GSK-3 mouse models to study neuronal apoptosis and neurodegeneration
- 1 Centro de Biología Molecular “Severo Ochoa”, Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid, Madrid, Spain
- 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, Instituto de Salud Carlos III, Madrid, Spain
Increased GSK-3 activity is believed to contribute to the etiology of chronic disorders like Alzheimer’s disease (AD), schizophrenia, diabetes, and some types of cancer, thus supporting therapeutic potential of GSK-3 inhibitors. Numerous mouse models with modified GSK-3 have been generated in order to study the physiology of GSK-3, its implication in diverse pathologies and the potential effect of GSK-3 inhibitors. In this review we have focused on the relevance of these mouse models for the study of the role of GSK-3 in apoptosis. GSK-3 is involved in two apoptotic pathways, intrinsic and extrinsic pathways, and plays opposite roles depending on the apoptotic signaling process that is activated. It promotes cell death when acting through intrinsic pathway and plays an anti-apoptotic role if the extrinsic pathway is occurring. It is important to dissect this duality since, among the diseases in which GSK-3 is involved, excessive cell death is crucial in some illnesses like neurodegenerative diseases, while a deficient apoptosis is occurring in others such as cancer or autoimmune diseases. The clinical application of a classical GSK-3 inhibitor, lithium, is limited by its toxic consequences, including motor side effects. Recently, the mechanism leading to activation of apoptosis following chronic lithium administration has been described. Understanding this mechanism could help to minimize side effects and to improve application of GSK-3 inhibitors to the treatment of AD and to extend the application to other diseases.
Glycogen synthase kinase-3 (GSK-3) was initially identified more than two decades ago as an enzyme involved in the control of glycogen metabolism (Cohen, 1979; Embi et al., 1980). In mammals two closely related isoenzymes, GSK-3α and GSK-3β are present (Woodgett, 1991). The more conserved isoform in evolution is GSK-3β with a widely expression throughout the animal kingdom, while GSK-3α is only found in vertebrates (Plyte et al., 1992; Alon et al., 2011). GSK-3α and βare ubiquitously expressed in all tissues, with particularly abundant levels in the brain (Woodgett, 1990; Yao et al., 2002; Perez-Costas et al., 2010).
GSK-3 is implicated in several signaling pathways like the insulin and insulin-like growth factor-1 (IGF-1)-mediated signal transduction or the wnt/wingless signaling pathway. GSK-3 phosphorylates and thereby regulates the functions of many metabolic, signaling, and structural proteins. Some metabolic and signaling proteins that are phosphorylated by GSK-3 are glycogen synthase, insulin receptor substrate-1 (IRS-1), cyclin D1, and eIF2B. Examples of structural proteins regulated by GSK-3 are microtubule associated proteins (MAPs) among others. Given the numerous targets and the implication in several signaling pathways, GSK-3 is involved in regulating many processes like cellular structure, function, and survival. Regarding the latter, as we will see below, GSK-3 plays a key role in apoptosis (Grimes and Jope, 2001; Jope and Johnson, 2004). Thus, GSK-3 is now recognized as an important regulator of a large number of cellular processes and, when deregulated, is thought to play a role in the etiology of various diseases. These include diabetes and/or insulin resistance (reviewed in Eldar-Finkelman, 2002), muscle hypertrophy (reviewed in Hardt and Sadoshima, 2002), cancer (reviewed in Manoukian and Woodgett, 2002), bipolar mood disorder (reviewed in Manoukian and Woodgett, 2002), schizophrenia (Emamian et al., 2004; reviewed in Jope and Roh, 2006), and neurodegenerative diseases like Alzheimer’s disease (AD; reviewed in Avila et al., 2004) and Huntington’s disease (Carmichael et al., 2002; Berger et al., 2005).
Dual Role of GSK-3 in Apoptosis
Programmed cell death is a major component of both normal development and disease. Apoptosis is a process that takes place with condensation of the nucleus, DNA fragmentation and, finally, disintegration of the cell in small apoptotic bodies that are destined to be phagocytized (Assuncao Guimaraes and Linden, 2004). Two opposing roles have been established for GSK-3 in apoptosis (Figure 1). Under certain conditions GSK-3 acts as a strong inhibitor of apoptosis and in other conditions plays a pro-apoptotic role. What determines whether GSK-3 acts in one way or the other is the type of apoptosis that occurs, which may be intrinsic (type I) or extrinsic (type II). Consequently, inhibitors of GSK-3 provide protection from intrinsic apoptosis signaling but potentiate extrinsic apoptosis signaling (Beurel and Jope, 2006). The concept that GSK-3 inhibits apoptosis came from the discovery that GSK-3β knockout mice died during embryonic development due to massive apoptosis of hepatocytes (Hoeflich et al., 2000), which demonstrated that GSK-3β is an important inhibitor of apoptosis. However, this observation appears to be in direct opposition to the finding that overexpression of GSK-3β is sufficient to induce apoptosis (Pap and Cooper, 1998). It is important to understand this duality, since, among the diseases in which GSK-3 is involved, some are characterized by excessive cell death (neurodegenerative diseases), while in others a deficient apoptosis is occurring (cancer or autoimmune diseases).
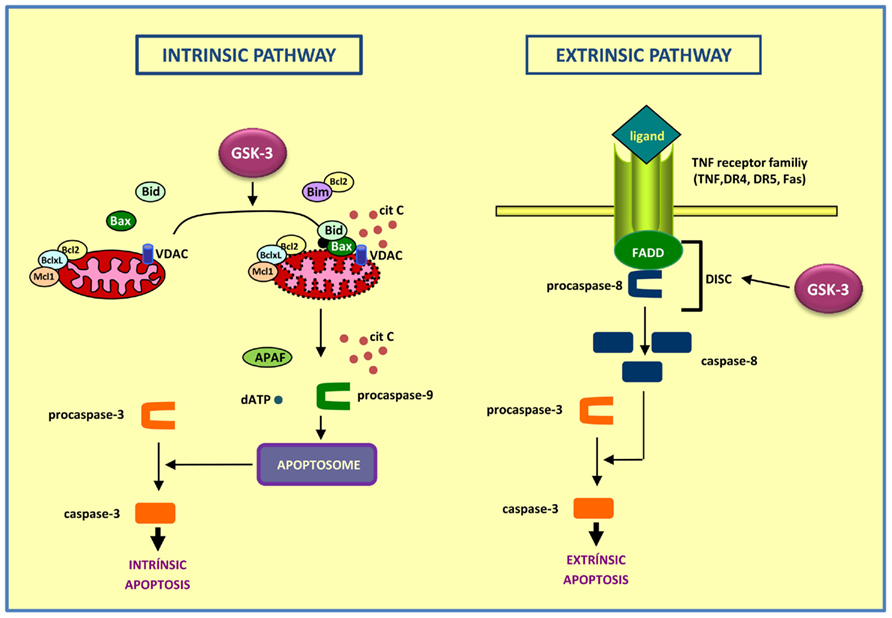
Figure 1. Diagram showing the intrinsic (left) and extrinsic (right) apoptotic pathways. In the intrinsic apoptotic pathway, GSK-3 exerts a pro-apoptotic role, acting on targets as Bax, Bim, and VDAC, thus contributing to the disintegration of mitochondria and the release of cytochrome c. On the contrary, the role of GSK-3 is anti-apoptotic in the extrinsic pathway, by preventing DISC complex formation.
(a) The intrinsic apoptosis involves the loss of integrity of mitochondria with release of cytochrome c leading to cell destruction. In the cytoplasm, released cytochrome c binds to APAF, ATP, and procaspase 9, thus forming the apoptosome and leading to activation of caspases which finally induces cell death. GSK-3 participates in the intrinsic apoptosis pathway by acting on targets such as Bax (Linseman et al., 2004), Bim (Hongisto et al., 2003), or VDAC (Pastorino et al., 2002), thus contributing to the disintegration of the mitochondria (Beurel and Jope, 2006).
