 Songlin Li1,2,3
Songlin Li1,2,3 Eric Y. Hayden4
Eric Y. Hayden4 Veronica J. Garcia5,6
Veronica J. Garcia5,6 Dieu-Trang Fuchs3Julia Sheyn3David A. Daley3Altan Rentsendorj3
Dieu-Trang Fuchs3Julia Sheyn3David A. Daley3Altan Rentsendorj3 Tania Torbati7
Tania Torbati7 Keith L. Black3
Keith L. Black3 Ueli Rutishauser3,6,8
Ueli Rutishauser3,6,8 David B. Teplow4Yosef Koronyo3
David B. Teplow4Yosef Koronyo3 Maya Koronyo-Hamaoui3,6*
Maya Koronyo-Hamaoui3,6*- 1Institute of Neuroscience and Chemistry, Wenzhou University, Wenzhou, China
- 2Institute of Life Sciences, Wenzhou University, Wenzhou, China
- 3Department of Neurosurgery, Cedars-Sinai Medical Center, Maxine-Dunitz Neurosurgical Institute, Los Angeles, CA, United States
- 4Department of Neurology, David Geffen School of Medicine at UCLA, Mary S. Easton Center for Alzheimer's Disease Research at UCLA, Brain Research Institute, Molecular Biology Institute, University of California, Los Angeles, Los Angeles, CA, United States
- 5Board of Governors Regenerative Medicine Institute, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 6Department of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 7College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA, United States
- 8Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, CA, United States
Impaired synaptic integrity and function due to accumulation of amyloid β-protein (Aβ42) oligomers is thought to be a major contributor to cognitive decline in Alzheimer's disease (AD). However, the exact role of Aβ42 oligomers in synaptotoxicity and the ability of peripheral innate immune cells to rescue synapses remain poorly understood due to the metastable nature of oligomers. Here, we utilized photo-induced cross-linking to stabilize pure oligomers and study their effects vs. fibrils on synapses and protection by Aβ-phagocytic macrophages. We found that cortical neurons were more susceptible to Aβ42 oligomers than fibrils, triggering additional neuritic arborization retraction, functional alterations (hyperactivity and spike waveform), and loss of VGluT1- and PSD95-excitatory synapses. Co-culturing neurons with bone marrow-derived macrophages protected synapses against Aβ42 fibrils; moreover, immune activation with glatiramer acetate (GA) conferred further protection against oligomers. Mechanisms involved increased Aβ42 removal by macrophages, amplified by GA stimulation: fibrils were largely cleared through intracellular CD36/EEA1+-early endosomal proteolysis, while oligomers were primarily removed via extracellular/MMP-9 enzymatic degradation. In vivo studies in GA-immunized or CD115+-monocyte-grafted APPSWE/PS1ΔE9-transgenic mice followed by pre- and postsynaptic analyses of entorhinal cortex and hippocampal substructures corroborated our in vitro findings of macrophage-mediated synaptic preservation. Together, our data demonstrate that activated macrophages effectively clear Aβ42 oligomers and rescue VGluT1/PSD95 synapses, providing rationale for harnessing macrophages to treat AD.
Introduction
Alzheimer's disease (AD) is a progressive, incurable and fatal neurodegenerative disorder and the most frequent cause of senile dementia (1, 2). AD is characterized by severe synaptic and neuronal loss resulting in gradual behavioral and cognitive decline (3, 4). Early pathological changes in AD involve accumulation of amyloid β-protein (Aβ) in the brain and retina (5–11), with the amyloidogenic 42-residue long alloform (Aβ42) viewed as the most pathognomonic (5, 12). These Aβ42 peptides have been shown to rapidly aggregate into insoluble fibrils that form the plaques in AD brains, but also to assemble into non-fibrillar soluble oligomers, believed to be highly neurotoxic (8, 13–20). While there is a weak correlation between cerebral plaque density and severity of AD, Aβ42 oligomers appear to closely associate with synaptopathy and cognitive impairment (21, 22). Specifically, growing evidence demonstrates the detrimental effects of Aβ42 oligomers on synaptic plasticity, spine morphology and density (13, 23), axonal transport (24–26), and cognition (8, 15, 17, 22, 27–29). These findings incentivize strategies that eradicate Aβ42 oligomers in order to preserve synapses and cognitive function (30–34).
