 Samuel I. Kim
Samuel I. Kim Christopher R. Cassella2
Christopher R. Cassella2 Katelyn T. Byrne
Katelyn T. Byrne- 1Program in Biochemistry, College of Arts and Sciences, University of Pennsylvania, Philadelphia, PA, United States
- 2Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 3Parker Institute for Cancer Immunotherapy, University of Pennsylvania, Philadelphia, PA, United States
Cancer immunotherapy has revolutionized the treatment landscape in medical oncology, but its efficacy has been variable across patients. Biomarkers to predict such differential response to immunotherapy include cytotoxic T lymphocyte infiltration, tumor mutational burden, and microsatellite instability. A growing number of studies also suggest that baseline tumor burden, or tumor size, predicts response to immunotherapy. In this review, we discuss the changes in immune profile and therapeutic responses that occur with increasing tumor size. We also overview therapeutic approaches to reduce tumor burden and favorably modulate the immune microenvironment of larger tumors.
Introduction
Nearly 50 years ago, Whitney and colleagues reported that mice bearing large carcinogen-induced sarcomas displayed defective spontaneous immune responses (1). These immunological defects were reversible with tumor resection, providing a link between tumor burden and dysfunctional immune responses. In the intervening decades, further interrogation into the immunobiology of the tumor microenvironment (TME) reveals significant local and systemic impacts on immune responses (2, 3). With the advent and success of immune checkpoint blockade (ICB) in patients with advanced disease (4), there is a need to understand biomarkers of response to improve outcomes in patients that remain resistant to immunotherapy. Despite the identification of several correlates of response to ICB, including CD8 T cell infiltration of the tumor site, tumor mutational burden, and cytolytic T cell gene expression profiles (5–7), these metrics are not universal predictors of response across all tumor types and patient subsets. Here, we review the literature with regard to total tumor burden as an important negative correlate of response and explore potential mechanisms to mitigate the local and systemic impact of high tumor burden on immune interventions.
Tumor Burden Impacts Baseline Immunity
The complex immunobiology of the TME is regulated by a number of factors, including tumor cell-intrinsic determinants of immune cell infiltration (8), the tissue in which the tumor is located (9), and the stromal and vasculature content of the tumor [reviewed elsewhere (10, 11)]. Immune cell intrinsic defects also contribute to tumor progression, including T cell exhaustion or tolerization to high avidity tumor antigens (12). This multitude of factors contribute to tumor progression, but preclude mechanistic insight in heterogeneous clinical samples, even across tumors harvested from a single patient (13). Mouse models provide crucial insight into the impact of tumor burden on immune features in the TME by allowing for isolation of the impact of tumor size as a variable regulating immune infiltration and immunotherapy sensitivity. Preclinical data support the clinical findings that large tumors are more immunosuppressive compared to small tumors on both the local and systemic level, directly impacting the ability of the host immune system to effectively mount natural or immunotherapy-induced immune responses.
The immunosuppressive nature of the tumor site is locally enforced via immune cell subsets recruited or induced within the TME. Pro-tumorigenic myeloid derived suppressor cells (MDSCs) mediate T cell suppression through a variety of mechanisms including depletion of arginine, oxidative stress of target cells, and release of the dicarbonyl radical methylglyoxal (14–16). MDSCs have been found to increasingly infiltrate the TME in a murine model of renal adenocarcinoma (RENCA) even as the populations of other immune cells (including T cells and dendritic cells; DCs) decreased (17, 18). Similarly, in a murine model of pancreatic ductal adenocarcinoma (PDAC), MDSCs and tumor-associated macrophages have been shown to increasingly infiltrate the TME as tumors progress (19). The same trends are observed in regulatory T cells (Tregs) in multiple tumor types, concurrent with a decrease or even absence of CD8 T cells, NK cells, and DCs during tumor progression (17, 19–22). These data suggest that targeting suppressive immune cell populations may help convert the TME of large tumors to more closely resemble the immune infiltrate of small tumors, augmenting the impact of immunotherapeutic interventions.
In addition to increased proportions of immunosuppressive cells in the TMEs, the cytokine production in large tumors is also skewed to a more suppressive profile as compared to small tumors. Transforming growth factor-β (TGF-β), a pleiotropic cytokine shown to exert anti-tumor effects in early-stage cancer but tumor-promoting effects in late-stage cancer (23), is amplified in large murine T cell leukemia and RENCA tumors (17, 24). IL-10 and nitric oxide synthase 2 (NOS2) similarly increase as tumors progress (17, 24). This likely reflects progressive increases of MDSCs and Tregs in growing tumors (19–22), as both cell populations are major producers of these suppressive cytokines.
However, not all TMEs follow the same positive correlation between suppressor cell infiltration and increased tumor size. In patients with colorectal cancer, CXCL9 – an IFN-γ inducible chemokine that recruits CD8 T cells – is not differentially expressed according to high or low tumor burden (25). In a mouse model of melanoma, B16 tumors show very little perturbation in the relative frequencies of effector or regulatory T cell subsets as tumors increase in size (17). Similarly, CD8 T cells are actually increased (while Tregs are decreased) in large vs. small tumors in a mouse model of colon carcinoma (17). However, chronic antigen exposure has the potential for tumor-specific T cell deletion, resulting in functional “holes” in the immune cell repertoire, a mechanism first reported in the context of chronic viral infections (26). High avidity T cell interactions also have the potential for deletion, as observed in a mouse model of lymphoma where tumor-specific T cells were rapidly lost in the context of a highly immunogenic tumor antigen when the number of target tumor cells was above a certain threshold (27). The failure of these effector cells in large tumors highlights the contribution of other suppressive mechanisms – beyond those mediated by sheer numbers of suppressive MDSC or Treg populations – in established and progressing tumors.
One such mechanism by which tumors escape T cell-mediated destruction is via negative immune checkpoints including programmed death-1 (PD-1) or cytotoxic T lymphocyte-associated protein 4 (CTLA-4). Ligation of immune checkpoints on T cells drives exhaustion and dysfunction, preventing autoimmunity and immunopathology in the context of extenuating immune activation, but usurped by the tumor to disable anti-tumor immunity. Immune checkpoint ligands such as programmed death ligand 1 (PD-L1) are abundant in the TME of many tumor types, independently of tumor mutation burden (28). Multiple tumor types exhibit increased PD-L1 expression as a direct correlate with tumor size including gastroenteropancreatic neuroendocrine tumors (29), advanced gastric cancer (30), and meningiomas (31). Furthermore, in primary and metastatic melanoma samples, PD-L1 levels are higher in primary and local metastases and lower in distant metastases (32) with similar findings in cutaneous squamous cell carcinoma (33) suggesting that expression of checkpoint ligands may be linked to tumor progression.
The immunosuppressive impacts of bearing a large tumor are often not limited to the local TME. Recently, Allen and colleagues revealed that the presence of a tumor negatively impacts the systemic immune landscape, including a global suppression of T cell responses to tumor-unrelated antigen challenges as a result of increased IL-1 and G-CSF (3). Similarly, conventional dendritic cells responsible for priming CD8+ T cells were found to be systematically and progressively dysregulated in mice bearing PDAC, leading to deficient T cell priming even in the presence of strong tumor neoantigens (34, 35). In murine PDAC, tumor cell-derived G-CSF impaired differentiation of type 1 conventional dendritic cells in the bone marrow and blood (36), and elevated serum levels of the cytokine IL-6 in tumor-bearing mice induced apoptosis of conventional dendritic cells (34). Thus the findings of failed systemic immunity in mice bearing large tumors have come full circle (1), revealing that both local and global suppression of the immune response is heightened in the context of increased tumor burden. Targeting both deficits may provide the signal necessary to drive immune responses in currently treatment-resistant tumor types.
Impact of Tumor Burden on Therapy-Induced Immune Responses
Spontaneous or natural cancer-immunity cycles have failed by the time a tumor is detected in the clinical setting, requiring therapeutic interventions to drive immune-mediated rejection of tumors. ICB has thus far garnered the most success and has shown remarkable clinical benefit in patients with aggressive disease (4), but gene therapy approaches such as chimeric antigen receptor (CAR) T cells have become standard-of-care for some patients with previously incurable malignancies (37), and oncolytic viruses have shown great promise in treatment-refractory brain tumors (38). Integrating knowledge about the impact of tumor burden on the efficacy of these therapies may enhance clinical outcomes.
Immune Checkpoint Blockade
Immune checkpoints such as PD-1/PD-L1 or CTLA-4 are negative regulators of activated T cells. Using monoclonal antibodies (mAbs) to disrupt the ligand/receptor pairing of these immune checkpoint molecules enables tumor-associated T cells to overcome immunosuppression and effectively perform anti-tumor functions (39). ICB has drastically improved clinical outcomes for patients with advanced disease including metastatic melanoma (40–42) and non-small cell lung cancer (NSCLC) (43–45).
Despite the immense positive impact of ICB in the clinical setting, many patients do not respond, and tumor burden is one metric that negatively correlates with ICB efficacy. Preclinical data reveals that PD-1 blockade is more effective in mice bearing smaller lung squamous cell tumors (22). Similarly, mice with advanced ovarian tumors are more resistant to PD-L1 blockade than mice with earlier stage tumors (21, 46). This negative correlation of tumor size and ICB sensitivity is borne out in patients where total tumor volume is predictive of response to αPD-1 with local or metastatic melanoma (47, 48). In patients with NSCLC, metabolic tumor volume was also a prognostic factor for sensitivity to PD-1 blockade in both retrospective and prospective studies (22, 49). Furthermore, dual ICB, which targets both PD-1 and CTLA-4 at the same time, has shown greater efficacy than single ICB in patients with metastatic NSCLC and melanoma (40, 43), and is more effective in melanoma patients with smaller baseline tumor diameters (50). Despite the plethora of studies correlating tumor size and ICB outcomes, the threshold of tumor burden at which ICB efficacy is reduced has not been defined. Given this, a dynamic metric, such as the ratio of proliferating T cells specifically reinvigorated by anti-PD-1 compared to the total tumor burden (51) may be the best approach for identifying patients that are resistant to ICB and would benefit from additional therapeutic interventions.