The stimuli able to trigger this type of cell death are diverse and they include, among others, DNA damage, oxidative stress or endoplasmic reticulum stress. Numerous pieces of evidence have shown that under conditions that activate the intrinsic pathway, GSK-3 plays a pro-apoptotic role. Initially it was found that GSK-3 was involved in apoptosis in response to inhibition of the PI3K pathway (Pap and Cooper, 1998). Later it was found that, in cultures of rat cortical neurons, GSK-3 not only was promoting apoptosis caused by inhibition of the PI3K pathway, but also it was involved in the apoptosis in response to a withdrawal of trophic factors (Hetman et al., 2000, 2002). Moreover, due to the withdrawal of trophic factors in PC12cells, phosphorylation of GSK-3β at Tyr 216 increases (activation), and can be reversed by lithium or insulin, which affects the phosphorylation at Ser 9 (inactivation) but not at Tyr 216 (Bhat et al., 2000). In response to thapsigargin, an inducer of endoplasmic stress, not only caspase-3 is activated, but it also significantly increases GSK-3 activity (Song et al., 2002). Three pro-apoptotic stimuli such as serum withdrawal, staurosporine, or heat shock produced increased nuclear localization of GSK-3β. This change in subcellular localization seemed to be independent of phosphorylation and prior to caspase-9 and -3 activation, thus indicating that nuclear accumulation of GSK-3 is an early event in the intrinsic apoptotic signaling. Nuclear GSK-3 or GSK-3 targets present in the nucleus, as transcription factors, may be promoting intrinsic apoptosis signaling (Bijur and Jope, 2001).
An interesting fact was described in Rat-1 and PC-12 cells where, in the absence of apoptotic stimuli, overexpression of GSK-3 resulted in cell death by itself, being able to reverse apoptosis with the expression of a dominant-negative form of GSK-3 (K85R-GSK-3; Pap and Cooper, 1998). In most of these studies induction of apoptosis can be reversed by lithium or other inhibitors of GSK-3, establishing a neuroprotective role with a potential therapeutic application of such inhibitors (Cohen and Goedert, 2004; Wada, 2009).
(b) The extrinsic apoptosis involves activation of death receptors of the TNF receptor family located in the plasma membrane, such as TNF-R, Fas (also called CD95), DR4, and DR5 (Ashkenazi and Dixit, 1998). Although each of these receptors is activated by its own ligand, they share a common mechanism. The stimulation by the ligand results in receptor trimerization to which the protein Fas-associated death domain (FADD) and procaspase-8 can bind. This promotes the formation of the death-inducing signaling complex (DISC; Peter and Krammer, 2003). Subsequently a self-activation of caspase-8 occurs, which leads to activation of effector caspases and completion of apoptosis (Beurel and Jope, 2006).
The role of GSK-3 as an anti-apoptotic agent of the extrinsic pathway has been less studied. Most of these studies were conducted in tumor cell models, following the hypothesis that inhibition of GSK-3 improves response to chemotherapy. The earliest evidence dating back to 1989 when Beyaert et al. found that LiCl enhanced the toxicity by TNF, in murine and human tumor cells, in a manner similar to the effects of cycloheximide, the actinomycin D, or interferon γ (Beyaert et al., 1989). As a result, a combination of both was proposed as a treatment to improve the response to chemotherapy in cancer patients.
Other studies show potentiation of the extrinsic pathway of apoptosis by selective inhibitors of GSK-3 in tumor cell lines (Liao et al., 2003; Ougolkov et al., 2007). A recent study, also in tumor cell lines, depth in this mechanism, and places GSK-3 complexed with death receptor, thus preventing the formation of DISC. When activated, the death receptor induces changes in the complex and inhibits GSK-3, favoring apoptosis (Sun et al., 2008). In 2004, also the group of R. S. Jope published the first study of GSK-3 as a component of the extrinsic pathway in neuronal cells. This study revealed that apoptosis induced by the extrinsic pathway, specifically by Fas receptor signaling, was enhanced by lithium and other GSK-3 inhibitors (Song et al., 2004).
Mouse Models with Genetic Modifications of GSK-3
In order to investigate the involvement of GSK-3 in cellular processes and to clarify its implications in diseases, a great effort was and is still being made to generate animal models that mimic pathological conditions through the deregulation of GSK-3. Up to date the main effort has been invested in creating animal models with altered expression of GSK-3, especially the evolutionary well conserved isoform GSK-3β.
To achieve this goal, different strategies have been applied: overexpression, suppression, and modulation of the expression of the enzyme. Many of these models direct expression or modification of GSK-3 to the central nervous system (CNS) for the generation of mouse models of AD. Nowadays several other species apart from the mouse are also used as model systems, which include Drosophila melanogaster and zebrafish. In this review we will focus on results obtained from mouse models.
Mouse Models with Increased Glycogen Synthase Kinase-3 Activity
One of the earliest diseases linked to dysfunctions of GSK-3β was AD. More recently, it has been proposed that the deregulation of GSK-3β might affect other tauopathies like frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17; Perez et al., 2000). Regarding AD, it has been demonstrated that GSK-3β mediates tau hyperphosphorylation, β-amyloid-induced neurotoxicity, and mutant presenilin-1 pathogenic effects (Jope and Johnson, 2004). Increased levels of GSK-3 have also been reported in postmortem analysis of brains from AD patients compared to age-matched control samples (Pei et al., 1997), where as a spatial and temporal pattern of increased active GSK-3β expression correlates with the progression of NFT and neurodegeneration (Leroy et al., 2002). Thus, the first GSK-3 mouse models focused on the study of the implications of GSK-3β overexpression in the CNS on the development of AD and related neuropathological diseases. To achieve this goal, promoters with known neuronal expression like NF-L, thy1, or the more region specific promoter CaMKIIα, were used.
In 1997, Brownlees et al. generated the first two GSK-3 mouse models. One carried the human wild-type GSK-3β isoform as the transgene and the other one the mutant form of GSK-3β in which serine-9 of GSK-3β was mutated to alanine (S9A), thus supposedly resulting in a more active form by preventing inactivation through the phosphorylation on Ser9 (reviewed in Grimes and Jope, 2001). These transgenes were under the transcriptional control of either the ubiquitous murine sarcoma virus (MSV) promoter or the neuronal specific promoter of the murine neurofilament light chain (NF-L; Brownlees et al., 1997b). In spite of detecting the two transgenes mRNAs through reverse-transcribed PCR, no substantial increase in total activity of GSK-3β was observed, neither regarding the wild-type GSK-3β nor the mutated form. They also observed that the expression of the mutated form of GSK-3β (S9A) was much lower, probably due to the fact of its increased activity. Despite no detectable GSK-3 overexpression, Western blot analysis of the two mouse lines with the highest levels of transgenic GSK-3β activity revealed that the phosphorylation status of tau was elevated at the AT8 epitope. The authors postulated that toxicity elicited by GSK-3β overexpression during embryonic and postnatal development of the CNS may explain why they were unable to generate mouse lines with detectable GSK-3β overexpression. Conversely, as it will be commented below, GSK-3β knockout mice die during embryonic life (Hoeflich et al., 2000). From this first model of GSK-3 increased activity we extract the notion of an embryonic lethality but no further analysis regarding the role of GSK-3 in apoptosis was achieved.