Multiple mechanisms mediate cerebral Aβ clearance, including phagocytosis and enzymatic degradation by innate immune cells such as microglia and bone marrow (BM)-derived monocytes (Mo) and macrophages (MΦ) (35, 36). However, under a chronic neuroinflammatory condition typical for AD, microglia exhibit a compromised phenotype, with diminished ability to clear Aβ (37, 38), further contributing to damaging inflammatory processes, and excessively pruning synapses (39–46). Nevertheless, unlike brain-resident microglia, BM-derived Mo/MΦ appear to have an increased capacity to remove Aβ fibrils, as shown by studies in both cell cultures and animal models (47–64). Indeed, various pre-clinical studies from our group and others have supported an emerging paradigm shift for the therapeutic potential of immunomodulation via enhanced cerebral recruitment of BM-derived Aβ-clearing macrophages (9, 35, 50, 52, 58, 62–70). In particular, we demonstrated that successful delivery of therapeutic Mo/MΦ to the brains of APPSWE/PS1dE9 double-transgenic (ADtg) mice can be achieved via immunization with altered myelin-derived antigens (e.g., glatiramer acetate – GA; also known as Copaxone®) or peripheral-blood enrichment with BM-derived CD115+-monocytes (MoBM) (50, 52, 58, 63, 70). In the immunized mice, cerebral MΦ were found to be highly-phagocytic, pro-healing, and anti-inflammatory, profoundly alleviating cerebral Aβ40 and Aβ42 burden, reducing microgliosis and astrocytosis, and ultimately improving cognitive functions. Further, GA induced macrophage-mediated recognition and phagocytosis of fibrillar Aβ42 (63, 70). However, the potential of clearing the non-fibrillar, soluble Aβ42 oligomeric forms by macrophages requires further investigation.
Experimental and clinical studies have indicated that the cognitive decline seen in AD patients and animal models is related to early pathological processes occurring in entorhinal cortex layers 2 and 3 (71–75). These layers play a crucial role in connecting brain cortical regions to the hippocampus. Our immunomodulation approach of recruiting Mo/MΦ to brain sites of Aβ deposits substantially reduced AD-related pathology within these regions (63, 70). Importantly, selective ablation of peripheral monocytes, either by diphtheria toxin-targeted depletion or Ccr2-mediated inhibition of cerebral monocyte infiltration, exacerbated cortical and hippocampal Aβ pathology in ADtg mice (52, 53). In addition, our preliminary data suggested a presynaptic preservation concomitant with Aβ-plaque reduction in hippocampi of these murine models (63). Yet, the underinvestigate role of peripheral Mo/MΦ in preservation of synaptic and neurite integrity, especially postsynaptic terminals within AD-relevant cortical regions, calls for broader exploration due to promising clinical implications of immunomodulation intervention strategies for AD.
Here, we applied photo-induced cross-linking of unmodified proteins (PICUP) to assess the impact of intrinsically metastable and polydisperse Aβ42 oligomers on neurons and to test the ability of macrophages to recognize, bind to, and remove them (76, 77). This technique allowed us to produce pure, stabilized assemblies of low-n Aβ42 oligomers (XL-oAβ42). The precise detrimental impact of these oligomeric conformers, as compared with preformed fibrils (fAβ42), was determined on synaptic density, neuritic arborization length, and neuronal function in primary (P1) cortical neurons. Furthermore, synaptic and neurite protection against Aβ42 oligomers was explored in co-cultures of cortical neurons accompanied with BM-derived MΦ or GA-activated MΦ. Next, the innate immune mechanisms of eliminating pathogenic Aβ42 assemblies and subsequently attaining synaptic rescue were studied. Finally, in ADtg mice, we determined synaptic loss as well as preservation of pre- and postsynaptic terminals following macrophage-mediated immunomodulation interventions in predefined hippocampal and cortical subregions.