T Cell Costimulation
In contrast to alleviating suppressive signals via ICB, direct co-stimulation of T cells using mAbs targeting molecules such as OX40 and 4-1BB has also been explored. OX40 is a member of the TNF receptor superfamily expressed by activated T cells, and agonistic OX40 mAbs have direct stimulatory activities on effector T cells with the benefit of inhibiting Treg function in the tumor site, showing early promise in clinical trials (52). Preclinical studies reveal that small MCA205 fibrosarcoma tumors and CT26 colon carcinomas are sensitive to OX40 agonism as a single agent, but larger tumors (50–120 mm2) require the addition of transforming growth factor-β (TGF-β) receptor antagonists to further reduce immune suppression (53). Similar to OX40, 4-1BB is a co-stimulatory receptor expressed on T cells and antigen-presenting cells, and 4-1BB agonists enhance the anti-tumor effector functions of cytotoxic T cells (54). In preclinical studies using an MC38 colon carcinoma model, 4-1BB agonist therapy had little impact on tumor progression when administered early (less than 48 h after tumor implantation), while the combination of 4-1BB and anti-CTLA-4 together was effective even in established tumors (14 days after implantation) (55). However, this approach failed in B16 melanoma tumors (55), suggesting that the unique tumor microenvironment, in addition to tumor size, dictates therapeutic efficacy.
Cancer Vaccines and Oncolytic Viruses
In contrast to ICB, where T cells are indiscriminately “rescued” from exhaustion regardless of antigen specificity, cancer vaccines aim to induce a tumor-specific adaptive immune response through delivery of whole tumor cells or tumor-derived antigens (56). Preclinical mouse models reveal cancer vaccine platforms are efficacious in small, but not large, tumors (21, 57). Coupling a bacteria Type II secretion protein with the model antigen ovalbumin (OVA), Binder and colleagues show that tumor rejection cannot be fully rescued in late stage B16 melanoma tumors, even with a strong OVA-specific CD8 T cell response (57). Similarly, two reports using killed tumor cells to vaccinate tumor-bearing mice find that late stage ovarian cancer or mammary tumors cannot be controlled, even with the addition of costimulatory or ICB mAbs (21, 58). These reports highlight the existence of a threshold of tumor burden below which the tumors are sensitive to immunotherapy, but above which the tumors are highly resistant.
Oncolytic vaccines offer the ability to prime T cell responses against tumor antigens via robust stimulation of the innate immune system. However, an adenovirus vaccine encoding tumor-specific somatic mutations (Gad-CT26-31) that works prophylactically or as a very early therapeutic intervention for small CT26 colon carcinomas was insufficient to eradicate large (>70mm3) established tumors (59). Similarly, a HPV vaccine resulted in the cures of small herpes poliovirus (HPV)-expressing TC-1 tumors whereas large tumors were more resistant, failing to achieve complete clearance (60). The inverse relationship between the success of viral vaccines or oncolytic viruses and high tumor burden holds true for patients with melanoma: treatment with the oncolytic virus talimogene laherparepvec, or T-VEC (which drives tumor cell killing and in situ priming against tumor antigens via cytokine release), results in better outcomes if tumor burden is low at the start of therapy (61). However, rational pairing of oncolytic viruses with ICB may hold promise: PD-1 blockade rescued anti-tumor immunity in mice bearing large tumors in both colon cancer and HPV models (59, 60). The potential beneficial impact of oncolytic viruses for diseases such as glioblastoma highlight the need to better understand the TME, to optimally pair treatment modalities for improved outcomes.
Direct Antigen-Presenting Cell Activation
Cancer vaccines are often utilizing ex vivo DC activation, or in vivo innate immune sensors for activation of antigen presenting cells (APCs) to improve T cell priming, with the underlying hypothesis that insufficient T cell priming is one reason for the failure of ICB or co-stimulation mAbs to “rescue” T cell responses in the TME (62). However, direct activation of APCs such as by using an agonist CD40 mAb is another option for driving newly primed T cell responses against tumors. Ligation of CD40, expressed on APCs, promotes the rapid licensing and maturation of dendritic cells and other APC subsets independently of CD4 T cell help (the natural source of CD40 ligand) (63–65) normally provided upstream of immune checkpoint expression. We have shown that CD40 stimulation drives T cell immunity in a genetically engineered mouse model of PDAC when used in combination with chemotherapy (66), ICB (67), or combinations of chemotherapy and radiotherapy with ICB (68, 69), and there are promising data from a clinical trial in patients with metastatic PDA receiving agonistic CD40 in combination with chemotherapy (70). To assess the impact of tumor size in response to agonistic CD40 mAb therapy, we segregated tumor-bearing mice from previous studies according to therapeutic response and assessed the baseline tumor size in each group (66, 67). As shown in Figure 1, mice that responded to anti-CD40 in combination with chemotherapy (gemcitabine, Gem; and nab-paclitaxel, nP) or dual ICB had smaller baseline tumors than mice that were resistant to treatment. Mice with smaller tumors also survived longer in both treatment cohorts, although treatment with CD40/ICB resulted in slightly larger tumors responding to therapy as compared to mice treated with chemotherapy and CD40, supporting findings in cancer vaccine studies where a multi-pronged approach is more effective. Thus, non-redundant methods of stimulation T cell priming—directly via costimulatory mAbs, or via activation of APCs via CD40 mAb—still reveal the baseline tumor size as a major determinant of response.
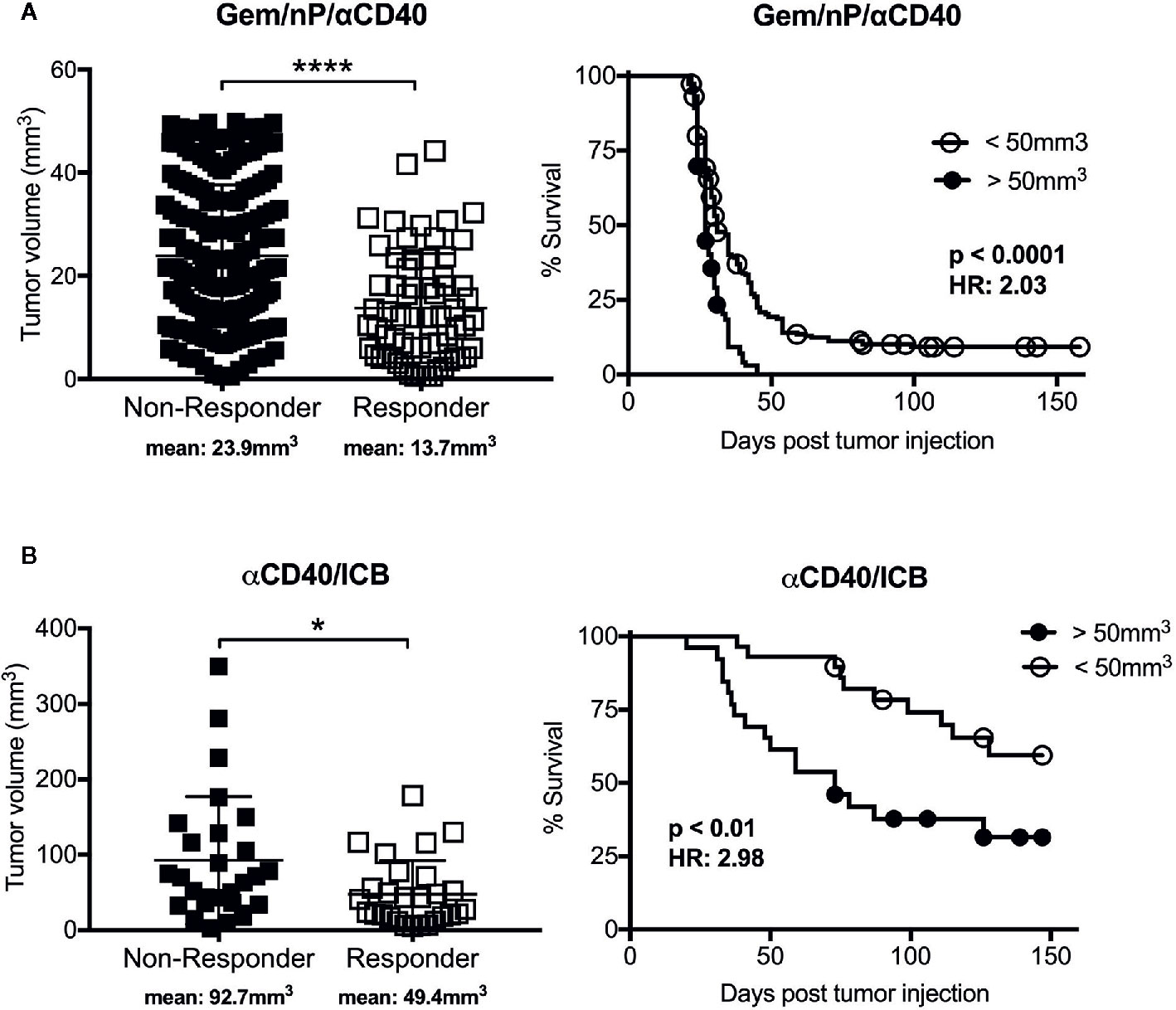
Figure 1 Efficacy of tumor rejection after combination treatment with agonistic CD40 antibody correlates with tumor size. Graphs from studies reported in (66) (A) and (67) (B), reported here stratified by tumor response (defined as cured of primary tumor; scatter plots) or baseline tumor size (survival curves). Briefly, C57Bl/6 mice were implanted subcutaneously with 2.5x105 4,662 pancreatic ductal adenocarcinoma (PDAC) cell lines and treated with (A) agonistic CD40 (αCD40) in combination with chemotherapy (gemcitabine, Gem; and nab-paclitaxel, nP) as described in (66), or treated with (B) αCD40 in combination with αPD-1 and αCTLA-4 (ICB) as reported in (67). Each symbol represents a single mouse, horizontal lines indicate mean (also denoted below graph), and error bars indicate standard deviation. **** indicates p value < 0.0001 and * p < 0.05 as determined by an unpaired t test. For survival curves analysis was performed using log-rank test with indicated p values, HR indicates Hazard Ratio according to the size cutoff of 50mm3, calculated using log-rank. Analyses were performed using Graphpad Prism.