The second transgenic animal published also expressed the S9A mutant form of the kinase, i.e., GSK-3β (S9A); this time under the control of the modified murine thy1 gene (Spittaels et al., 2000). The engineered thy1 construct used in this model drives the expression of the transgene only postnatally and only in neurons. This mouse shows a twofold increase in GSK-3β activity. Accordingly, an increase in phosphorylated tau was demonstrated, but only in older transgenic mice (7–8 months). In a second round of characterization of this mouse line, Spittaels et al. also observed in these mice a significant decrease in overall brain weight and volume, with the largest reduction occurring in the cerebral cortex. This reduction was not due to apoptosis, since there was no increase in TUNEL or in cleaved caspase-3 positive neurons (Spittaels et al., 2002). The resulting compacted architecture was further demonstrated by an increased neuronal density, by reduced neuronal cell bodies and of the somatodendritic compartment of pyramidal neurons in the cortex. The overall reduction in size of the entire CNS induced by constitutive active GSK-3β caused only very subtle changes in the psychomotor ability of adult and aging GSK-3β transgenic mice. Overexpression of GSK-3β in this model also has been shown to up- or down-regulate the expression of numerous proteins in neurons, including down-regulation of MAP2 (Tilleman et al., 2002). In spite of increased tau phosphorylation and decreased brain weight these GSK-3β transgenic mice performed normally in the Morris water maze test (Spittaels et al., 2002).
One of the characteristics of persons affected by FTDP-17 is an increased expression of the longest human protein tau isoform containing the four repeat regions in the carboxyl-terminal part (Heutink, 2000). For this reason Spittaels et al. (1999), in another approach, cross-bred their GSK-3β (S9A) mice with transgenic mice that carry the longest human protein tau isoform expressed under the same promoter (thy1). The authors observed a reduction of the number of axonal enlargements and motor impairments typical of these tau transgenic animals (Spittaels et al., 2000). In these mice, an increase in phosphorylation of human tau was demonstrated, although neither an increase in insoluble tau aggregates nor the presence of paired helical filaments was observed. Spittaels et al. concluded that the improvement of axonopathy and motor problems found in htau40 mice when mixed with GSK-3β (S9A) mice was due to phosphorylation of tau induced by GSK-3β which decreases its affinity for microtubules and consequently rescues axonal transport. The same group crossed this mutant GSK-3β (S9A) mice with a tau model overexpressing Tau (P301L; Terwel et al., 2008) observing an increase in forebrain tau pathology although conversely the bigenic mice survived longer than the parental Tau (P301L).
In a similar approach Li et al. (2004) used the isoform GSK-3β (S9A), isoform resistant to inhibition by phosphorylation, under control of the neuron specific human platelet-derived growth factor (PDGF) β-chain promoter. This promoter drove the expression mainly to the cortex and hippocampus of the transgenic brain. They observed an increase in GSK-3 activity without changes in the total level of GSK-3β. They also observed an increase in tau phosphorylation detected by the AT8 antibody by Western blot analysis. This approach did not show any evidence of apoptosis although they show increased pretangle morphology which could be indicating a neurodegenerative process.
Overall, the GSK-3β (S9A) mice generated by Spittaels and co-workers show decreased brain weight and volume, more pronounced in the cortex that cannot be attributed to an apoptotic effect as there is no evidence of increased TUNEL or caspase-3 levels.
In view of the postulated lethality of embryonic GSK-3β overexpression (Brownlees et al., 1997a) as well as the known role of GSK-3β in development, a further GSK-3β transgenic mouse was generated (Lucas et al., 2001) by using the conditional tetracycline-regulated system. In these mice transgene expression was under the control of the CaM kinase II α-promoter to achieve substantial overexpression of wild-type GSK-3β in forebrain neurons and, therefore, in regions more relevant for AD.
In this transgenic line (Tet/GSK-3β mice), GSK-3β overexpression was restricted to certain cortical neurons and hippocampal neurons. It was in the hippocampal region were a 30% increase in GSK-3β activity was observed by enzymatic assay on tissue homogenate (Hernandez et al., 2002). Hippocampal overexpression of GSK-3β resulted in an increase in the phosphorylation of tau in Tet/GSK-3β animals, as detected with antibodies raised against the phosphorylated tau modified in AD, like PHF-1/AD2. This hyperphosphorylated tau was found in the somatodendritic compartment, not because of an increase in the total amount of tau protein, but exclusively due to the hyperphosphorylation of tau by GSK-3β. In spite of a substantial increase in hyperphosphorylated tau, the aberrant tau aggregation found in AD was not observed in these GSK-3β transgenic mice. In this model, β-catenin, another GSK-3 substrate relevant to AD (Zhang et al., 1998), was also analyzed. GSK-3 is a key enzyme involved in β-catenin stabilization and nuclear translocation (reviewed in Barth et al., 1997; Anderton, 1999). Increased GSK-3β in Tet/GSK-3β mice was seen effective in modifying β-catenin in neurons of the CNS in vivo, since nuclear β-catenin was reduced by ~75% in hippocampal granular cells (Lucas et al., 2001). Considering that the genes transactivated by β-catenin are poorly characterized, these mice may serve as a good tool to identify such genes. Tet/GSK-3β mice also demonstrate neuronal stress and neuronal death as revealed the presence of reactive glia, TUNEL labeling, and cleaved caspase-3 staining. These data are in agreement with the known role of GSK-3β in the survival pathway, as well as supporting the neuroprotective effect of lithium (reviewed in Chuang et al., 2002). The hyperphosphorylation of hippocampal tau in transgenic Tet/GSK-3β mice, despite the lack of filament formation, results in a behavioral impairment that can be measured in the Morris water maze test (Hernandez et al., 2002) and in the object recognition test (Engel et al., 2006a).
Animal models generated by using the tetracycline-regulated system make it possible to explore any potential reversal of their phenotype (Mayford et al., 1996; Yamamoto et al., 2000). Thus the Tet/GSK-3β should be a good tool to test the neuroprotective effect of forthcoming GSK-3β specific inhibitors. In 2006 Engel and co-workers published a study of the effect of silencing transgene expression by the administration of tetracycline to the Tet/GSK-3β mice (Tet-OFF system). They show that transgene shutdown in symptomatic mice leads to normal GSK-3 activity, normal phospho-tau levels, diminished neuronal death, and suppression of the cognitive deficit, thus further supporting the potential of GSK-3 inhibitors for AD therapeutics (Engel et al., 2006b).
The tetracycline-regulated system also gives us the opportunity to overexpress the GSK-3β transgene in other regions than the CNS, using another promoter for the tetracycline-activator. This would provide the great opportunity to study the implications of a deregulation of GSK-3β in other cell types, other tissues, and to generate animal models for other diseases.
The implication of wild-type GSK-3β in tau pathology was further analyzed by combining this transgenic model with transgenic mice expressing tau with a triple FTDP-17 mutation which develop prefibrillar tau aggregates (VLW mice; Lim et al., 2001). In the VLW single transgenic mice, which express the longest human tau isoform with three FTDP-17 point mutations (G272V, P301L, and R406W, hTauVLW) under the control of the modified thy1 mouse promoter, thin filaments with a width of ~5 nm can be observed. The triple transgenic mice Tet/GSK-3β/VLW showed an increase in tau phosphorylation in the hippocampus, the area where the two transgenes, GSK-3β and hTauVLW were co-expressed and where GSK-3 activity was increased. In addition, hyperphosphorylated tau with an aberrant high molecular weight was present in these triple transgenic mice but not in the single transgenic mice VLW or the Tet/GSK-3β mice. Similarly, thioflavine-S positive neurons were only observed in the hippocampus of Tet/GSK-3β/VLW mice and filaments with a width wider than 10 nm (a width similar to that found in tauopathies) could be purified only from Tet/GSK-3β/VLW mice. This model demonstrates that an increase in GSK-3β activity is a key factor able to induce tau aggregation (sarkosyl-insoluble material as well as tau filaments). Neurodegeneration studies in these mice showed a decrease in the volume of the dentate gyrus of Tet/GSK-3β/VLW mice at 5 months, not seen in Tet/GSK-3β mice of the same age. At the advanced age of 18 months both Tet/GSK-3β and Tet/GSK-3β/VLW exhibited a severe atrophy of the dentate gyrus at a similar level in both transgenic mice. Thus, the neurodegenerative process occurring in the dentate gyrus of Tet/GSK-3β transgenic mice is accelerated by the presence of mutated tau.