Materials and Methods
Mice
Alzheimer's disease double-transgenic (ADtg) B6.Cg-Tg (APPswe, PSEN1ΔE9) 85Dbo/J mice and their age-matched wildtype (WT) C57BL/6J littermates were purchased from Jackson Laboratories (MMRRC stock 34832-JAX) and then bred and maintained at Cedars-Sinai Medical Center. All animals in this study have a C57BL/6 congenic background. Two cohorts of 10-month-old males, WT and ADtg (n = 6 per experimental group), were used. The first cohort underwent weekly s.c. GA immunization or monthly i.v. CD115+-MoBM injections for a duration of 2 months. Control groups were comprised of either naïve WT mice or monthly i.v. PBS injected ADtg mice. At the completion of the experiment, mice underwent behavioral testing at 12 months of age, then were perfused, under deep anesthesia, with ice-cold 0.9% saline solution containing 0.5 mM EDTA, and brains were collected for analyses. Harvested brains were cut in half. One hemisphere was post-fixed overnight in phosphate-buffered saline (PBS) containing 2.5% paraformaldehyde. Tissue was cryoprotected and stored in PBS containing 30% sucrose and 0.1% sodium azide. Brains were cut coronally in 30 μm serial sections for histology. The second hemisphere was further snap-frozen for biochemical and molecular assays as described previously (63). The second cohort underwent perfusion as described above for tissue collection.
Culture and Co-culture of Primary Cortical Neurons and Macrophages
Cortical neuronal cultures were prepared from postnatal day 1 C57BL/6 mice (78, 79). Neonates were decapitated, and their brains were removed. The cerebral cortex was dissected, stripped of meninges, dissociated with a combination of calcium and magnesium free Hank's balanced salt solution (HBSS; Life Technologies) containing 0.2% w/v papain suspension (Worthington Biochemical), and digested for 12 min at 37°C. The triturated cells were passed through a 70 μm strainer and counted. The cells were plated in laminin and poly-D-lysine-coated coverslips (BD Biosciences) at a density of 8 × 104 cells per ml (in 24-well plates) in NbActiv4 (BrainBits), then supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin. The purity of primary neurons was about 92–94% (78). Bone marrow-derived macrophages were obtained as described previously (63). Briefly, bone marrow cells were isolated from the femurs and tibiae of 8- to 16-week-old C57BL/6 mice, differentiated into macrophages by incubation in complete RPMI-1640 (Life Technologies), and supplemented with 10% fetal bovine serum (Life Technologies) and 20 ng/ml macrophage colony stimulating factor (M-CSF; PeproTech) for 5 days. Primary cultures of macrophages were then suspended in NbActiv4 and co-cultured with primary cortical neurons (ratio of 1:1) per well in 24-well tissue-culture plates for 2 days. Primary neurons and/or macrophages were incubated overnight with 100nM of scrambled (s), fibrillar (f), or oligomeric (o) Aβ42 (see below “Preparation of Aβ1−42 oligomers, fibrils and Scrambled Aβ42”). Additional groups included those with macrophages treated with glatiramer acetate (GA; 30 μg/ml, TEVA Neuroscience) for 24 h. Experiments were carried out at day 9 of seeding cortical neurons starting with synapse formation and maturation (78). Cultures were washed in PBS and then fixed in 4% formaldehyde for 20 min at room temperature for further experimental procedures.
Microelectrode Array Recording
Cortical neuronal cultures (as described above) were plated in a M768-GLx 12-, 24-, or 48-well plate (Axion Biosystems). Preformed Aβ42 (100 nM) were added to the culture on day 9. Simultaneous recordings from 64 extracellular electrodes per well were made using the Maestro (Axion BioSystems) microelectrode array (MEA) system at a constant temperature of 37°C. Data were sampled at 12.5 kHz, digitized and analyzed using Axion Integrated Studio software (Axion BioSystems) where spike events were detected using an adaptive spike detection threshold of 5.5 SD for each electrode with 1 s binning. Detected waveforms were then further sorted usingOffline Sorter v4 software (Plexon) and exported through NeuroExplorer (Nex Technologies) into MatLab for waveform analysis using custom code. All data is presented as means ± s.e.m. and include recordings from 5 independent experiments.