Adoptive and CAR T Cell Therapies
Given the systemic inhibition of immune responses in the presence of large tumors, adoptive cell therapy (ACT) is an appealing therapeutic option that bypasses the requirement for in vivo T cell priming or reactivation. Tumor-specific T cells are enriched via ex vivo expansion and activation before reinjection into the patient, and ACT of tumor infiltrating lymphocytes (TILs) has shown promise in metastatic melanoma (71, 72). However, tumor burden presents an issue even with antigen specific T cell therapies, as shown in a mouse model of lymphoma where ACT is effective only against small tumors (27). In large tumors, the authors found functional impairment and rapid deletion of the transferred CD8+ T cells, which was reversed via pretreatment with chemotherapy to reduce tumor size (27). Similar observations were made in a mouse model of sarcoma, where small tumors regressed after ACT, but large tumors on the contralateral flank did not (73). Recent studies show that the impairment and deletion of ACTs observed in large (but not small) tumors is reflective of suppressive local TME, as T cell affinity and expression of the tumor specific antigen was similar in both tumor sizes (74).
In addition to TILs, genetically modified T cells can also be used for ACT, including CAR T cells, where success has been made in many hematological malignancies, but solid tumors remain a more difficult problem (37). Hematological cancers such as acute lymphoblastic leukemia have a direct correlation between total tumor burden and response to CD19 CAR T cell treatment (75). In a mouse model of hepatocellular carcinomas, CAR T cells targeting glypican 3 were sufficient to drive regressions of small tumors but had no effect on large tumors, which was reversed upon the addition of sorafenib as a result of increased IL-12 production from intratumoral macrophages (76). In contrast, CAR T cells targeting glypican 1 were sufficient to drive tumor regressions in both large and small murine colorectal tumors (77). Thus the local TME can impact treatment efficacy (and can be targeted with rational therapeutic combinations) but the systemic impact on the immune response as a result of bearing large tumors is a significant barrier to treatment outcomes.
Strategies to Reduce Tumor-Burden Associated Impacts on Immunity
Given that large tumors are more resistant to immunotherapy and harbor greater populations of suppressive cells, targeting tumor burden-associated alterations in the tumor microenvironment may exert therapeutic effects. Here, we briefly cover two major categories of approaches that may improve immune responses against tumors: directly reducing tumor size and modulating the tumor immune microenvironment.
Traditional Interventions
Surgery
Tumor resection is the oldest method of treating cancer, and one of the original methods of restoring defective tumor surveillance in mouse models (1). The use of tumor resection prior to administration of immunotherapy improves control of tumor growth in multiple mouse models (3, 22, 78). This “resetting” of the immune response has been linked to the alleviation of immune suppression by tumor-derived IL-1 and granulocyte colony-stimulating factor (G-CSF) and could be phenocopied by mAb blockade of either cytokine (3). However, the trauma of surgical resection and the ensuing wound-healing process can lead to the outgrowth of otherwise immune-controlled metastases (79–81), which can be reversed by neoadjuvant administration of immunotherapies in some settings (81, 82). For example, patients with metastatic renal cell cancer benefited when surgery was combined with IFNα-2b treatment, resulting in a three-month increase in median survival with combination treatment compared to IFN treatment alone (83). In addition to immunotherapies, anti-inflammatory medication also prevents surgery-induced outgrowth of metastases by reducing the proportion of suppressive tumor-associated macrophages (82). Thus, surgical resection with careful management of the immunosuppressive wound-healing process could be considered prior to adjuvant immunotherapy.
Chemotherapy
Cytotoxic chemotherapy is another viable strategy to reduce tumor size that has been employed in the clinic for decades. Moreover, some chemotherapy regimens drive immunogenic cell death of tumor cells, leading to release of tumor antigens that can be presented by APCs to prime cytotoxic T cells (84). In this way, chemotherapy can exert both a local anti-tumor effect as well as systemic positive pressure on the immune system; these two outcomes are not mutually exclusive but highly interdependent. For example, a pre-clinical study showed that administering a low dose of the chemotherapy cyclophosphamide (CTX) one day before ACT of tumor-specific CD8 T cells is effective in reducing tumor burden and thereby prevents deletion of the transferred CD8 T cells in a murine lymphoma model (27). In the clinic, combination chemo-immunotherapy regimens have shown increased efficacy over chemotherapy alone in patients with advanced NSCLC (44) and metastatic melanoma (85).
However, given the cytotoxic nature of chemotherapy on any proliferating cell including T cells (86, 87), the impact of combination chemo-immunotherapy has variable results. The use of CTX followed by αCTLA-4 resulted in significant tumor regressions in a mouse model of colon cancer, while reversing the order resulted in apoptosis of tumor-reactive T cells (88), and we and others have observed similar detrimental impacts on sequencing gemcitabine with αCD40 in a mouse model of PDAC (89, 90). Furthermore, we have reported that the addition of the chemotherapy to agonistic CD40 and dual ICB significantly reduced the long-term survival of tumor bearing mice compared to immunotherapy alone (67). These findings suggest that chemotherapy, while important to restrain tumor growth, may not synergize with some types of immunotherapy. Additional investigations are necessary to determine optimal sequencing strategies such that chemotherapy can be employed to shrink large tumors without negatively impacting a developing immune response.
Radiation Therapy
Radiotherapy delivers high-energy rays directly to tumor sites as a form of curative control locally at the tumor, and the resultant abscopal effect can be beneficial for immune responses. The administration of radiation treatment completely ablates the tumor and surrounding stroma, effectively debulking the tumors. The immune impact is therefore complex—immune suppression may be alleviated by the eradication of the local tumor, while increased tumor cell death enhances systemic anti-tumor T cell response by generating novel tumor antigens presented on major histocompatibility complex (MHC)-I (91). In preclinical and clinical studies, the addition of radiotherapy to immunotherapy treatments such as ICB (92–94) and/or αCD40 (68) significantly improved immune response and survival. Mechanistically, radiation can upregulate expression of neoantigens (94), diversify the T cell repertoire (92), and enhance antigen presentation and co-stimulation on dendritic cells (68). Thus, in contrast to the detrimental immune impact of chemotherapy, radiation may promote immune cell activation while reducing tumor burden as a rational therapy partner for many immunotherapies.
Simulating a “Small” Immune Microenvironment
Physical Barriers to Immune Infiltration
Some of the highest hurdles for immune cell infiltration of the TME are the physical barriers of entry to the tumor site. Large tumors have reduced vascularization which creates regions of hypoxia that are particularly prominent in large tumors (95, 96), but reduced vascularization also compromises the delivery of a number of different types of therapies into the TME itself (97). This is further compromised when the TME is highly fibrotic, as is the case with PDAC and reviewed elsewhere in greater detail (98). While several mechanisms to target the stroma, such as inhibition of fibroblast activation protein (99), inhibition of Hedgehog signaling (100), or the use of chemotherapeutic agents such as nab-paclitaxel (101–103) have been devised, immune therapies can also be modified to overcome this barrier. For example, Maute et al. engineered a PD-1 ectodomain capable of blocking PD-1:PD-L1 interactions that is an order of magnitude smaller than anti-PDL1, and able to more effectively penetrate the TME (104). As a result, the PD-1 ectodomain was more effective against both large (150 mm3) and small (50 mm3) mouse CT26 colon carcinomas as compared to anti-PD-L1, which was only effective against small tumors (104). Thus, designing drugs with a higher degree of tumor penetration is a feasible approach for improved delivery of some immunotherapies.
Overcoming Tumor-Associated Suppressive Cells
As discussed in Part I, the proportions of suppressive cell populations such as MDSCs are increased in large and progressing tumors. Blocking MDSC recruitment to or activity in the TME renders the tumor sensitive to immunotherapy [reviewed in depth elsewhere (105)]. We and others have shown that MDSCs are rapidly recruited to the PDAC TME via multiple chemokine and cytokine axes including CXCR2 (8, 106, 107), GM-CSF (108, 109), and G-CSF (8). Perturbation of these pathways reduces MDSC recruitment to the tumor site (8, 107, 108), increases CD8 T cells in the TME (8, 107, 108), and converts the tumor to immunotherapy-sensitive (8). MDSC reduction can be achieved via depletion, using CD33-specific immunotoxin Gemtuzumab ozogamicin (110) or ADH-503, a small-molecule agonist for CD11b (111), while the immunosuppressive activity of MDSCs can be blunted using anti-TIGIT (112) or CD73 blockade (113).
Tumor-associated macrophages (TAMs) play a similarly suppressive role in the TME and can also be targeted using depletion strategies. Colony-stimulating factor-1 (CSF-1), a key regulator of macrophage differentiation, can be blocked via CSF-1R mAb or antagonists, resulting in a sensitization of the TME to immune cell killing (114). Skewing TAMs to an anti-tumor functional profile also reduces the immunosuppression in hosts bearing large tumors. For example, pharmacological inhibition of PI3K-γ, which is highly expressed on myeloid cells, shifted TAMs from a M2-like immunosuppressive phenotype to an inflammatory M1-like phenotype in the murine 4T1 mammary carcinoma and B16 melanoma models (115). Paradoxically, reducing TAMs can result in a compensatory increase of MDSCs (116, 117). However, this effect can be overcome by blocking both TAMs and gMDSCs, which augments the efficacy of PD-1 blockade in a mouse model of cholangiocarcinoma (116) and bolsters chemotherapy in a mouse model of PDAC (117). The suppressive myeloid and macrophages subsets in the TME are thus a major immune cell barrier that must be overcome to generate effective anti-tumor responses.