As a proof of concept that tau modification by GSK-3 plays a role in neurodegeneration found in the previously exposed models, GSK-3 overexpressing mice were brought to tau knockout (Tau−/−) background. Interestingly in Tet/GSK-3β + Tau−/− mice the toxic effect of GSK-3 overexpression is milder and slower in the absence of tau (Gomez de Barreda et al., 2010).
What we can extract from studies in Tet/GSK-3β is that an increase in GSK-3 activity correlates with an increase in apoptosis which seems to involve tau modification, as seen when mice were combined with models of tau modifications.
Another approach used to study the deregulation of GSK-3 is the generation of knock-in mice with a modified GSK-3 transgene. In 2005, McManus et al. have generated a GSK-3 homozygous knock-in mice in which the protein kinase B (PKB/AKT) phosphorylation sites on GSK-3α (Ser21) and GSK-3β (Ser9) were changed to a non-phosphorylatable Ala residue (McManus et al., 2005). This protein modification would enable studying the role played by the inactivation of GSK-3 through these phosphorylation events in insulin and Wnt signaling pathways. These knock-in mice develop and reproduce normally and were not diabetic, despite insulin being unable to stimulate glycogen synthase in muscle. In double GSK-3 knock-in mice the GSK-3 isoforms are present at normal levels. Analyzing this model it was demonstrated that insulin regulates muscle glycogen synthase mainly through GSK-3β rather than GSK-3α. Regarding the CNS, this model shows altered neurogenesis as well as a deficient production of the supporting vascular endothelial growth factor (VEGF) in the hippocampus (Eom and Jope, 2009). In vivo chronic co-administration of lithium and fluoxetine increases proliferation in the dentate gyrus of wild-type but not GSK-3α/β (S21A/S21A/S9A/S9A) knock-in mice. Following these results it was suggested that blockade of the inhibitory control of GSK-3 results in impaired neurogenesis (Eom and Jope, 2009). At this respect, it has been described that GSK-3 is a master regulator of neuronal progenitor homeostasis during embryonic development (Kim et al., 2009). In the specific case of adult neurogenesis taking place in the subgranular zone of the dentate gyrus, a role for tau phosphorylation (by GSK-3) has been found in the migration and differentiation of neuronal precursors into mature neurons (Fuster-Matanzo et al., 2009; Hong et al., 2010). Interestingly, Tet/GSK-3 mouse model of GSK-3 overexpression showed an impairment in proliferation and maturation that together with increased apoptosis, contribute to the atrophy of the dentate gyrus (Sirerol-Piquer et al., 2011).
Overall GSK-3 increased activity induced in homozygous GSK-3α/β (S21A/S21A/S9A/S9A) knock-in mice results in altered neurogenesis, without clear implications in apoptosis.
An additional transgenic model overexpressing wild-type GSK-3β was generated to be crossed with mice overexpressing FAD-mutant APP (Rockenstein et al., 2007). These mice showed an improvement in Morris water maze compared with single mutant APP mice, and strikingly, they had a decrease in tau phosphorylation as well as a decrease in APP phosphorylation and Aβ levels. To explain these unexpected results, the authors argued that, although GSK-3β was overexpressed, this resulted in an increase in the inhibitory domain of the kinase. That was the reason to name this animal model, in a confusing way, as a dominant-negative GSK-3β model. Overall the manuscript demonstrates that GSK-3 is able to modulate the amyloid aspect of AD in vivo, but has no implications in apoptosis.
In summary, the role of GSK-3 in apoptosis when explored in mouse models with increased GSK-3 activity seems to be pro-apoptotic. Even if apoptosis has not been studied in detail, a toxic effect seems to be common to these mouse models.
Animal Models with Reduced Glycogen Synthase Kinase-3 Activity
Several genetic approaches have been used to generate mice with a decrease in GSK-3 levels. The first study to suppress its expression in a mouse model was the generation of GSK-3β-KO mice by Hoeflich et al. (2000). In this study the GSK-3β gene has been disrupted by targeted deletion. These investigators showed that GSK-3β is absolutely essential for survival. The generated GSK-3β-knockout mice developed normally to mid-gestation, but died around embryonic day 14 following massive tumor necrosis factor-α (TNF-α)-induced hepatocyte apoptosis, which could be prevented by the injection of antibodies that block the function of TNF-α. Nuclear factor κB (NF-κB) activation is known to counteract TNF-α- induced apoptotic signaling to promote survival by turning on a set of anti-apoptotic genes (Pomerantz and Baltimore, 2000). The intriguing finding made by Hoeflich et al. is that GSK-3β is required for the NF-κB-mediated survival response. This study therefore demonstrates for the first time an isoform-specific GSK-3 function since GSK-3α was unable to compensate for the loss of GSK-3β.
Although the GSK-3β knockout mice died during mid-gestation, heterozygotes mice were viable and appeared morphologically normal. Mouse embryonic fibroblasts derived from the GSK-3β heterozygotes knockout mice expressed reduced levels of GSK-3β protein without a compensatory increase in GSK-3α protein levels, indicating a partial loss of function in GSK-3β heterozygous. The authors also observed a reduced GSK-3β activity in the brains of the heterozygous mice (Hoeflich et al., 2000).
These GSK-3β heterozygous knockout mice were used by Beaulieu et al. and O’Brien et al. (2004) in two subsequent studies. In the first study Beaulieu et al. (2004) demonstrated in dopamine transporter knockout mice that dopamine can exert its behavioral effects by acting on a lithium-sensitive signaling cascade involving Akt/PKB and GSK-3. In this study increased dopamine neurotransmission resulted in inactivation of Akt and concomitant activation of GSK-3α and GSK-3β. These biochemical changes were effectively reversed by the administration of the GSK-3 inhibitor lithium or when combined with the GSK-3β heterozygous knockout mice, thus establishing this cascade as an important mediator of dopamine action in vivo. Interestingly, these DAT-KO mice, with increased GSK-3 activity, also show increased microgliosis, and a small percentage of apoptotic neurons associated with increased tau phosphorylation (Cyr et al., 2003).
O’Brien et al. (2004) compared the behavioral effects of chronic lithium treatment on mice with the behavioral phenotype of the GSK-3β heterozygous knockout mice. In this study the authors observed that lithium treated mice spent less time immobile in the forced swimming test, test widely used as a predictor of antidepressant efficacy. The same result was obtained when they used the GSK-3β heterozygotes knockout mice. In the exploratory test, both, lithium treated and GSK-3β heterozygotes knockout mice, acted in the same way with less exploratory activity as compared to wild-type mice. Molecular targets of GSK-3 dependent signaling, such as β-catenin, are also affected similarly by lithium and GSK-3β haploinsufficience with a substantial increase of ~30%. These behavioral and molecular correlations strongly support the hypothesis that GSK-3 is an important target for the behavioral effects of lithium.
However, another heterozygous mice generated by homologous recombination in Takashima’s lab have no defect regarding learning and memory in Morris water maze test although they show impaired memory reconsolidation in fear conditioning test (Kimura et al., 2008).
In summary, GSK-3β knockout mice present an embryonic lethality promoted by massive apoptosis in hepatocytes. Liver degeneration correlates with excessive TNF-α toxicity. As TNF-α is a member of death receptor family, we could conclude that apoptosis in hepatocytes is taking place by extrinsic pathway of apoptosis. Studies in fibroblasts from −/− embryos revealed decreased NFκB activation which seems to be GSK-3β dependent. No further analysis of apoptosis in CNS has been made in the heterozygous mice generated by Hoeflich et al. (2000) nor in the heterozygous made by Kimura et al. (2008).
Mice without GSK-3α are viable and show an increase in glucose and insulin sensitivity accompanied by liver glycogen accumulation and a reduction in fat mass (MacAulay et al., 2007). Respect to CNS these animals show an increase in cerebellar volume, although normal brain volume, suggesting a role for GSK-3α in this area. In addition, GSK-3α KO mice present alterations in a wide range of behavior tests involving this isoenzyme in normal brain function. Some defects found in exploratory activity, decreased immobility time and anti-aggression behavior remind to those found in GSK-3β ± mice. Abnormal behaviors as decreased locomotion, decreased social motivation, impaired sensorimotor gating, associative memory, and coordination are specific for GSK-3α −/− mice (Kaidanovich-Beilin et al., 2009).
