Chem. 279, 25219–25225. doi: 10.1074/jbc.M310381200
Penzo, D., Tagliapietra, C., Colonna, R., Petronilli, V., and Bernardi, P. (2002). Effects of fatty acids on mitochondria: implications for cell death. Biochim. Biophys. Acta 1555, 160–165. doi: 10.1016/S0005-2728(02)00272-4
Phielix, E., Meex, R., Ouwens, D. M., Sparks, L., Hoeks, J., Schaart, G., et al. (2012). High oxidative capacity due to chronic exercise training attenuates lipid-induced insulin resistance. Diabetes 61, 2472–2478. doi: 10.2337/db11-1832
Pulinilkunnil, T., Kienesberger, P. C., Nagendran, J., Waller, T. J., Young, M. E., Kershaw, E. E., et al. (2013). Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes 62, 1464–1477. doi: 10.2337/db12-0927
Ramsay, R. R., and Tubbs, P. K. (1976). The effects of temperature and some inhibitors on the carnitine exchange system of heart mitochondria. Eur. J. Biochem. 69, 299–303. doi: 10.1111/j.1432-1033.1976.tb10886.x
Rijzewijk, L. J., van der Meer, R. W., Smit, J. W., Diamant, M., Bax, J. J., Hammer, S., et al. (2008). Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 52, 1793–1799. doi: 10.1016/j.jacc.2008.07.062
Roden, M. (2005). Muscle triglycerides and mitochondrial function: possible mechanisms for the development of type 2 diabetes. Int. J. Obes. (Lond.) 29(Suppl. 2), S111–S115. doi: 10.1038/sj.ijo.0803102
Roe, C. R., and Ding, J. H. (2001). “Mitochondrial fatty acid oxidation disorders,” in The Metabolic and Molecular Bases of Inherited Disease, eds C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York, NY: McGraw-Hill), 2297–2326.
Rolfe, D. F., and Brown, G. C. (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758.
Romijn, J. A., Coyle, E. F., Sidossis, L. S., Gastaldelli, A., Horowitz, J. F., Endert, E., et al. (1993). Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am. J. Physiol. 265, E380–E391.
Rossignol, D. A., and Frye, R. E. (2014). Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 5:150. doi: 10.3389/fphys.2014.00150
Schafer, F. Q., and Buettner, G. R. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212. doi: 10.1016/S0891-5849(01)00480-4
Schilling, J. D., and Mann, D. L. (2012). Diabetic cardiomyopathy: bench to bedside. Heart Fail. Clin. 8, 619–631. doi: 10.1016/j.hfc.2012.06.007
Schonfeld, P., and Reiser, G. (2006). Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J. Biol. Chem. 281, 7136–7142. doi: 10.1074/jbc.M513198200
Schonfeld, P., Schild, L., and Bohnensack, R. (1996). Expression of the ADP/ATP carrier and expansion of the mitochondrial (ATP + ADP) pool contribute to postnatal maturation of the rat heart. Eur. J. Biochem. 241, 895–900. doi: 10.1111/j.1432-1033.1996.00895.x
Schonfeld, P., and Wojtczak, L. (2007). Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim. Biophys. Acta 1767, 1032–1040. doi: 10.1016/j.bbabio.2007.04.005
Schonfeld, P., and Wojtczak, L. (2008). Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic. Biol. Med. 45, 231–241. doi: 10.1016/j.freeradbiomed.2008.04.029
Schrauwen, P., Schrauwen-Hinderling, V., Hoeks, J., and Hesselink, M. K. (2010). Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 1801, 266–271. doi: 10.1016/j.bbalip.2009.09.011
Scorrano, L., Penzo, D., Petronilli, V., Pagano, F., and Bernardi, P. (2001). Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha apoptotic signaling. J. Biol. Chem. 276, 12035–12040. doi: 10.1074/jbc.M010603200
Shaw, C. S., Clark, J., and Wagenmakers, A. J. (2010). The effect of exercise and nutrition on intramuscular fat metabolism and insulin sensitivity. Annu. Rev. Nutr. 30, 13–34. doi: 10.1146/annurev.nutr.012809.104817
Singh, R., and Cuervo, A. M. (2012). Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012:282041. doi: 10.1155/2012/282041
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Sivitz, W. I., and Yorek, M. A. (2010). Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 12, 537–577. doi: 10.1089/ars.2009.2531
Skulachev, V. P. (1991). Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 294, 158–162. doi: 10.1016/0014-5793(91)80658-P
Solaini, G., and Harris, D. A. (2005). Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem. J. 390, 377–394. doi: 10.1042/BJ20042006
Stanley, B. A., Sivakumaran, V., Shi, S., McDonald, I., Lloyd, D., Watson, W. H., et al. (2011). Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J. Biol. Chem. 286, 33669–33677. doi: 10.1074/jbc.M111.284612
Stanley, K. K., and Tubbs, P. K. (1974). The occurrence of intermediates in mitochondrial fatty acid oxidation. FEBS Lett. 39, 325–328. doi: 10.1016/0014-5793(74)80141-9
Stanley, K. K., and Tubbs, P. K. (1975). The role of intermediates in mitochondrial fatty acid oxidation. Biochem. J. 150, 77–88.