Suppression in the TME is also closely regulated by Tregs, including both natural Tregs and tumor-induced Tregs. Selective depletion of Tregs by αCD25 shows promise in preclinical tumor-bearing animal models [reviewed in (118)], but can have unintended consequences of deleting effector CD25+ CD4 T cells. While effector CD4 T cell depletion does not negatively impact all immunotherapies (119), there is a growing appreciation of the contributions of CD4 T cells in mediating anti-tumor responses (67, 120–122). Some data suggest that αCTLA-4 specifically depletes CTLA-4+ Tregs in mouse models (123), but not in patients (124). Similarly, targeting glucocorticoid-induced TNFR family related protein (GITR) via agonistic antibody has also been shown to deplete Tregs in mouse models (125), while agonistic CD40 administration results in a drastic reduction in Tregs in the murine PDAC TME (66). These findings support targeting Tregs as well as other suppressive cell populations in the large TME to render the ratio of effector cell to suppressive cell more in alignment with ratios found in small tumor sites.
Stimulating Immune Responses
In contrast to removing suppression, many immunotherapies specifically aim to promote immune activation and, to a certain extent, inflammation. The stimulation of innate immune signaling pathways is the original immunotherapy approach, used by William Coley in the early 1900s. “Coley’s Toxins” included a mix of bacterial products that he applied to the tumor lesions of patients, and he observed striking tumor regressions (126). Over the next century, immunologists deconstructed the immune response, resulting in the identification of cell types, receptors, and ligands that can be utilized to mount immune reactions.
Toll-like receptors (TLRs) are canonical pattern recognition receptors, and ligation by toll-like agonists stimulate the innate immune sensing pathways. In the context of the cancer-immunity cycle, providing TLR agonists licenses APCs to enhance antigen presentation and production of inflammatory cytokines (127). TLR agonists have significant anti-tumor roles by driving polarization of TAMs toward an anti-tumor, pro-inflammatory M1 phenotype (128). This conversion of TAMs to an M1 phenotype renders the TME more permissive to downstream activation of CD8+ T cells and infiltration to the tumor site naturally or via orthogonal combinations (129). Furthermore, TLR2 agonists alleviate the immunosuppression mediated by Tregs by reprogramming the Tregs to effector Th17 cells (130).
Despite these positive changes in the TME after TLR agonist administration, there are some tumor cells that express—and are activated—by TLR ligation (127, 131), which promotes tumor growth and chemoresistance in TLR7/TLR8 overexpressing human pancreatic cancer cells (132) and production of immunosuppressive cytokines (TGF-β, vascular endothelial growth factor (VEGF), IL-8) and resistance to apoptosis in human lung cancer cells (133). Implementation of TLR agonists may therefore have a number of benefits to reducing tumor suppression, but the type of TLR stimulated and the downstream impact may be regulated by the immunobiology of a specific TME.
A second class of innate immune sensor stimulation bypasses the binding of TLRs and instead directly stimulates the cyclic GMP-AMP synthase (cGAS) - stimulator of IFN genes (STING) pathway, which is triggered in the presence of cytosolic DNA (134). Activation of the cGAS-STING pathway in cancer cells leads to secretion of pro-inflammatory molecules that attract immune cells and restrict tumorigenesis (135, 136). Furthermore, tumor-cell derived DNA is transferred to APCs, activating the cGAS-STING pathway and production of type I interferons, leading to improved CD8 T cell priming (137, 138). STING activation has also been shown to act on tumor endothelial cells, leading to potent production of type I IFNs (139), further enhancing the anti-tumor response. STING agonists therefore offer an effective method of increasing CD8 T cell infiltration in the TME of large tumors, and have the potential to synergize in combination with a number of treatment modalities including ICB (140), VEGF receptor 2 blockade (141), and chemotherapy (142).
The TLR and STING agonist pathways are dependent downstream on signaling via type I IFNs. In contrast, CD40 stimulation bypasses the use of innate immune sensors and Type I IFN signaling, thus presenting an alternate bridge between innate and adaptive immunity (66, 67). Agonistic CD40 can drive the priming of a robust anti-tumor T cell response dependent on IFN-γ and Batf3 expression (8, 66, 67), while CD40 ligation on TAMs drives a tumoricidal response that depletes the stroma in the TME (143). As such CD40 activation is an important mechanism of increasing effector T cell trafficking to immunologically cold tumors, a finding that can be extended to treatment of large tumors that harbor more immunosuppressive cells and fewer effector immune cells.
Oncolytic Viruses
Oncolytic viruses (OVs), originally designed to specifically kill tumor cells, have more recently been utilized as potent activators of local and systemic immune responses. OVs drive tumor cell death, resulting in the release of inflammatory signals (cell stress danger-associated molecular patterns or DAMPS) and the liberation of tumor-associated antigens to promote anti-tumor immunity. Most recently, clinical trials in patients with brain tumors have shown promising results using OVs (144), such as PVSRIPO (a modified poliovirus OV) in patients with glioblastoma, where some patients have shown complete and durable remissions (145). Future investigations into combinatorial approaches may further enhance the outcomes of OVs in tumors and help delineate which patients with brain tumors may require additional resections vs. orthogonal treatment combinations.
The immunostimulatory impacts of OVs can also be modulated by “cytokine-arming,” whereby the virus also contains a cytokine payload that re-educates, recruits, or stimulates immune cells locally in the TME [reviewed in (146)]. One highly successful example of a cytokine-armed OV is T-VEC, which selectively targets tumor cells while also producing GM-CSF (147). Mechanistically, T-VEC modulates the TME by activating and attracting tumor-specific CD8 T cells (148) via tumor cell lysis (resulting in the liberation of tumor antigens and DAMPs) and the recruitment of DCs [reviewed in (149)]. T-VEC has shown remarkable efficacy in clinical trials for patients with advanced melanoma both as monotherapy (150) and in combination with anti-CTLA-4 (151, 152). OVs thus have the potential to specifically target tumor cells for destruction while delivering a payload that promotes immune activation, a multi-pronged approach that may be ideal for large tumors that are otherwise not amenable to traditional tumor debulking approaches.
Tumor Cell Intrinsic Mechanisms of Immune Regulation
In addition to contributing to aberrant tumor cell growth, oncogenic signaling pathways have the potential to regulate immune responses within the TME, suggesting that precision medicine in combination with immunotherapy should be explored for improved outcomes. For example, the Wnt/β-catenin signaling in a mouse model of melanoma hindered T-cell infiltration and established an immune “cold” TME by downregulating the expression of the chemokine CCL4 (153). Targeting the gene encoding β-catenin via RNA interference synergizes with dual ICB in a mouse model of breast cancer mice and results in complete tumor regressions (154), and small molecule inhibitors of Wnt signaling are being explored for potential application in tumor-bearing hosts (155). Similarly, we found increased expression of MYC in immune “cold” tumors, resulting in increased CXCL1 production and MDSC recruitment concomitant with a lack of T cells in the TME (8). JQ1 is a small molecule that binds competitively to bromodomains and inhibits MYC gene transcription by displacing bromodomain 4 (BRD4) (156), and has the added benefit of reducing PD-L1 expression by tumor cells and APCs (157) while also directly promoting T cell persistence and effector functions in the TME (157, 158). Although JQ1 shows promise in driving tumor responses when used in combination with ACT and ICB (158, 159), it has also been shown to activate and promote tumor invasion and metastasis pathways in mouse models of prostate cancer (160), suggesting further investigation is necessary for optimal application in the clinical setting.
There has been significant effort to develop effective inhibitors against the mitogen-activated protein kinase (MAPK) signaling pathway, given that it is one of the most commonly mutated pathways across all types of cancer (161). RAS proteins are activated upstream of MAPK and direct the establishment of the immunosuppressive TME via GM-CSF production (108) and upregulation of PD-L1 (162), but have proved difficult to target [reviewed elsewhere in detail (163)]. Interestingly, using ACT of T cells specific for an MHC I epitope derived from mutated Kras controlled tumor growth in a patient with metastatic colorectal cancer (164) and is being further investigated preclinically (165) and via Kras-directed cancer vaccine trials (163). Recently, the clinical development of Sotorasib, an inhibitor to mutant G12C KRAS, has yielding promising results in a phase I clinical study performed on patients with advanced solid tumors harboring the KRAS p.G12C mutation (166). It has also been shown to increase T cell, dendritic cell, and macrophage infiltration into the tumor site of CT26 KRAS G12C colon carcinoma-bearing mice, either alone or in combination with anti-PD-1 therapy (167). However, it is likely that tumor burden will continue to be a major barrier to these approaches as discussed above, and thus targeting RAS via immune-mediated mechanisms may require combinatorial approaches for optimal outcomes.
Targeting proteins downstream of RAS in the MAPK pathway has been more successful. BRAFV600E is one of the most common mutations, and a class of RAF monomer inhibitors specifically target this mutation to block mutant BRAF signaling (161). Vemurafenib is one such small molecule and administration significantly enhanced the survival of patients with metastatic melanoma harboring the BRAFV600E mutation (168). Critically, vemurafenib directly alters immune-associated features of malignant cells, such that blocking mutant BRAF signaling results in the reduction of suppressive cytokine production and increased MHC expression by tumor cells (169). This alters the local TME, resulting in decreased recruitment and survival of suppressive MDSCs (170) and Tregs (20), with a concomitant increase in MHC expression of melanocyte differentiation antigens [known targets of the endogenous T cell response against melanoma (171)] and with some BRAFi, T cell infiltration in to the TME (172). Thus BRAF inhibition controls tumor progression while also altering the tumor site toward a “small” TME, suggesting combination with other immune interventions may be highly effective.