Amyloid-β42 Determination by Sandwich ELISA
At day 7, macrophages were incubated for 30 min or 24 h with fibrillar (f) or oligomeric (o) Aβ42 and thereafter culture medium was collected. Total protein levels in the supernatant were determined using the Pierce BCA Protein Assay Kit (#23227; Thermo Scientific). The Aβ42 levels in the supernatant were assessed using a sandwich ELISA for an anti-human Aβ42 end-specific kit (KHB3442, Invitrogen; which does not recognize mouse Aβ nor human Aβ40/Aβ43). The kit was used according to the manufacturer's instructions. This assay is based on a combination of two antibodies specific for the N- and the COOH-termini of Aβ42 sequences. The bound rabbit anti-COOH-terminus was detected with a horseradish peroxidase-labeled anti-rabbit antibody and was read at 450 nm using a microplate reader (Spectra Max 384 plus, Molecular Devices). Concentrations detected at 24 h incubation were normalized to basal levels at 30 min incubation.
Immunocytohistochemistry and Quantification of Synapses and Neurites
Primary neurons/macrophages in coverslips and brain coronal cryosections at bregma −2.50, −2.65, and −2.80 mm (80) per animal were washed in PBS and then treated with a permeabilization/blocking solution containing 20% normal horse serum (Invitrogen) and 0.05% Triton X-100 (Sigma-Aldrich). Cells or tissue sections were stained overnight at 4°C with combinations of presynaptic marker VGluT1 (Chemicon, 1:6,000), postsynaptic marker PSD95 (Abcam, 1:600; for synapses), or neuron-specific class III beta-tubulin/Tuj1 (Abcam) or Tubb3 (BioLegend; for neurites; Supplementary Table 1) in 2% blocking solution in PBS. Secondary antibodies of Cy3 donkey anti-guinea pig or anti-mouse or Cy5 donkey anti-rabbit (Supplementary Table 1) were incubated for 1 h at room temperature. The samples were washed in PBS and mounted using ProLong® Gold with DAPI (Life Technologies). All groups were immunostained using identical procedures. Negative controls were processed using the same protocol with the omission of the primary or secondary antibodies to assess nonspecific labeling. There was no specific staining with these negative controls. Microphotographs were shot using a Carl Zeiss Axio Imager Z1 fluorescence microscope equipped with ApoTome (AxioVision 4.6.3, Carl Zeiss). The parameters for scanning were consistent across all groups.
Synaptic and neuritic quantification of primary cultures in vitro was carried out from 16 images, each coverslipped at a 40 × objective lens. At least 2 coverslips, 32 images, and 150 neurons for each condition were analyzed. For synaptic analysis in vivo and to cover the hippocampal area, 3 of the same rectangular fields (90 × 70 μm) under 100 × oil objective lens were precisely selected in the lateral and medial blade molecular layer (ML) of the dentate gyrus (DG), the stratum lacunosum-moleculare (SLM), the stratum radium (SR) and the stratum oriens (SO) of cornu ammonis 1 (CA1) in each condition, respectively. In addition, 2 of the same fields were carefully chosen in layers 2 and 3 of the entorhinal cortex (ENT). Fifteen optical sections per field, 15 fields per hippocampal area, 4 fields per entorhinal cortex per section, and 855 total images per brain were analyzed. Single optical section images at 0.25 μm intervals and 3.75 μm Zeiss ApoTome high-resolution scans were performed. Synaptic puncta number and synaptic immunoreactive (IR) area were quantified using Puncta Analyzer (81, 82) and ImageJ (NIH) macro and batch process. Total neurite length was measured using the NeuriteTracer program (83). Briefly, the cultures were immunostained with Tuj1 for neurite and NeuN for the neuronal nucleus. For each condition, at least 150 primary neurons, 32 images in random fields from 2 coverslips in 2 independent experiments were analyzed. The NeuriteTracer was utilized to detect the neurites strongly stained for Tuj1. Following optimization of parameters to separate neurites from the neuronal cell body and tracing the neurite through skeletonization, positively labeled neurites and respective lengths were quantified (Figure 3B). The observer was blind to the treatment conditions. Average puncta number, synaptic area, and percentage of the area per image or per neuron were calculated for each condition.