In order to delete GSK-3 specifically from neurons, deletion of GSK-3α/β was carried out. Thus, mice with a GSK-3α null background and also carrying the construction GSK-3β (loxP/loxP) were crossed with nestin-cre line (Kim et al., 2009). These animals showed bigger heads and died at P0. The main finding was a massive hyperproliferation of neural progenitors by expansion of the radial progenitor pool. These effects were linked with β-catenin deregulation, Sonic Hedgehog, Notch, and fibroblast growth factor signaling. This model mainly demonstrates the role of GSK-3β in brain development (reviewed in Hur and Zhou, 2010). Besides, it has been recently demonstrated that GSK-3β is involved not only in embryonic neural development but also in adult neurogenesis. Thus, GSK-3 is able to phosphorylate tau protein in doublecortin positive cells in adult dentate gyrus (Fuster-Matanzo et al., 2009; Hong et al., 2010). In addition, GSK-3α/β (S21A/S21A/S9A/S9A) knock-in mice, described previously (Eom and Jope, 2009), present a drastic impairment in adult neurogenesis in vivo. In good agreement, adult neurogenesis is also altered in Tet/GSK-3β mice (Sirerol-Piquer et al., 2011).
From these studies we can extract that decreasing GSK-3 activity would translate in diminished apoptosis as indicated by the phenotype of bigger heads in both mouse models which is similar to the exencephaly observed in knockout mice for caspase-9 and caspase-3 (Haydar et al., 1999).
The study of a long term genetic suppression of the GSK-3 activity was difficult to achieve because of the embryonic lethality reported by Hoeflich et al. (2000). An alternative approach to study neurological consequences of sustained GSK-3 inhibition has been to generate mice with conditional expression of a canonical dominant-negative form of GSK-3(Gomez-Sintes et al., 2007). More precisely the K85R mutant form of GSK-3β (DN-GSK-3) was chosen in view of its previously shown efficacy in decreasing GSK-3 activity (Dominguez et al., 1995). The mouse design is similar to the one used for generating Tet/GSK-3β mice (Lucas et al., 2001). It consists in a transgenic mouse with conditional tetracycline-controlled system (Tet-OFF system), in which the expression of the tTA transgene is driven by CaMKIIα promoter. This promoter addresses the expression of tTA, and consequently of the Myc-K85R-GSK-3β construction, specifically to adult neurons in the forebrain. The transgene expression was evident in striatum, cortex, and hippocampus of Tet/DN-GSK-3 mice. GSK-3 activity measured in cortex and striatum homogenates revealed decreased GSK-3 activity. Subsequently, in these regions both GSK-3α and β showed increased phosphorylation in Ser 9 and 21 respectively and decreased phosphorylation in tau epitopes, PHF-1 and AT8. As a neurological consequence of decreased activity in postnatal neurons of the forebrain they showed impaired motor coordination. The motor deficit correlates with an increase of neuronal apoptosis, detected by activated caspase-3 immunostaining and TUNEL selectively in areas responsible of motor control, like cortex and striatum. Interestingly these consequences were restored after shutdown expression (doxycycline administration). In this model, an increased apoptosis as a consequence of decreased GSK-3 activity was found. Regarding the paradoxical dual role of GSK-3 in apoptosis exposed previously, these results suggest that apoptosis induced by GSK-3 inhibition is occurring via the extrinsic pathway.
Models with increased GSK-3 activation had revealed a common effect in increasing apoptosis/toxicity. Intriguingly, studies in mouse models with decreased GSK-3 have raised different results regarding the role of GSK-3 in apoptosis. These controversial data are probably dependent on the isoform affected α or β, the cellular type or the developmental state in which GSK-3 activity has been decreased. If the isoform lacking during development is GSK-3α, the consequence is an increase in cerebellar volume which could be indicating a defect in apoptosis. When GSK-3β is decreased at early embryonic stage as in the two knockout mice of GSK-3β, it results in increased apoptosis/lethality. On the contrary, if GSK-3β is deleted specifically in neurons over a GSK-3α −/− background, then has an opposed effect of bigger heads which again indicates decreased apoptosis. Finally, mice with decreased GSK-3β activity occurring once the CNS is formed as Tet/DN-GSK-3β revealed an increased in apoptosis in striatum and cortex where expression of the transgene is more remarkable. This apoptosis seems to act through extrinsic signaling pathway. However, despite these experimental data, the contribution and the physiological role of both GSK-3 isoenzymes in relation to apoptosis are still not clear. Thus, it will be necessary to understand their relative contribution to apoptotic signaling cascades and, as a consequence, their relative contribution to neurodegeneration.
Correlate with Pharmacological Inhibition
Deregulation of GSK-3 has been shown to be involved in all mechanisms described for AD neuropathology (Grimes and Jope, 2001). Interestingly, there is indirect evidence of increased activity of GSK-3 in AD patients (reviewed in Imahori and Uchida, 1997; Pei et al., 1997; Blalock et al., 2004; Hye et al., 2005; Leroy et al., 2007; Hooper et al., 2008). Some studies have also implicated GSK-3 in the pathology of type 2 diabetes (Eldar-Finkelman and Krebs, 1997; Eldar-Finkelman, 2002; Jope and Johnson, 2004). Thus, regarding the previous data, GSK-3 inhibitors were presented as a promising therapeutic tool for the treatment of AD, type 2 diabetes and possibly other neurodegenerative diseases (Cohen and Goedert, 2004; Meijer et al., 2004; Martinez and Perez, 2008).
Moreover, taking into account that cancer is characterized by a defect in apoptotic mechanism, inhibition of GSK-3 has been postulated as useful pharmacological approach to improve response to chemotherapy (Beyaert et al., 1989; Beurel et al., 2004; Luo, 2009). This therapeutic strategy would be only effective when directed to certain types of cancer, since directed to other types may have the opposite effect.
A reliable approach to test the validity of GSK-3 inhibitors for the treatment of AD would be the restoration of normal levels of GSK-3 in the Tet/GSK-3β mouse model (Lucas et al., 2001). These mice permit to explore whether the biochemical, histopathological, and behavioral consequences of increased GSK-3 activity are susceptible to revert after the shutdown of transgene expression. In this study Engel et al. (2006b) found that 6 weeks of doxycycline administration were sufficient to lower GSK-3 activity and tau phosphorylation to normal levels, to diminish neuronal cell death, and to improve cognitive deficit, thus further supporting the potential of GSK-3 inhibitors for AD therapy. However, GSK-3 inhibition leading to levels of GSK-3 activity below normal could be counterproductive. This was evidenced by the characterization of Tet/DN-GSK-3 mice carried out in our lab that showed increased levels of apoptosis and motor deficits as a consequence of sustained inhibition of GSK-3 (Gomez-Sintes et al., 2007). Fortunately this toxicity can be reverted when restoring normal activity of GSK-3, which suggests that GSK-3 should be maintained within certain physiological levels. The study performed by Frautschy et al. support this notion. They perform intracerebroventricular infusions of Aβ42 oligomer, which produces an increase in GSK-3 activity, and/or SB 216763, a selective inhibitor of GSK-3. Their results show a beneficial effect of SB 216763 to revert toxicity induced by Aβ42 oligomer. Intriguingly, they have also found a certain toxicity of SB when administered to control animals with normal levels of GSK-3 activity (Hu et al., 2009).
In view of the potential use of GSK-3 inhibitors for the treatment of AD and maybe other diseases in which deregulation of GSK-3 has been involved, many efforts have been directed to the development of selective and effective GSK-3 inhibitors (reviewed in Cohen and Goedert, 2004; Frame and Zheleva, 2006; Gould et al., 2006; Martinez and Perez, 2008). Indeed, two GSK-3 selective inhibitors from the pharmaceutical company Noscira are at the present under clinical trials phase II (Medina and Castro, 2008; Sereno et al., 2009; Del Ser, 2010). Nowadays this company is in the most advanced stage of the development of GSK-3 inhibitors. We must still wait for future studies in analysis of postmortem tissue to evaluate the effect of these inhibitors regarding apoptosis.