Similar to BRAFi, mitogen/extracellular signal regulated kinase (MEK) inhibition controls tumor progression, but has been shown to negatively impact T cell priming and proliferation in vitro and in lymph nodes (173). However, MEKi appears to have a negligible negative impact on the local immune response against the tumor when administered in vivo (173). Indeed, MEKi increases infiltration of antigen-specific CD8 T cells into CT26 colon carcinoma tumors and protects intratumoral T cells from TCR-driven exhaustive apoptosis (174). These findings support the rational combination of MEKi with ICB, and accordingly, MEKi augments PD-1 or PD-LI blockade in mouse models of colon carcinoma and melanoma (174–176).
Despite the positive immune impacts of inhibiting mutant BRAF or MEK, many patients still progress after a period of tumor control, leading to the combination of BRAF/MAPKi in the clinical setting (177). This combination rendered tumors more sensitive to ICB, and adding PD-1 blockade further potentiated BRAF/MEKi in a mouse model of melanoma (178), and has been translated to the clinic for patients with mutant BRAF (179). Given the potential negative impact of MEKi on T cell priming in tumor-draining lymph nodes, rational immunotherapy combinations are crucial – e.g., pairing with CD40 stimulation may not be ideal while ICB or certain types of ACT may work very well with MEKi and these early combination trials are very promising.
Epigenomic Modifiers
Abnormal gene expression is a hallmark of cancer progression, and histone deacetylases (HDAC), which regulate chromatin remodeling, are aberrantly expressed in many cancer types. HDACi results in cytostatic or cytotoxic effects on tumor cells, but this approach has been more effective in hematological malignancies than in solid tumors when used as a monotherapy or combined with other drugs including chemotherapy, reviewed by Suraweera and colleagues (180). Despite this, HDACi does reverse some of the gene repression found in tumor cells, thereby increasing expression of MHC and costimulatory molecules for both T and NK cells on tumor cells in the TME (181). In addition to tumor cell intrinsic effects, HDACi can also have direct effects on lymphocytes in the TME, similar to MEKi. Here the data are somewhat contradictory, as HDACi has differential impacts on T cell subsets, alternatively promoting IFN-γ production (182) and survival (183), while also restraining proliferation and cytotoxic functions in vitro (184). Importantly, tumor-associated Tregs are sensitive to Class I HDACi, with reductions in frequencies and suppressive function in a dose-dependent manner in a mouse model of renal cell carcinoma or prostate cancer (185). The impact of HDACi on APC subsets in the TME remains less clear, with some TAM subsets becoming less suppressive, other subsets acquiring an alternatively activated phenotype and DCs losing licensing and activation markers (181). These studies support the use of HDACi to help remodel the TME and associated immune response, but preclinical modeling will be crucial to elucidate the impact of HDACi on the initiation and maintenance of tumor immunity.
Autophagy Inhibition
Autophagy is a mechanism by which cancer cells (and other cell types) degrade intracellular organelles as a source of nutrients under both basal and stress-induced conditions (186). Two FDA-approved drugs developed to treat malaria – chloroquine (CQ) and hydroxychloroquine (HCQ) – have been used extensively in the clinical setting but are also potent autophagy inhibitors. Initial in vitro studies revealed a cytotoxic impact after blocking autophagy in cancer cells and the first clinical trial was run in glioblastoma where patient survival was significantly extended when CQ was combined with radiation and chemotherapy (187), and CQ or HCQ have now been studied in the context of a number of different tumor types with somewhat variable results (186). In a mouse model of pancreatic cancer, a recent study highlighted the autophagy-mediated downregulation of MHC expression by tumor cells, which was reversible upon autophagy inhibition (188). Combining CQ with dual ICB drove potent tumor regressions and responses (188), revealing autophagy as a determinant of tumor cell immunogenicity. While this alone helps skew the TME toward a “small” TME phenotype with increased T cell infiltration (188), the benefits of systemic autophagy inhibition are much broader (186). CD8 T cells display enhanced anti-tumor effector function (189), while DCs upregulate MHC I expression after autophagy inhibition. However, tumor-intrinsic autophagy may be required for chemotherapy-induced tumor cell death, which can be a potent immune stimulator (190), highlighting the need for preclinical investigations to identify rational therapeutic partners for autophagy inhibition and successful immune responses against cancer.
Concluding Remarks
Clinical and pre-clinical data indicate that tumor burden negatively correlates with response to a range of immunotherapies that include ICB, adoptive T cell therapy, and activation of antigen-presenting cells. Mechanistically, large tumors exert greater local and systemic changes to the immune system, and harbor more immunosuppressive cells and molecules that dampen antitumor activity. Many of the alterations locally and systemically reflect a more immunosuppressive tumor microenvironment; however, integrating this within the context of tumor burden may help stratify patients for optimally designed combination therapy approaches. Targeting features of the “large” TME can skew the immune microenvironment more toward phenotype of a “small” TME, rendering the tumor more sensitive to immunotherapeutic interventions as highlighted in Figure 2. A patient presenting with a large tumor burden that is amenable to debulking or size reduction to help alleviate system immunosuppression may require one approach, while a patient presenting with a relatively small but highly immunosuppressive tumor may require an entirely different therapeutic intervention. Small tumors that display an immunosupportive phenotype and are, at baseline, more amenable to immune interventions may require less invasive treatments than large tumors of the same type. Further studies interrogating the impact of bearing a large vs. small tumor are warranted, including investigations into the systemic impact on the immune response, the spatio-temporal localization of immune cell subsets within the TME as tumors progress, and opportunities to harness precision medicine with personalized immunotherapies. These investigations may reveal novel combinations of interventions that have the greatest impact on therapeutic outcome for patients with the greatest unmet clinical need.
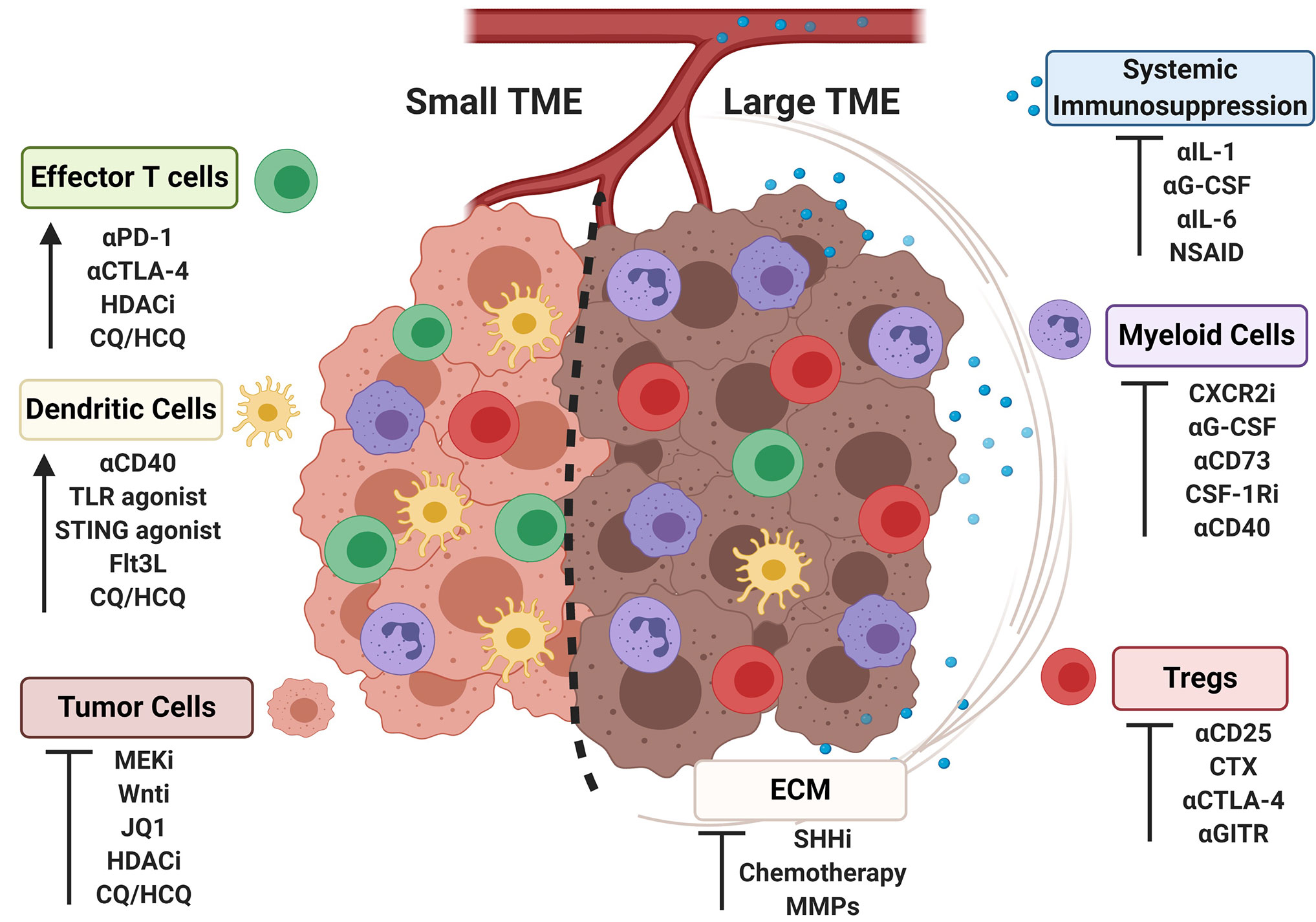
Figure 2 Strategies for skewing immune aspects of the large tumor microenvironment to a small tumor microenvironment. Representative cell populations frequencies found in large or small tumor microenvironments (TMEs) are shown (cell identities labeled on figure according to colors as shown). Strategies to target the TME are listed under cell populations known to response to specific interventions, with the outcomes indicated by an arrow or an inhibitory symbol.