Primary Cultures of Bone Marrow-Derived Macrophages and the Phagocytosis Assay
To test amyloid-β phagocytosis by macrophages, in repeated experiments monocytes were isolated from the bone marrow of wild-type mice (n = 18 mice) and differentiated into macrophages by 7-day cultivation in complete RPMI-1640 medium (#21870; Life Technologies) with 10% serum and 20 ng/ml MCSF (#315-02; PeproTech). Macrophage primary cultures were then plated at 1.2 × 105 cells per well (3–4 wells for each condition) in 24-well tissue-culture plates on glass coverslips overnight. Next, macrophages were either treated with 30 μg/ml GA (Copaxone® TEVA Neuroscience) for the duration of 1, 3, or 24 h, or not treated (control group). Before addition of f/o/sAβ42 or vehicle, the cells were chilled in a 4°C ice bath for 5 min; immediately after addition of the preformed Aβ42 (100 nM), the plates were centrifuged at 515g in 25°C followed by incubation at 37°C for 30 or 60 min. The cells were then rinsed with Aβ-free medium to remove non-incorporated Aβ and later washed twice with PBS. Methanol (99.8%) at −20°C for 20 min or 4% paraformaldehyde at room temperature for 12 min were used for fixation of the cells followed by repeated washes with PBS. For immunostaining, the cells were first stained using the mouse anti-human amyloid-β mAb clone 6E10 (1:100; SIG-39320; Covance), rat anti-CD36 mAb clone MF3 (1:200; ab80080; Abcam), rat anti-CD204 scavenger receptor type I/II (SCARA1) mAb (1:100; MCA1322; AbD Serotec), rabbit anti-EEA1 pAb (1:100; Millipore #07-1820) and goat anti-MMP-9 pAb (1:100; AF909; R&D systems; Supplementary Table 1). Secondary polyclonal antibodies included donkey anti-mouse, anti-rat, anti-rabbit, and anti-goat conjugated with Cy2, Cy3, or Cy5 (1:200; Jackson ImmunoResearch Laboratories). The cells were mounted using ProLong® Gold with DAPI (Molecular Probes, Life Technologies; Supplementary Table 1). Several fields (minimum n = 5 randomly selected per group) were obtained from each well using a Carl Zeiss Axio Imager Z1 ApoTome-equipped microscope (an average of 120 cells in each field). Images were obtained using the same exposure time on each occasion. The fluorescent signal and its total area were determined and quantified by the conversion of individual images to greyscale and standardizing to baseline using histogram-based thresholds with NIH ImageJ software. The “mean area per cell” was a result of a numerical average of the individual cell's immunoreactive area per field. The “area/cell” measures the total fluorescent signal (area) divided by the total number of cells (DAPI count) of the same field (image). For all experiments, the investigators were blinded to the treatment condition. Colocalization of EEA1-6E10 (puncta analysis) was performed as synaptic puncta number analysis described above (81, 82).
Preparation of Aβ1−42 Oligomers, Fibrils, and Scrambled Aβ42
Filtration was used to prepare LMW Aβ42 as described (84). Microcon YM-30 filters (EMD Millipore) were washed in 200 μl of distilled deionized water. Aβ was dissolved in 10% (v/v) 60 mM NaOH and 90% (v/v) 10 mM phosphate buffer, pH 7.4, at a concentration of 1 mg/ml. The solution was sonicated for 1 min and then placed into the washed filter. Following centrifugation at 14,000 × g for 20 min, the filtrate, LMW Aβ42, was collected. This LMW Aβ was stabilized using photo-induced cross-linking of unmodified proteins (PICUP) (76, 85–87) to yield a distribution of “oligomeric Aβ.” Briefly, 2 mM Tris(2,2′-bipyridyl)di- chlororuthenium(II) hexahydrate (Ru(Bpy); Aldrich) and 40 mM ammonium persulfate (APS; Sigma) were prepared in distilled deionized water. An 18 μl aliquot of 80 μM LMW Aβ42 was placed in a PCR tube, followed by 1 μl of Ru(Bpy) and 1 μl of APS. The sample was irradiated (150 W incandescent lamp) for 1 s and the reaction was quenched immediately with 1 M dithiothrietol. Cross-linking reagents were removed by dialysis using 3.5 kDa MWCO Slide-A-Lyzer cassettes (Pierce) against 10 mM sodium phosphate pH 7.4. More than 5 changes of buffer were completed, and protein purity and concentration were confirmed with optical absorption spectroscopy and SDS-PAGE.