A classical non-selective GSK-3 inhibitor, lithium, has been used in clinics for decades (Klein and Melton, 1996; Stambolic et al., 1996). Since its introduction into psychiatric pharmacotherapy 60 years ago, lithium remains the most effective agent in the treatment and prophylaxis of major mood disorders, particularly bipolar disorder (BD; Manji et al., 1999; Baldessarini et al., 2002; Grof and Muller-Oerlinghausen, 2009; Kovacsics and Gould, 2010). Despite the obvious advantages of chronic lithium therapy, its clinical use is often curtailed by its narrow therapeutic index and its devastating overdose-induced toxicity (Macritchie and Young, 2004). Accordingly, patients must be closely monitored not only at the beginning of treatment, but also during treatment maintenance, to keep serum lithium concentrations within a therapeutic window of 0.6–1.4 mM. Even within this therapeutic range, mild neurological side effects such as hand tremor are common, and progressive toxicity to marked neurological impairment correlates with increasing serum levels above 1.5 mM (Macritchie and Young, 2004). The biochemical and cellular basis for lithium’s therapeutic efficacy and the precise molecular mechanisms through which it exerts its unwanted neurological side effects remain to be fully elucidated. In 2008 a study in AD patients was accomplished to test the feasibility of chronic lithium treatment for this disease. It showed that side effects, although mild and reversible, difficult the application to elder people and the discontinuation rates are high (Macdonald et al., 2008).
As lithium inhibits GSK-3 in vivo (Klein and Melton, 1996; Stambolic et al., 1996) and we recently reported neuronal apoptosis and motor deficits in dominant-negative GSK-3 transgenic mice (Gomez-Sintes et al., 2007), we hypothesized that therapeutic levels of lithium could also induce neuronal loss through GSK-3 inhibition and that maybe this is the cause of the known extrapyramidal side effects, as hand tremor, produced by chronic treatment with lithium in BD patients. To test this hypothesis chronic lithium was administered to wild-type mice and it was found that apoptosis and motor coordination impairment were occurring, similarly as in Tet/DN-GSK-3β mice (Gomez-Sintes and Lucas, 2010). In this case GSK-3 inhibition was not restricted to forebrain areas as it was not dependent of CaMKII promoter. Thus, increased apoptosis was noticeable in areas like cortex, striatum, globus pallidus, hippocampus, and cerebellum, all of them involved in motor control. Interestingly chronic lithium resulted in a poor performance of motor tasks as evidenced by vertical pole test or DigiGait apparatus, which measures footprint pattern and other parameters of walking regularity. To depth into the mechanism of lithium toxicity, we elaborated a hypothesis involving NFAT/Fas signaling that we proceed to test (Figure 2). The results demonstrated that GSK-3 inhibition by lithium increase translocation of nuclear factor of activated T cells c3/4 (NFATc3/4) transcription factors to the nucleus, leading to increased Fas ligand (FasL) levels, and Fas receptor activation, which finally gives rise to extrinsic apoptosis signaling (death receptor dependent; Gomez-Sintes and Lucas, 2010).
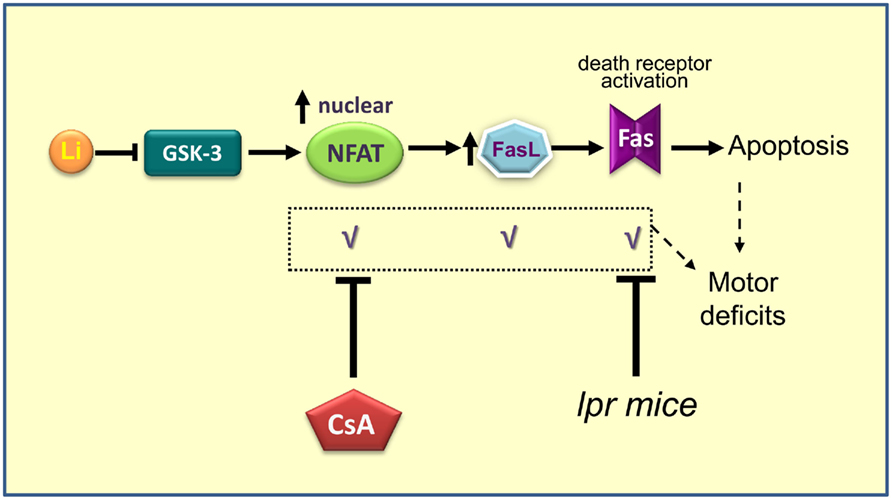
Figure 2. Scheme summarizing the proposed mechanism for lithium induced neuronal apoptosis (Gomez-Sintes and Lucas, 2010). Prolonged administration of lithium produces inhibition of GSK-3, which promotes entry and accumulation of NFAT into the nucleus. Once in the nucleus, NFAT activates the production of FasL. When FasL is secreted outside the cell binds to Fas receptor present in the membrane of the same cell or cells nearby, which triggers death by apoptosis. When NFAT/Fas signaling is blocked by co-administration of Cyclosporin A or when lithium is administered to Fas-deficient mice (lpr) motor deficits and apoptosis are absent.
Interestingly, apoptosis and motor deficits were prevented when cyclosporin A, that blocks NFAT nuclear translocation, was co-administered with lithium. These side effects could be also reverted by blocking Fas signaling. This was tested by treating lpr mice (naturally Fas receptor-deficient) with chronic lithium. These findings leave an open way to combined therapies. To test the validity of these results but providing a genetic inhibition of GSK-3, experiments with Tet/DN-GSK-3 mice in lpr background are being undertaken.
All the mouse models with increased or decreased GSK-3 activity described in this review have helped to understand the implication of GSK-3 on neurodegenerative or psychiatric diseases and also to depth in the physiology of GSK-3. Although there is still a lot of work to do in this direction, these models constitute a good tool to achieve these studies.
Understanding the role of GSK-3 in apoptosis would improve the application of GSK-3 inhibitors to diseases as ADs, as apoptosis seems to be responsible of motor side effectsGSK-3 inhibition. The combination of GSK-3 inhibitors and NFAT/Fas signaling blockers could be a good clinical strategy to minimize side effects and extend the use of GSK-3 inhibitors to other neurodegenerative and neuropsychiatric diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alon, L. T., Pietrokovski, S., Barkan, S., Avrahami, L., Kaidanovich-Beilin, O., Woodgett, J. R., Barnea, A., and Eldar-Finkelman, H. (2011). Selective loss of glycogen synthase kinase-3alpha in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 585, 1158–1162.
Ashkenazi, A., and Dixit, V. M. (1998). Death receptors: signaling and modulation. Science 281, 1305–1308.
Assuncao Guimaraes, C., and Linden, R. (2004). Programmed cell deaths. Apoptosis and alternative deathstyles. Eur. J. Biochem. 271, 1638–1650.
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384.
Baldessarini, R. J., Tondo, L., Hennen, J., and Viguera, A. C. (2002). Is lithium still worth using? An update of selected recent research. Harv. Rev. Psychiatry 10, 59–75.
Barth, A. I., Nathke, I. S., and Nelson, W. J. (1997). Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr. Opin. Cell Biol. 9, 683–690.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Berger, Z., Ttofi, E. K., Michel, C. H., Pasco, M. Y., Tenant, S., Rubinsztein, D. C., and O’kane, C. J. (2005). Lithium rescues toxicity of aggregate-prone proteins in Drosophila by perturbing Wnt pathway. Hum. Mol. Genet. 14, 3003–3011.
Beurel, E., and Jope, R. S. (2006). The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 79, 173–189.