Author Contributions
SK and KB wrote the manuscript; all authors edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Roy and Diana Vagelos Scholars Program in the Molecular Life Sciences at the University of Pennsylvania (SK) and Parker Institute for Cancer Immunotherapy (KB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Robert Vonderheide, Alexander Morrison, and members of the Byrne and Vonderheide laboratories for helpful discussions pertaining to this review. Figure 2 was generated using Biorender.com.
References
1. Whitney RB, Levy JG, Smith AG. Influence of tumor size and surgical resection on cell-mediated immunity in mice. J Natl Cancer Inst (1974) 53(1):111–6. doi: 10.1093/jnci/53.1.111
2. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
3. Allen BM, Hiam KJ, Burnett CE, Venida A, DeBarge R, Tenvooren I, et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med (2020) 26(7):1125–34. doi: 10.1038/s41591-020-0892-6
4. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
5. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
6. Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
7. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (2018) 362(6411):eaar3593. doi: 10.1126/science.aar3593
8. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity (2018) 49(1):178–93.e7. doi: 10.1016/j.immuni.2018.06.006
9. Horton BL, Fessenden TB, Spranger S. Tissue Site and the Cancer Immunity Cycle. Trends Cancer (2019) 5(10):593–603. doi: 10.1016/j.trecan.2019.07.006
10. Yamauchi M, Barker TH, Gibbons DL, Kurie JM. The fibrotic tumor stroma. J Clin Invest (2018) 128(1):16–25. doi: 10.1172/JCI93554
11. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy article. Cell Death Dis (2018) 9(115):1–14. doi: 10.1038/s41419-017-0061-0
12. Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer Res (2008) 68(8):2993–3000. doi: 10.1158/0008-5472.CAN-07-5008
13. Stanta G, Bonin S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front Med (2018) 5:85:85. doi: 10.3389/fmed.2018.00085
14. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
15. Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol (2018) 200(2):422–31. doi: 10.4049/jimmunol.1701019
16. Baumann T, Dunkel A, Schmid C, Schmitt S, Hiltensperger M, Lohr K, et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol (2020) 21(5):555–66. doi: 10.1038/s41590-020-0666-9
17. Yu JW, Bhattacharya S, Yanamandra N, Kilian D, Shi H, Yadavilli S, et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PloS One (2018) 13(11):e0206223. doi: 10.1371/journal.pone.0206223
18. Najjar YG, Rayman P, Jia X, Pavicic PG Jr., Rini BI, Tannenbaum C, et al. Myeloid-Derived Suppressor Cell Subset Accumulation in Renal Cell Carcinoma Parenchyma Is Associated with Intratumoral Expression of IL1β, IL8, CXCL5, and Mip-1α. Clin Cancer Res (2017) 23(9):2346–55. doi: 10.1158/1078-0432.CCR-15-1823
19. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res (2007) 67(19):9518–27. doi: 10.1158/0008-5472.CAN-07-0175
20. Shabaneh TB, Molodtsov AK, Steinberg SM, Zhang P, Torres GM, Mohamed GA, et al. Tumor Biology and Immunology Oncogenic BRAF V600E Governs Regulatory T-cell Recruitment during Melanoma Tumorigenesis. Cancer Res (2018) 78(17):5038–49. doi: 10.1158/0008-5472.CAN-18-0365
21. Duraiswamy J, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors-response. Cancer Res (2014) 74)2:633–4. doi: 10.1158/0008-5472.CAN-13-2752
22. Guisier F, Cousse S, Jeanvoine M, Thiberville L, Salaun M. A rationale for surgical debulking to improve anti-PD1 therapy outcome in non small cell lung cancer. Sci Rep (2019) 9(1):16902. doi: 10.1038/s41598-019-52913-z
23. Colak S, Ten Dijke P. Targeting TGF-beta Signaling in Cancer. Trends Cancer (2017) 3(1):56–71. doi: 10.1016/j.trecan.2016.11.008
24. Hsieh CL, Chen DS, Hwang LH. Tumor-induced immunosuppression: A barrier to immunotherapy of large tumors by cytokine-secreting tumor vaccine. Hum Gene Ther (2000) 11(5):681–92. doi: 10.1089/10430340050015581
25. Wu Z, Huang X, Han X, Li Z, Zhu Q, Yan J, et al. The chemokine CXCL9 expression is associated with better prognosis for colorectal carcinoma patients. BioMed Pharmacother (2016) 78:8–13. doi: 10.1016/j.biopha.2015.12.021
26. Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci (2009) 106(21):8623–8. doi: 10.1073/pnas.0809818106
27. Prato S, Zhan Y, Mintern JD, Villadangos JA. Rapid Deletion and Inactivation of CTLs upon Recognition of a Number of Target Cells over a Critical Threshold. J Immunol (2013) 191(7):3534–44. doi: 10.4049/jimmunol.1300803
28. Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight (2019) 4(6):e126908. doi: 10.1172/jci.insight.126908
29. Sampedro-Nunez M, Serrano-Somavilla A, Adrados M, Cameselle-Teijeiro JM, Blanco-Carrera C, Cabezas-Agricola JM, et al. Analysis of expression of the PD-1/PD-L1 immune checkpoint system and its prognostic impact in gastroenteropancreatic neuroendocrine tumors. Sci Rep (2018) 8(1):17812. doi: 10.1038/s41598-018-36129-1
30. Wang Y, Zhu C, Song W, Li J, Zhao G, Cao H. PD-L1 Expression and CD8(+) T Cell Infiltration Predict a Favorable Prognosis in Advanced Gastric Cancer. J Immunol Res (2018) 2018:4180517. doi: 10.1155/2018/4180517
31. Karimi S, Mansouri S, Mamatjan Y, Liu J, Nassiri F, Suppiah S, et al. Programmed death ligand-1 (PD-L1) expression in meningioma; prognostic significance and its association with hypoxia and NFKB2 expression. Sci Rep (2020) 10(1):14115. doi: 10.1038/s41598-020-70514-z
32. Chapon M, Randriamampita C, Maubec E, Badoual C, Fouquet S, Wang SF, et al. Progressive upregulation of PD-1 in primary and metastatic melanomas associated with blunted TCR signaling in infiltrating T lymphocytes. J Invest Dermatol (2011) 131(6):1300–7. doi: 10.1038/jid.2011.30
33. Slater NA, Googe PB. PD-L1 expression in cutaneous squamous cell carcinoma correlates with risk of metastasis. J Cutan Pathol (2016) 43(8):663–70. doi: 10.1111/cup.12728
34. Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med (2020) 217(8):e20190673. doi: 10.1084/jem.20190673
35. Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell (2020) 37(3):289–307.e9. doi: 10.1016/j.ccell.2020.02.008
36. Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat Commun (2018) 9(1):1250. doi: 10.1038/s41467-018-03600-6
37. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMra1706169
38. Martikainen M, Essand M. Virus-Based Immunotherapy of Glioblastoma. Cancers (2019) 11(2):186. doi: 10.3390/cancers11020186
39. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov (2018) 8(9):1069–86. doi: 10.1158/2159-8290.CD-18-0367
40. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
41. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
42. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi: 10.1056/NEJMoa1305133
43. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med (2019) 381(21):2020–31. doi: 10.1056/NEJMoa1910231
44. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378(22):2078–92. doi: 10.1056/NEJMoa1801005
45. Parikh PM, Vaid A, Advani SH, Digumarti R, Madhavan J, Nag S, et al. Randomized, double-blind, placebo-controlled phase II study of single-agent oral talactoferrin in patients with locally advanced or metastatic non-small-cell lung cancer that progressed after chemotherapy. J Clin Oncol (2011) 29(31):4129–36. doi: 10.1200/JCO.2010.34.4127
46. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi: 10.1158/0008-5472.CAN-13-1550
47. Joseph RW, Elassaiss-Schaap J, Kefford R, Hwu WJ, Wolchok JD, Joshua AM, et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res (2018) 24(20):4960–7. doi: 10.1158/1078-0432.CCR-17-2386
48. Lee JHJ, Lyle M, Menzies AM, Chan MMK, Lo S, Clements A, et al. Metastasis-specific patterns of response and progression with anti-PD-1 treatment in metastatic melanoma. Pigment Cell Melanoma Res (2018) 31(3):404–10. doi: 10.1111/pcmr.12675
49. Chardin D, Paquet M, Schiappa R, Darcourt J, Bailleux C, Poudenx M, et al. Baseline metabolic tumor volume as a strong predictive and prognostic biomarker in patients with non-small cell lung cancer treated with PD1 inhibitors: a prospective study. J Immunother Cancer (2020) 8(2):e000645. doi: 10.1136/jitc-2020-000645
50. Nishino M, Giobbie-Hurder A, Ramaiya NH, Hodi FS. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J Immunother Cancer (2014) 2(1):40. doi: 10.1186/s40425-014-0040-2
51. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature (2017) 545:60–5. doi: 10.1038/nature22079
52. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J Hematol Oncol (2018) 11(1):39. doi: 10.1186/s13045-018-0582-8
53. Triplett TA, Tucker CG, Triplett KC, Alderman Z, Sun L, Ling LE, et al. STAT3 signaling is required for optimal regression of large established tumors in mice treated with anti-OX40 and TGFβ Receptor Blockade. Cancer Immunol Res (2015) 3(5):526–35. doi: 10.1158/2326-6066.CIR-14-0187
54. Bartkowiak T, Curran MA. 4-1BB agonists: Multi-potent potentiators of tumor immunity. Front Oncol (2015) 5:117:117. doi: 10.3389/fonc.2015.00117
55. Kocak E, Lute K, Chang X, May KF Jr., Exten KR, Zhang H, et al. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res (2006) 66(14):7276–84. doi: 10.1158/0008-5472.CAN-05-2128
56. Vermaelen K. Vaccine strategies to improve anticancer cellular immune responses. Front Immunol (2019) 10:8:8. doi: 10.3389/fimmu.2019.00008
57. Binder DC, Engels B, Arina A, Yu P, Slauch JM, Fu YX, et al. Antigen-specific bacterial vaccine combined with anti-PD-L1 rescues dysfunctional endogenous T cells to reject long-established cancer. Cancer Immunol Res (2013) 1(2):123–33. doi: 10.1158/2326-6066.CIR-13-0058
58. Cuadros C, Dominguez AL, Lollini P-L, Croft M, Mittler RS, Borgström P, et al. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4-1BB monoclonal antibodies induces T cell-mediated protective immunity in Her-2/neu transgenic mice. Int J Cancer (2005) 116(6):934–43. doi: 10.1002/ijc.21098