Steinberg, S. F. (2013). Oxidative stress and sarcomeric proteins. Circ. Res. 112, 393–405. doi: 10.1161/CIRCRESAHA.111.300496
Stellingwerff, T., Boon, H., Jonkers, R. A., Senden, J. M., Spriet, L. L., Koopman, R., et al. (2007). Significant intramyocellular lipid use during prolonged cycling in endurance-trained males as assessed by three different methodologies. Am. J. Physiol. Endocrinol. Metab. 292, E1715–E1723. doi: 10.1152/ajpendo.00678.2006
Stewart, H. B., Tubbs, P. K., and Stanley, K. K. (1973). Intermediates in fatty acid oxidation. Biochem. J. 132, 61–76.
Suh, J. H., Heath, S. H., and Hagen, T. M. (2003). Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic. Biol. Med. 35, 1064–1072. doi: 10.1016/S0891-5849(03)00468-4
Sumegi, B., Sherry, A. D., Malloy, C. R., Evans, C., and Srere, P. A. (1991). Is there tight channelling in the tricarboxylic acid cycle metabolon? Biochem. Soc. Trans. 19, 1002–1005.
Tansey, J. T., Sztalryd, C., Gruia-Gray, J., Roush, D. L., Zee, J. V., Gavrilova, O., et al. (2001). Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 98, 6494–6499. doi: 10.1073/pnas.101042998
Tarnopolsky, M. A., Rennie, C. D., Robertshaw, H. A., Fedak-Tarnopolsky, S. N., Devries, M. C., and Hamadeh, M. J. (2007). Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1271–R1278. doi: 10.1152/ajpregu.00472.2006
Tocchetti, C. G., Caceres, V., Stanley, B. A., Xie, C., Shi, S., Watson, W. H., et al. (2012). GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes 61, 3094–3105. doi: 10.2337/db12-0072
Ussher, J. R., Koves, T. R., Jaswal, J. S., Zhang, L., Ilkayeva, O., Dyck, J. R., et al. (2009). Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes 58, 1766–1775. doi: 10.2337/db09-0011
van Loon, L. J., Greenhaff, P. L., Constantin-Teodosiu, D., Saris, W. H., and Wagenmakers, A. J. (2001). The effects of increasing exercise intensity on muscle fuel utilisation in humans. J. Physiol. 536, 295–304. doi: 10.1111/j.1469-7793.2001.00295.x
van Loon, L. J., Koopman, R., Stegen, J. H., Wagenmakers, A. J., Keizer, H. A., and Saris, W. H. (2003). Intramyocellular lipids form an important substrate source during moderate intensity exercise in endurance-trained males in a fasted state. J. Physiol. 553, 611–625. doi: 10.1113/jphysiol.2003.052431
Walther, T. C., and Farese, R. V. Jr. (2009). The life of lipid droplets. Biochim. Biophys. Acta 1791, 459–466. doi: 10.1016/j.bbalip.2008.10.009
Walther, T. C., and Farese, R. V. Jr. (2012). Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714. doi: 10.1146/annurev-biochem-061009-102430
Wang, H., Sreenevasan, U., Hu, H., Saladino, A., Polster, B. M., Lund, L. M., et al. (2011). Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 52, 2159–2168. doi: 10.1194/jlr.M017939
Wang, H., Sreenivasan, U., Gong, D. W., O'Connell, K. A., Dabkowski, E. R., Hecker, P. A., et al. (2013). Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J. Lipid Res. 54, 953–965. doi: 10.1194/jlr.M032466