For preparation of fibrillar Aβ42, LMW Aβ42 was prepared as described above, and the peptide preparation was incubated at 100 μM with orbital shaking at 37°C until fibril formation occurred, approximately 2 weeks. The presence of fibrils was confirmed by electron microscopy. We used a previously designed “scrambled” Aβ42 peptide that: (1) had an amino acid composition identical to that of wild type Aβ42, and (2) did not display amphipathicity (as does the wild type peptide). We accomplished this by using word scrambler software (for example: https://www.wordunscrambler.net/word-scrambler.aspx) to randomly permute the Aβ amino acid sequence. We then used Kyte-Doolittle analysis (88) to determine the hydropathy profiles of the permuted sequences. This sequence met the 2 criteria for scrambled Aβ42: YHAGVDKEVVFDEGAGAEHGLAQK -IVRGFGVSDVSMIHINLF.
Both WT and scrambled Aβ42 were synthesized using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry, purified by reverse phase-high performance liquid chromatography, and characterized by mass spectrometry and amino acid analysis, as described previously (89). Quantitative amino acid analysis and mass spectrometry yielded the expected compositions and molecular weights for each peptide. Purified peptides were stored as lyophilizates at −20°C. During fibril assembly studies, scrambled Aβ42 has been shown to maintain a statistical coil secondary structure, bind thioflavin T poorly, and exhibit oligomerization characteristics distinct from those of wild type Aβ42 (90). Scrambled Aβ42 was dissolved in 10% (v/v) 60 mM NaOH and 90% (v/v) 10 mM phosphate buffer, pH 7.4, at a concentration of 1 mg/ml.
Transmission Electron Microscopy
Ten microliters of Aβ were spotted onto 400 mesh carbon-coated Formvar grids (Electron Microscopy Sciences) and incubated for 2 min. Each grid was then negatively stained with 1% (v/v) filtered (0.2 μm) uranyl acetate (Ted Pella), which was immediately wicked off. Electron microcopy analysis was performed using JEOL 1200 EX at 80 KV (91).
All other experimental protocols, including genotyping, GA immunization, isolation and adoptive transfer of bone-marrow CD115+ monocytes, quantification and stereological counting, and Barnes maze behavioral tests were previously described (63).
Statistics
GraphPad Prism 5.0b (GraphPad Software) was used to analyze the data. A comparison of three or more groups was performed using two-way or one-way ANOVA followed by the Tukey's or Bonferonni's (92) post hoc multiple comparison test for paired groups. Two-group comparisons were analyzed using two-tailed paired t-tests (StatPlus). Correlation analyses were performed using the Pearson's coefficient (r) tests (Prism). Results are expressed as mean ± standard error of the mean (s.e.m.). A p value less than 0.05 was considered significant.