59. D’Alise AM, Leoni G, Cotugno G, Troise F, Langone F, Fichera I, et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat Commun (2019) 10(1):2688. doi: 10.1038/s41467-019-10594-2
60. Rice AE, Latchman YE, Balint JP, Lee JH, Gabitzsch ES, Jones FR. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther (2015) 22(9):454–62. doi: 10.1038/cgt.2015.40
61. Kaufman H, Amatruda T, Nemunaitis JJ, Chesney JA, Delman KA, Spitler LE, et al. Tumor size and clinical outcomes in melanoma patients treated with talimogene laherparepvec (T-VEC). J Clin Oncol (2015) 33(15_suppl):9074. doi: 10.1200/jco.2015.33.15_suppl.9074
62. Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell (2018) 33(4):563–9. doi: 10.1016/j.ccell.2018.03.008
63. French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med (1999) 5(5):548–53. doi: 10.1038/8426
64. Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med (1999) 5(7):774–9. doi: 10.1038/10495
65. Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393(6684):480–3. doi: 10.1038/31002
66. Byrne KT, Vonderheide RH. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep (2016) 15(12):2719–32. doi: 10.1016/j.celrep.2016.05.058
67. Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Nat Acad Sci (2020) 117(14):8022–31. doi: 10.1073/pnas.1918971117
68. Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, et al. Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res (2018) 78(15):4282–91. doi: 10.1158/0008-5472.CAN-17-3821
69. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer ARL, Bajor DL, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res (2015) 3(4):399–411. doi: 10.1158/2326-6066.CIR-14-0215
70. O’Hara MH, O’Reilly EM, Mick R, Varadhachary G, Wainberg ZA, Ko A, et al. A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine and nab-paclitaxel with or without nivolumab in untreated metastatic ductal pancreatic adenocarcinoma patients. Cancer Res (2019) 79(13):CT004. doi: 10.1158/1538-7445.SABCS18-CT004
71. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi: 10.1158/1078-0432.CCR-11-0116
72. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin Cancer Res (2016) 22(15):3734–45. doi: 10.1158/1078-0432.CCR-15-1879
73. Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity (2000) 13(2):265–76. doi: 10.1016/S1074-7613(00)00026-1
74. Segal G, Prato S, Zehn D, Mintern JD, Villadangos JA. Target Density, Not Affinity or Avidity of Antigen Recognition, Determines Adoptive T Cell Therapy Outcomes in a Mouse Lymphoma Model. J Immunol (2016) 196(9):3935–42. doi: 10.4049/jimmunol.1502187
75. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):449–59. doi: 10.1056/NEJMoa1709919
76. Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined Antitumor Effects of Sorafenib and GPC3-CAR T Cells in Mouse Models of Hepatocellular Carcinoma. Mol Ther (2019) 27(8):1483–94. doi: 10.1016/j.ymthe.2019.04.020
77. Kato D, Yaguchi T, Iwata T, Katoh Y, Morii K, Tsubota K, et al. GPC1 specific CAR-T cells eradicate established solid tumor without adverse effects and synergize with anti-PD-1 Ab. Elife (2020) 9:e49392. doi: 10.7554/eLife.49392
78. Shimizu T, Fuchimoto Y, Okita H, Fukuda K, Kitagawa Y, Ueno S, et al. A curative treatment strategy using tumor debulking surgery combined with immune checkpoint inhibitors for advanced pediatric solid tumors: An in vivo study using a murine model of osteosarcoma. J Pediatr Surg (2018) 53(12):2460–4. doi: 10.1016/j.jpedsurg.2018.08.023
79. Demicheli R, Retsky MW, Hrushesky WJ, Baum M. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat Clin Pract Oncol (2007) 4(12):699–710. doi: 10.1038/ncponc0999
80. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell (2013) 155(4):750–64. doi: 10.1016/j.cell.2013.10.029
81. Bakos O, Lawson C, Rouleau S, Tai LH. Combining surgery and immunotherapy: turning an immunosuppressive effect into a therapeutic opportunity. J Immunother Cancer (2018) 6(1):86. doi: 10.1186/s40425-018-0398-7
82. Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med (2018) 10(436):eaan3464. doi: 10.1126/scitranslmed.aan3464
83. Flanigan RC, Salmon SE, Blumenstein BA, Bearman SI, Roy V, McGrath PC, et al. Nephrectomy Followed by Interferon Alfa-2b Compared with Interferon Alfa-2b Alone for Metastatic Renal-Cell Cancer. N Engl J Med (2001) 345(23):1655–9. doi: 10.1056/NEJMoa003013
84. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17(2):97–111. doi: 10.1038/nri.2016.107
85. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
86. Mackall CL. T-cell immunodeficiency following cytotoxic antineoplastic therapy: a review. Oncologist (1999) 4(5):370–8. doi: 10.1634/theoncologist.4-5-370
87. McCoy MJ, Lake RA, van der Most RG, Dick IM, Nowak AK. Post-chemotherapy T-cell recovery is a marker of improved survival in patients with advanced thoracic malignancies. Br J Cancer (2012) 107(7):1107–15. doi: 10.1038/bjc.2012.362
88. Iida Y, Harashima N, Motoshima T, Komohara Y, Eto M, Harada M. Contrasting effects of cyclophosphamide on anti-CTL-associated protein 4 blockade therapy in two mouse tumor models. Cancer Sci (2017) 108(10):1974–84. doi: 10.1111/cas.13337
89. Byrne KT, Leisenring NH, Bajor DL, Vonderheide RH. CSF-1R-Dependent Lethal Hepatotoxicity When Agonistic CD40 Antibody Is Given before but Not after Chemotherapy. J Immunol (2016) 197(1):179–87. doi: 10.4049/jimmunol.1600146
90. Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFN and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov (2016) 6(4):400–13. doi: 10.1158/2159-8290.CD-15-1032
91. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med (2006) 203(5):1259–71. doi: 10.1084/jem.20052494
92. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520(7547):373–7. doi: 10.1038/nature14292
93. Maity A, Mick R, Huang AC, George SM, Farwell MD, Lukens JN, et al. A phase I trial of pembrolizumab with hypofractionated radiotherapy in patients with metastatic solid tumours. Br J Cancer (2018) 119(10):1200–7. doi: 10.1038/s41416-018-0281-9
94. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med (2018) 24(12):1845–51. doi: 10.1038/s41591-018-0232-2
95. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83–92. doi: 10.2147/HP.S93413
96. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis (2018) 7(1):10. doi: 10.1038/s41389-017-0011-9
97. Lee CM, Tannock IF. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer (2010) 10:255. doi: 10.1186/1471-2407-10-255
98. Oberstein PE, Olive KP. Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol (2013) 6(4):321–37. doi: 10.1177/1756283X13478680
99. Lo A, Li CP, Buza EL, Blomberg R, Govindaraju P, Avery D, et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight (2017) 2(19):e92232. doi: 10.1172/jci.insight.92232
100. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324(5933):1457–61. doi: 10.1126/science.1171362
101. Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer (2013) 109(4):926–33. doi: 10.1038/bjc.2013.415
102. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol (2011) 29(34):4548–54. doi: 10.1200/JCO.2011.36.5742
103. Neesse A, Frese KK, Chan DS, Bapiro TE, Howat WJ, Richards FM, et al. SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut (2014) 63(6):974–83. doi: 10.1136/gutjnl-2013-305559
104. Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Nat Acad Sci (2015) 112(47):e6506–e14. doi: 10.1073/pnas.1519623112
105. Kramer ED, Abrams SI. Granulocytic Myeloid-Derived Suppressor Cells as Negative Regulators of Anticancer Immunity. Front Immunol (2020) 11:1963:1963. doi: 10.3389/fimmu.2020.01963
106. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
107. Chao T, Furth EE, Vonderheide RH. CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol Res (2016) 4(11):968–82. doi: 10.1158/2326-6066.CIR-16-0188
108. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell (2012) 21(6):822–35. doi: 10.1016/j.ccr.2012.04.025
109. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
110. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
111. Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Trans Med (2019) 11(499):eaau9240. doi: 10.1126/scitranslmed.aau9240
112. Wu L, Mao L, Liu JF, Chen L, Yu GT, Yang LL, et al. Blockade of TIGIT/CD155 signaling reverses t-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res (2019) 7(10):1700–13. doi: 10.1158/2326-6066.CIR-18-0725
113. Roh M, Wainwright DA, Wu JD, Wan Y, Zhang B. Targeting CD73 to augment cancer immunotherapy. Curr Opin Pharmacol (2020) 53:66–76. doi: 10.1016/j.coph.2020.07.001
114. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med (2018) 10(436):eaan3311. doi: 10.1126/scitranslmed.aan3311
115. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
116. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy CB, et al. Targeting tumor-associated macrophages and granulocytic-myeloid-derived suppressor cells augments pd-1 blockade in cholangiocarcinoma. J Clin Invest (2020) 130(10):5380–96. doi: 10.1172/JCI137110
117. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
118. Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. Eur J Immunol (2019) 49(8):1140–6. doi: 10.1002/eji.201847659
119. Cote AL, Byrne KT, Steinberg SM, Zhang P, Turk MJ. Protective CD8 memory T cell responses to mouse melanoma are generated in the absence of CD4 T cell help. PloS One (2011) 6(10):e26491. doi: 10.1371/journal.pone.0026491
120. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature (2019) 574(7780):696–701. doi: 10.1038/s41586-019-1671-8
121. Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature (2020) 584(7822):624–9. doi: 10.1038/s41586-020-2611-3
122. Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4(+) T Cells. Immunity (2020) 52(1):151–66 e6. doi: 10.1016/j.immuni.2019.12.007
123. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
124. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer Res (2019) 25(4):1233–8. doi: 10.1158/1078-0432.CCR-18-0762
125. Mahne AE, Mauze S, Joyce-Shaikh B, Xia J, Bowman EP, Beebe AM, et al. Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res (2017) 77(5):1108–18. doi: 10.1158/0008-5472.CAN-16-0797
127. Urban-Wojciuk Z, Khan MM, Oyler BL, Fåhraeus R, Marek-Trzonkowska N, Nita-Lazar A, et al. The role of tlrs in anti-cancer immunity and tumor rejection. Front Immunol (2019) 10:2388:2388. doi: 10.3389/fimmu.2019.02388
128. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat BioMed Engl (2018) 2(8):578–88. doi: 10.1038/s41551-018-0236-8
129. Mullins SR, Vasilakos JP, Deschler K, Grigsby I, Gillis P, John J, et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer (2019) 7(1):244. doi: 10.1186/s40425-019-0724-8
130. Nyirenda MH, Sanvito L, Darlington PJ, O’Brien K, Zhang G-X, Constantinescu CS, et al. TLR2 Stimulation Drives Human Naive and Effector Regulatory T Cells into a Th17-Like Phenotype with Reduced Suppressive Function. J Immunol (2011) 187(5):2278–90. doi: 10.4049/jimmunol.1003715
131. Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol (2013) 93(6):847–63. doi: 10.1189/jlb.1012501
132. Grimmig T, Matthes N, Hoeland K, Tripathi S, Chandraker A, Grimm M, et al. TLR7 and TLR8 expression increases tumor cell proliferation and promotes chemoresistance in human pancreatic cancer. Int J Oncol (2015) 47(3):857–66. doi: 10.3892/ijo.2015.3069
133. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol (2007) 44(11):2850–9. doi: 10.1016/j.molimm.2007.01.022
134. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339(6121):786–91. doi: 10.1126/science.1232458
135. Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov (2020) 10(1):26–39. doi: 10.1158/2159-8290.CD-19-0761
136. Yum S, Li M, Frankel AE, Chen ZJ. Roles of the cGAS-STING pathway in cancer immunosurveillance and immunotherapy. Ann Rev Cancer Biol (2019) 3(1):323–44. doi: 10.1146/annurev-cancerbio-030518-055636
137. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41(5):830–42. doi: 10.1016/j.immuni.2014.10.017
138. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208(10):1989–2003. doi: 10.1084/jem.20101158
139. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Nat Acad Sci (2015) 112(50):15408–13. doi: 10.1073/pnas.1512832112
140. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Nat Acad Sci (2017) 114(7):1637–42. doi: 10.1073/pnas.1621363114
141. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest (2019) 129(10):4350–64. doi: 10.1172/JCI125413
142. Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep (2016) 6:19049. doi: 10.1038/srep19049
143. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (2011) 331(6024):1612–6. doi: 10.1126/science.1198443
144. Zhang Q, Liu F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis (2020) 11(6):485. doi: 10.1038/s41419-020-2696-5
145. Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med (2018) 379(2):150–61. doi: 10.1056/NEJMoa1716435
146. de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev (2018) 41:28–39. doi: 10.1016/j.cytogfr.2018.03.006
147. Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther (2003) 10(4):292–303. doi: 10.1038/sj.gt.3301885
148. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell (2017) 170(6):1109–19 e10. doi: 10.1016/j.cell.2017.08.027
149. Kohlhapp FJ, Kaufman HL. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin Cancer Res (2016) 22(5):1048–54. doi: 10.1158/1078-0432.CCR-15-2667
150. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol (2015) 33(25):2780–8. doi: 10.1200/JCO.2014.58.3377
151. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol (2016) 34(22):2619–26. doi: 10.1200/JCO.2016.67.1529
152. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J Clin Oncol (2018) 36(17):1658–67. doi: 10.1200/JCO.2017.73.7379
153. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
154. Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. RNAi-Mediated beta-Catenin Inhibition Promotes T Cell Infiltration and Antitumor Activity in Combination with Immune Checkpoint Blockade. Mol Ther (2018) 26(11):2567–79. doi: 10.1016/j.ymthe.2018.09.005
155. Qu Y, Olsen JR, Yuan X, Cheng PF, Levesque MP, Brokstad KA, et al. Small molecule promotes beta-catenin citrullination and inhibits Wnt signaling in cancer. Nat Chem Biol (2018) 14(1):94–101. doi: 10.1038/nchembio.2510
156. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature (2010) 468(7327):1067–73. doi: 10.1038/nature09504
157. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep (2016) 16(11):2829–37. doi: 10.1016/j.celrep.2016.08.032
158. Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest (2016) 126(9):3479–94. doi: 10.1172/JCI86437
159. Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S, et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol Res (2018) 6(10):1234–45. doi: 10.1158/2326-6066.CIR-18-0077
160. Wang L, Xu M, Kao CY, Tsai SY, Tsai MJ. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J Clin Invest (2020) 130(4):1782–92. doi: 10.1172/JCI126327
161. Yaeger R, Corcoran RB. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov (2019) 9(3):329–41. doi: 10.1158/2159-8290.CD-18-1321
162. Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity (2017) 47(6):1083–99. doi: 10.1016/j.immuni.2017.11.016
163. Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol (2018) 15(11):709–20. doi: 10.1038/s41571-018-0105-0
164. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375(23):2255–62. doi: 10.1056/NEJMoa1609279
165. Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol Res (2016) 4(3):204–14. doi: 10.1158/2326-6066.CIR-15-0188
166. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
167. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
168. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364(26):2507–16. doi: 10.1056/NEJMoa1103782
169. Reddy SM, Reuben A, Wargo JA. Influences of BRAF Inhibitors on the Immune Microenvironment and the Rationale for Combined Molecular and Immune Targeted Therapy. Curr Oncol Rep (2016) 18(7):42. doi: 10.1007/s11912-016-0531-z
170. Steinberg SM, Zhang P, Malik BT, Boni A, Shabaneh TB, Byrne KT, et al. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer Immunol Res (2014) 2(11):1044–50. doi: 10.1158/2326-6066.CIR-14-0074
171. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res (2010) 70(13):5213–9. doi: 10.1158/0008-5472.CAN-10-0118
172. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res (2012) 18(5):1386–94. doi: 10.1158/1078-0432.CCR-11-2479
173. Kuske M, Westphal D, Wehner R, Schmitz M, Beissert S, Praetorius C, et al. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for Melanoma therapy. Pharmacol Res (2018) 136:151–9. doi: 10.1016/j.phrs.2018.08.019
174. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
175. Yarchoan M, Mohan AA, Dennison L, Vithayathil T, Ruggieri A, Lesinski GB, et al. MEK inhibition suppresses B regulatory cells and augments anti-tumor immunity. PloS One (2019) 14(10):e0224600. doi: 10.1371/journal.pone.0224600
176. Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology (2016) 5(12):e1238557. doi: 10.1080/2162402X.2016.1238557
177. Eroglu Z, Ribas A. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther Adv Med Oncol (2016) 8(1):48–56. doi: 10.1177/1758834015616934
178. Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med (2015) 7(279):279ra41. doi: 10.1126/scitranslmed.aaa4691
179. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med (2019) 25(6):936–40. doi: 10.1038/s41591-019-0476-5
180. Suraweera A, O’Byrne KJ, Richard DJ. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front Oncol (2018) 8:92:92. doi: 10.3389/fonc.2018.00092
181. Shen L, Orillion A, Pili R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics (2016) 8(3):415–28. doi: 10.2217/epi.15.118
182. Chang S, Collins PL, Aune TM. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol (2008) 181(12):8372–81. doi: 10.4049/jimmunol.181.12.8372
183. Lisiero DN, Soto H, Everson RG, Liau LM, Prins RM. The histone deacetylase inhibitor, LBH589, promotes the systemic cytokine and effector responses of adoptively transferred CD8+ T cells. J Immunother Cancer (2014) 2:8. doi: 10.1186/2051-1426-2-8
184. Moreira JM, Scheipers P, Sorensen P. The histone deacetylase inhibitor Trichostatin A modulates CD4+ T cell responses. BMC Cancer (2003) 3:30. doi: 10.1186/1471-2407-3-30
185. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PloS One (2012) 7(1):e30815. doi: 10.1371/journal.pone.003081
186. Mulcahy Levy JM, Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ (2020) 27(3):843–57. doi: 10.1038/s41418-019-0474-7
187. Briceno E, Reyes S, Sotelo J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus (2003) 14(2):e3. doi: 10.3171/foc.2003.14.2.4
188. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581(7806):100–5. doi: 10.1038/s41586-020-2229-5
189. DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy Regulation of Metabolism Is Required for CD8(+) T Cell Anti-tumor Immunity. Cell Rep (2019) 27(2):502–13 e5. doi: 10.1016/j.celrep.2019.03.037
Keywords: tumor burden, immunotherapy, immune infiltrate, tumor microenvionment, immunosuppression
Citation: Kim SI, Cassella CR and Byrne KT (2021) Tumor Burden and Immunotherapy: Impact on Immune Infiltration and Therapeutic Outcomes. Front. Immunol. 11:629722. doi: 10.3389/fimmu.2020.629722
Received: 15 November 2020; Accepted: 18 December 2020;
Published: 01 February 2021.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Limin Zheng, Sun Yat-Sen University, ChinaZong Sheng Guo, University of Pittsburgh, United States
Copyright © 2021 Kim, Cassella and Byrne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katelyn T. Byrne, byrnek@upenn.edu