Beurel, E., Kornprobst, M., Blivet-Van Eggelpoel, M. J., Ruiz-Ruiz, C., Cadoret, A., Capeau, J., and Desbois-Mouthon, C. (2004). GSK-3beta inhibition by lithium confers resistance to chemotherapy-induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Exp. Cell Res. 300, 354–364.
Beyaert, R., Vanhaesebroeck, B., Suffys, P., Van Roy, F., and Fiers, W. (1989). Lithium chloride potentiates tumor necrosis factor-mediated cytotoxicity in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 86, 9494–9498.
Bhat, R. V., Shanley, J., Correll, M. P., Fieles, W. E., Keith, R. A., Scott, C. W., and Lee, C. M. (2000). Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 11074–11079.
Bijur, G. N., and Jope, R. S. (2001). Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J. Biol. Chem. 276, 37436–37442.
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., and Landfield, P. W. (2004). Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178.
Brownlees, J., Irving, N. G., Brion, J. P., Gibb, B. J., Wagner, U., Woodgett, J., and Miller, C. C. (1997a). Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3beta transgenes. Neuroreport 8, 3251–3255.
Brownlees, J., Irving, N. G., Brion, J. P., Gibb, B. J. M., Wagner, U., Woodgett, J., and Miller, C. C. J. (1997b). Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3 beta transgenes. Neuroreport 8, 3251–3255.
Carmichael, J., Sugars, K. L., Bao, Y. P., and Rubinsztein, D. C. (2002). Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 277, 33791–33798.
Chuang, D. M., Chen, R. W., Chalecka-Franaszek, E., Ren, M., Hashimoto, R., Senatorov, V., Kanai, H., Hough, C., Hiroi, T., and Leeds, P. (2002). Neuroprotective effects of lithium in cultured cells and animal models of diseases. Bipolar Disord. 4, 129–136.
Cohen, P. (1979). The hormonal control of glycogen metabolism in mammalian muscle by multivalent phosphorylation. Biochem. Soc. Trans. 7, 459–480.
Cohen, P., and Goedert, M. (2004). GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 3, 479–487.
Cyr, M., Beaulieu, J. M., Laakso, A., Sotnikova, T. D., Yao, W. D., Bohn, L. M., Gainetdinov, R. R., and Caron, M. G. (2003). Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 11035–11040.
Del Ser, T. (2010). Phase IIa clinical trial on Alzheimer’s disease with NP12, a GSK-3 inhibitor. Alzheimers Dement. 6, S147.
Dominguez, I., Itoh, K., and Sokol, S. Y. (1995). Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proc. Natl. Acad. Sci. U.S.A. 92, 8498–8502.
Eldar-Finkelman, H. (2002). Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol. Med. 8, 126–132.
Eldar-Finkelman, H., and Krebs, E. G. (1997). Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc. Natl. Acad. Sci. U.S.A. 94, 9660–9664.
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137.
Embi, N., Rylatt, D. B., and Cohen, P. (1980). Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527.
Engel, T., Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernandez, F. (2006a). Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455.
Engel, T., Hernandez, F., Avila, J., and Lucas, J. J. (2006b). Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 26, 5083–5090.
Eom, T. Y., and Jope, R. S. (2009). Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 66, 494–502.
Frame, S., and Zheleva, D. (2006). Targeting glycogen synthase kinase-3 in insulin signalling. Expert Opin. Ther. Targets 10, 429–444.
Fuster-Matanzo, A., De Barreda, E. G., Dawson, H. N., Vitek, M. P., Avila, J., and Hernandez, F. (2009). Function of tau protein in adult newborn neurons. FEBS Lett. 583, 3063–3068.
Gomez de Barreda, E., Perez, M., Gomez Ramos, P., De Cristobal, J., Martin-Maestro, P., Moran, A., Dawson, H. N., Vitek, M. P., Lucas, J. J., Hernandez, F., and Avila, J. (2010). Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 37, 622–629.
Gomez-Sintes, R., Hernandez, F., Bortolozzi, A., Artigas, F., Avila, J., Zaratin, P., Gotteland, J. P., and Lucas, J. J. (2007). Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J. 26, 2743–2754.
Gomez-Sintes, R., and Lucas, J. J. (2010). NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J. Clin. Invest. 120, 2432–2445.
Gould, T. D., Picchini, A. M., Einat, H., and Manji, H. K. (2006). Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr. Drug Targets 7, 1399–1409.
Grimes, C. A., and Jope, R. S. (2001). The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65, 391–426.
Grof, P., and Muller-Oerlinghausen, B. (2009). A critical appraisal of lithium’s efficacy and effectiveness: the last 60 years. Bipolar Disord. 11(Suppl. 2), 10–19.
Hardt, S. E., and Sadoshima, J. (2002). Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ. Res. 90, 1055–1063.
Haydar, T. F., Kuan, C. Y., Flavell, R. A., and Rakic, P. (1999). The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb. Cortex 9, 621–626.
Hernandez, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533.
Hetman, M., Cavanaugh, J. E., Kimelman, D., and Xia, Z. (2000). Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J. Neurosci. 20, 2567–2574.
Hetman, M., Hsuan, S. L., Habas, A., Higgins, M. J., and Xia, Z. (2002). ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. J. Biol. Chem. 277, 49577–49584.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hong, X. P., Peng, C. X., Wei, W., Tian, Q., Liu, Y. H., Yao, X. Q., Zhang, Y., Cao, F. Y., Wang, Q., and Wang, J. Z. (2010). Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 20, 1339–1349.
Hongisto, V., Smeds, N., Brecht, S., Herdegen, T., Courtney, M. J., and Coffey, E. T. (2003). Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 23, 6027–6036.
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439.
Hu, S., Begum, A. N., Jones, M. R., Oh, M. S., Beech, W. K., Beech, B. H., Yang, F., Chen, P., Ubeda, O. J., Kim, P. C., Davies, P., Ma, Q., Cole, G. M., and Frautschy, S. A. (2009). GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 33, 193–206.
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551.
Hye, A., Kerr, F., Archer, N., Foy, C., Poppe, M., Brown, R., Hamilton, G., Powell, J., Anderton, B., and Lovestone, S. (2005). Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 373, 1–4.
Imahori, K., and Uchida, T. (1997). Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J. Biochem. 121, 179–188.
Jope, R. S., and Johnson, G. V. (2004). The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102.
Jope, R. S., and Roh, M. S. (2006). Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets 7, 1421–1434.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., Van Eede, M., Hattori, S., Laliberte, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., Macaulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2, 35.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Kimura, T., Yamashita, S., Nakao, S., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2008). GSK-3beta is required for memory reconsolidation in adult brain. PLoS ONE 3, e3540. doi:10.1371/journal.pone.0003540
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Kovacsics, C. E., and Gould, T. D. (2010). Shock-induced aggression in mice is modified by lithium. Pharmacol. Biochem. Behav. 94, 380–386.
Leroy, K., Boutajangout, A., Authelet, M., Woodgett, J. R., Anderton, B. H., and Brion, J. P. (2002). The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer’s disease. Acta Neuropathol. 103, 91–99.
Leroy, K., Yilmaz, Z., and Brion, J. P. (2007). Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 33, 43–55.
Li, B., Ryder, J., Su, Y., Moore, S. A. Jr., Liu, F., Solenberg, P., Brune, K., Fox, N., Ni, B., Liu, R., and Zhou, Y. (2004). Overexpression of GSK3betaS9A resulted in tau hyperphosphorylation and morphology reminiscent of pretangle-like neurons in the brain of PDGSK3beta transgenic mice. Transgenic Res. 13, 385–396.
Liao, X., Zhang, L., Thrasher, J. B., Du, J., and Li, B. (2003). Glycogen synthase kinase-3beta suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand resistance in prostate cancer. Mol. Cancer Ther. 2, 1215–1222.
Lim, F., Hernandez, F., Lucas, J. J., Gomez-Ramos, P., Moran, M. A., and Avila, J. (2001). FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci. 18, 702–714.
Linseman, D. A., Butts, B. D., Precht, T. A., Phelps, R. A., Le, S. S., Laessig, T. A., Bouchard, R. J., Florez-Mcclure, M. L., and Heidenreich, K. A. (2004). Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 24, 9993–10002.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39.