Results
Excitatory Synapses Are More Vulnerable to Aβ42 Oligomers Than Fibrils
To assess the effect of Aβ42 oligomers vs. fibrils on synaptic integrity and neuronal structure, postnatal day 1 (P1) primary cortical neurons (CN) were incubated for 12 h with 100 nM of either defined and stabilized oligomers (XL-oAβ42), preformed fibrils (fAβ42), or non-aggregated scrambled Aβ control peptides (sAβ42)(90), compared to medium alone without Aβ (vehicle; Figures 1A–D). The pure populations of Aβ42 fibrils and stabilized low-n oligomers were verified by SDS-PAGE gel and transmission electron microscopy (Figures 1A–C). We observed no statistical difference in synaptic puncta number [analyzed by colocalized presynaptic VGluT1 (vesicular glutamate transporter 1) and PSD95 (postsynaptic density protein 95)] or neurite length between vehicle and sAβ42 groups (Supplementary Figures 1A,B). There was also no significant difference between CN co-cultured for 24 h with BM-derived MΦ for 24 h supplemented with either medium alone or sAβ42 (Supplementary Figures 1A,B). Interestingly, there was a small but significant increase in VGluT1/PSD95 synaptic number in cortical neurons following 24 h co-culture with GA-treated MΦ (Supplementary Figure 1A). While sAβ42 did not induce loss of synapses or neuritic length retraction in CN, incubation with fAβ42, and moreover, with XL-oAβ42 (Figure 1D), decreased presynaptic VGluT1+ number by 63 and 81%, respectively, as compared to vehicle (Figure 1E; P < 0.001). Similarly, but to a lesser extent, postsynaptic PSD95+ puncta numbers were significantly reduced by 48 and 62% after exposure to fAβ42 or XL-oAβ42, respectively (Figure 1E; P < 0.001). Analysis of co-localized VGluT1 and PSD95 puncta number indicated a 50 and 70% reduction in synaptic density after 12 h exposure to fAβ42 or XL-oAβ42, respectively (Figure 1F; P < 0.001). Importantly, cortical excitatory synapses were substantially more vulnerable to low-n Aβ42 oligomers than to their fibril counterparts (Figure 1F; P < 0.001).
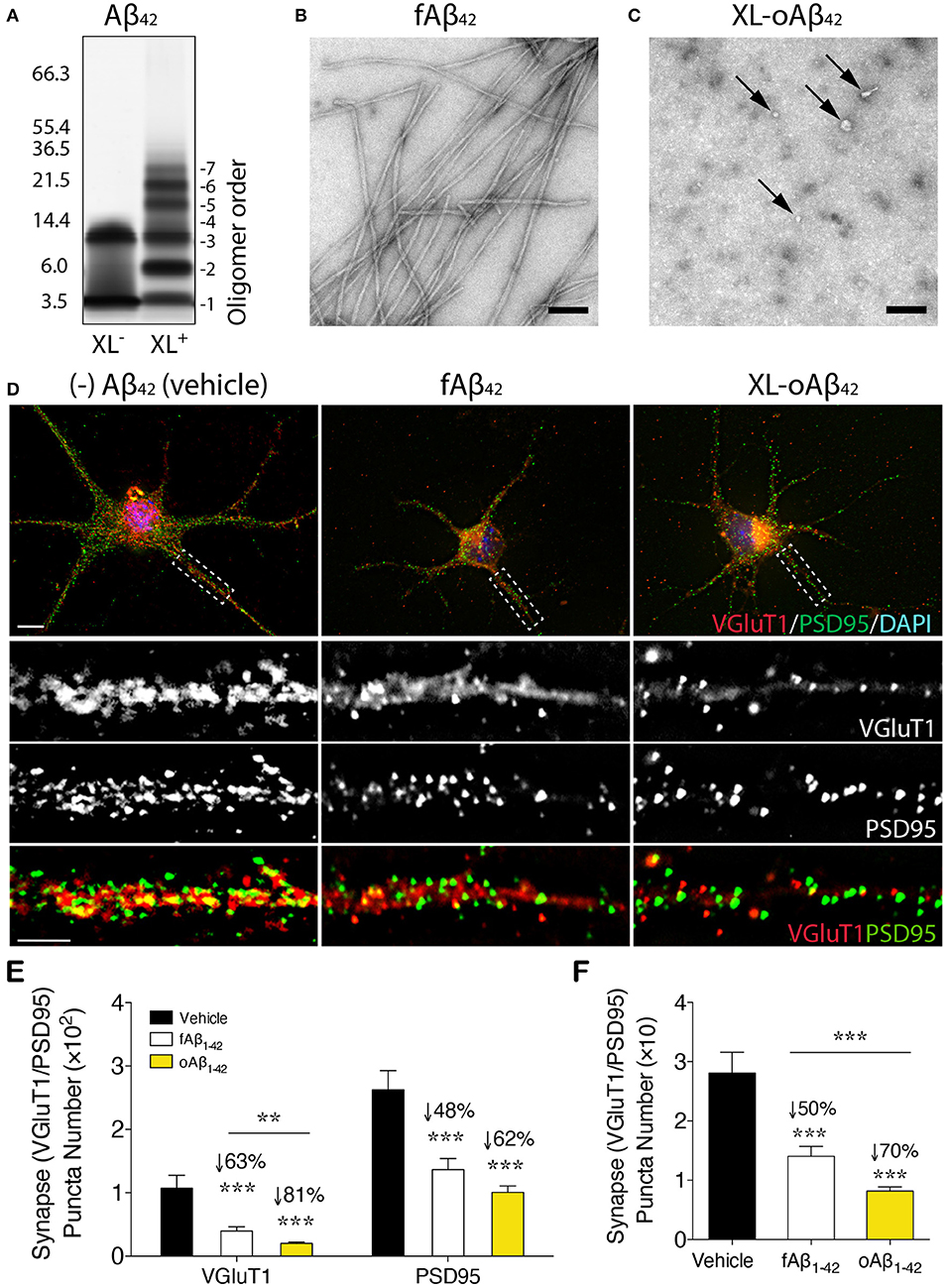