Wang, H., and Sztalryd, C. (2011). Oxidative tissue: perilipin 5 links storage with the furnace. Trends Endocrinol. Metab. 22, 197–203. doi: 10.1016/j.tem.2011.03.008
Wang, Y., Mohsen, A. W., Mihalik, S. J., Goetzman, E. S., and Vockley, J. (2010). Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 285, 29834–29841. doi: 10.1074/jbc.M110.139493
Watt, M. J., and Spriet, L. L. (2010). Triacylglycerol lipases and metabolic control: implications for health and disease. Am. J. Physiol. Endocrinol. Metab. 299, E162–E168. doi: 10.1152/ajpendo.00698.2009
Welch, G. R. (1977). On the role of organized multienzyme systems in cellular metabolism: a general synthesis. Prog. Biophys. Mol. Biol. 32, 103–191. doi: 10.1016/0079-6107(78)90019-6
Wende, A. R., Symons, J. D., and Abel, E. D. (2012). Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 14, 517–531. doi: 10.1007/s11906-012-0307-2
Wojtczak, L. (1976). Effect of long-chain fatty acids and acyl-CoA on mitochondrial permeability, transport, and energy-coupling processes. J. Bioenerg. Biomembr. 8, 293–311.
Wojtczak, L., and Schonfeld, P. (1993). Effect of fatty acids on energy coupling processes in mitochondria. Biochim. Biophys. Acta 1183, 41–57. doi: 10.1016/0005-2728(93)90004-Y
Wojtczak, L., and Zaluska, H. (1967). The inhibition of translocation of adenine nucleotides through mitochondrial membranes by oleate. Biochem. Biophys. Res. Commun. 28, 76–81. doi: 10.1016/0006-291X(67)90409-3
Yang, B. Z., Mallory, J. M., Roe, D. S., Brivet, M., Strobel, G. D., Jones, K. M., et al. (2001). Carnitine/acylcarnitine translocase deficiency (neonatal phenotype): successful prenatal and postmortem diagnosis associated with a novel mutation in a single family. Mol. Genet. Metab. 73, 64–70. doi: 10.1006/mgme.2001.3162
Zammit, V. A. (1999). Carnitine acyltransferases: functional significance of subcellular distribution and membrane topology. Prog. Lipid Res. 38, 199–224. doi: 10.1016/S0163-7827(99)00002-8
Zima, A. V., and Blatter, L. A. (2006). Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 71, 310–321. doi: 10.1016/j.cardiores.2006.02.019
Keywords: palmitoyl CoA, lipid droplet, perilipin, beta-oxidation, redox environment, energetics, reactive oxygen species
Citation: Aon MA, Bhatt N and Cortassa SC (2014) Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 5:282. doi: 10.3389/fphys.2014.00282
Received: 07 April 2014; Accepted: 10 July 2014;
Published online: 31 July 2014.
Edited by:
Paolo Bernardi, University of Padova, ItalyReviewed by:
Nina Kaludercic, National Research Council of Italy, ItalyChristian Frezza, Hutchison/MRC Research Institute, UK
Paolo Bernardi, University of Padova, Italy
Copyright © 2014 Aon, Bhatt and Cortassa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Aon, Division of Cardiology, Department of Medicine, Johns Hopkins University School of Medicine, 720 Rutland Avenue, Ross Bldg. 1059, Baltimore, MD 21205, USA e-mail: maon1@jhmi.edu