Luo, J. (2009). Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 273, 194–200.
MacAulay, K., Doble, B. W., Patel, S., Hansotia, T., Sinclair, E. M., Drucker, D. J., Nagy, A., and Woodgett, J. R. (2007). Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 6, 329–337.
Macdonald, A., Briggs, K., Poppe, M., Higgins, A., Velayudhan, L., and Lovestone, S. (2008). A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 23, 704–711.
Macritchie, K. A. N., and Young, A. H. (2004). “Adverse syndromes associated with lithium,” in Adverse Syndromes and Psychiatric Drugs: A Clinical Guide, eds P. Haddad, S. Dursun and B. Deakin (New York, NY: Oxford University Press), 89–109.
Manji, H. K., Moore, G. J., and Chen, G. (1999). Lithium at 50: have the neuroprotective effects of this unique cation been overlooked? Biol. Psychiatry 46, 929–940.
Manoukian, A. S., and Woodgett, J. R. (2002). Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv. Cancer Res. 84, 203–229.
Martinez, A., and Perez, D. I. (2008). GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer’s disease? J. Alzheimers Dis. 15, 181–191.
Mayford, M., Bach, M. E., Huang, Y. Y., Wang, L., Hawkins, R. D., and Kandel, E. R. (1996). Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678–1683.
McManus, E. J., Sakamoto, K., Armit, L. J., Ronaldson, L., Shpiro, N., Marquez, R., and Alessi, D. R. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583.
Medina, M., and Castro, A. (2008). Glycogen synthase kinase-3 (GSK-3) inhibitors reach the clinic. Curr. Opin. Drug Discov. Devel. 11, 533–543.
Meijer, L., Flajolet, M., and Greengard, P. (2004). Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25, 471–480.
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
Ougolkov, A. V., Bone, N. D., Fernandez-Zapico, M. E., Kay, N. E., and Billadeau, D. D. (2007). Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 110, 735–742.
Pap, M., and Cooper, G. M. (1998). Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932.
Pastorino, J. G., Shulga, N., and Hoek, J. B. (2002). Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618.
Pei, J. J., Tanaka, T., Tung, Y. C., Braak, E., Iqbal, K., and Grundke-Iqbal, I. (1997). Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 56, 70–78.
Perez, M., Lim, F., Arrasate, M., and Avila, J. (2000). The FTDP-17-linked mutation R406W abolishes the interaction of phosphorylated tau with microtubules. J. Neurochem. 74, 2583–2589.
Perez-Costas, E., Gandy, J. C., Melendez-Ferro, M., Roberts, R. C., and Bijur, G. N. (2010). Light and electron microscopy study of glycogen synthase kinase-3beta in the mouse brain. PLoS ONE 5, e8911. doi:10.1371/journal.pone.0008911
Peter, M. E., and Krammer, P. H. (2003). The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10, 26–35.
Plyte, S. E., Hughes, K., Nikolakaki, E., Pulverer, B. J., and Woodgett, J. R. (1992). Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim. Biophys. Acta 1114, 147–162.
Pomerantz, J. L., and Baltimore, D. (2000). Signal transduction. A cellular rescue team. Nature 406, 26–27, 29.
Rockenstein, E., Torrance, M., Adame, A., Mante, M., Bar-On, P., Rose, J. B., Crews, L., and Masliah, E. (2007). Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27, 1981–1991.
Sereno, L., Coma, M., Rodriguez, M., Sanchez-Ferrer, P., Sanchez, M. B., Gich, I., Agullo, J. M., Perez, M., Avila, J., Guardia-Laguarta, C., Clarimon, J., Lleo, A., and Gomez-Isla, T. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis 35, 359–367.
Sirerol-Piquer, M. A., Gomez-Ramos, P., Hernández, F., Perez, M., Morán, M. A., Fuster-Matanzo, A., Lucas, J. J., Avila, J., and García-Verdugo, J. M. (2011). GSK3β overexpression induces neuronal death and depletion of neurogenic niches in dentate gyrus. Hippocampus 21, 910–922.
Song, L., De Sarno, P., and Jope, R. S. (2002). Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 277, 44701–44708.
Song, L., Zhou, T., and Jope, R. S. (2004). Lithium facilitates apoptotic signaling induced by activation of the Fas death domain-containing receptor. BMC Neurosci. 5, 20. doi:10.1186/1471-2202-5-20
Spittaels, K., Van Den Haute, C., Van Dorpe, J., Geerts, H., Mercken, M., Bruynseels, K., Lasrado, R., Vandezande, K., Laenen, I., Boon, T., Van Lint, J., Vandenheede, J., Moechars, D., Loos, R., and Van Leuven, F. (2000). Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 275, 41340–41349.
Spittaels, K., Van Den Haute, C., Van Dorpe, J., Terwel, D., Vandezande, K., Lasrado, R., Bruynseels, K., Irizarry, M., Verhoye, M., Van Lint, J., Vandenheede, J. R., Ashton, D., Mercken, M., Loos, R., Hyman, B., Van Der Linden, A., Geerts, H., and Van Leuven, F. (2002). Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 113, 797–808.
Spittaels, K., Vandenhaute, C., Vandorpe, J., Bruynseels, K., Vandezande, K., Laenen, I., Geerts, H., Mercken, M., Sciot, R., Vanlommel, A., Loos, R., and Vanleuven, F. (1999). Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am. J. Pathol. 155, 2153–2165.
Stambolic, V., Ruel, L., and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668.
Sun, M., Song, L., Li, Y., Zhou, T., and Jope, R. S. (2008). Identification of an antiapoptotic protein complex at death receptors. Cell Death Differ. 15, 1887–1900.
Terwel, D., Muyllaert, D., Dewachter, I., Borghgraef, P., Croes, S., Devijver, H., and Van Leuven, F. (2008). Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 172, 786–798.
Tilleman, K., Stevens, I., Spittaels, K., Haute, C. V., Clerens, S., Van Den Bergh, G., Geerts, H., Van Leuven, F., Vandesande, F., and Moens, L. (2002). Differential expression of brain proteins in glycogen synthase kinase-3 transgenic mice: a proteomics point of view. Proteomics 2, 94–104.
Wada, A. (2009). GSK-3 inhibitors and insulin receptor signaling in health, disease, and therapeutics. Front. Biosci. 14, 1558–1570.
Woodgett, J. R. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438.
Woodgett, J. R. (1991). cDNA cloning and properties of glycogen synthase kinase-3. Meth. Enzymol. 200, 564–577.
Yamamoto, A., Lucas, J. J., and Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66.
Yao, H. B., Shaw, P. C., Wong, C. C., and Wan, D. C. (2002). Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 23, 291–297.
Zhang, Z., Hartmann, H., Do, V. M., Abramowski, D., Sturchler-Pierrat, C., Staufenbiel, M., Sommer, B., Van De Wetering, M., Clevers, H., Saftig, P., De Strooper, B., He, X., and Yankner, B. A. (1998). Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395, 698–702.
Keywords: GSK-3, apoptosis, mouse models, Alzheimer’s disease, neurodegenerative disorder
Citation: Gómez-Sintes R, Hernández F, Lucas JJ and Avila J (2011) GSK-3 mouse models to study neuronal apoptosis and neurodegeneration. Front. Mol. Neurosci. 4:45. doi: 10.3389/fnmol.2011.00045
Received: 31 August 2011; Accepted: 31 October 2011;
Published online: 16 November 2011.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Jean-Martin Beaulieu, Centre de recherche Universite Laval Robert Giffard, CanadaOksana Kaidanovich-Beilin, Samuel Lunenfeld Research Institute, Canada
Copyright: © 2011 Gómez-Sintes, Hernández, Lucas and Avila. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jesús Avila, Centro de Biología Molecular “Severo Ochoa”, Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid, Campus UAM de Cantoblanco, 28049 Madrid, Spain. e-mail: javila@cbm.uam.es