Figure 1. Impact of defined and stabilized Aβ42 oligomers vs. purified fibrils on synaptic integrity in primary cortical neurons. (A) SDS-PAGE image of cross-linked (XL+) and non-XL (XL−) Aβ42 oligomers. (B) Electron microscopy (EM) of preformed fibrillar (f)Aβ42 and (C) XL-oAβ42, consisting of a defined distribution of covalently linked oligomers that were characterized by negative EM stain. Spherical (arrows) and amorphous structures were observed for XL-oAβ42, whereas typical amyloid fibrils were seen in fAβ42. (D) Representative high-resolution images of pre-VGluT1 and post-PSD95 synapses in primary cortical neurons, treated with no (–)Aβ42 (vehicle), fAβ42 or XL-oAβ42. Areas within dashed boxes are magnified below. (E,F) Quantification of pre- and postsynapses in postnatal day 1 (P1) mouse primary cortical neurons. (E) VGluT1- and PSD95-immunoreactive area per neuron and (F) colocalized VGluT1/PSD95 synaptic puncta number in P1 cortical neurons treated with vehicle, fAβ42 or XL-oAβ42. ***P < 0.001, fAβ42 vs. vehicle or XL-oAβ42 vs. vehicle; **P < 0.01, fAβ42 vs. XL-oAβ42, by one-way ANOVA and Tukey's post-test. Data expressed as mean ± s.e.m.; n = 32 fields analyzed from 2 independent experiments; Scale bars = 100 nm (B,C) and 5 μm (D).
Spontaneous Neuronal Hyperactivity and Altered Spike Waveforms Induced by Aβ42 Oligomers
To examine the functional consequences of exposing neurons to Aβ42 oligomers vs. fibrils, we recorded the spontaneous extracellular activity of P1 cortical neurons using Microelectrode arrays (MEA) after incubation with vehicle, fAβ42, or XL-oAβ42 for either 24 or 48 h (Figures 2A–G and Supplementary Figures 2A–D). We first compared the spontaneous activity levels across all neurons with a level of activity of at least 0.2 Hz. We found that after incubation with Aβ42 oligomers for 24 h, spontaneous activity levels were significantly larger compared to incubation with vehicle or fibrils (Figure 2A; P < 0.05), whereas activity levels for incubation with fibrils was not significantly different from incubation with vehicle (Figure 2A; P < 0.05). After 48 h, neurons incubated with either fAβ42 or XL-oAβ42 had significantly elevated levels of spontaneous activity compared to vehicle (Figure 2B; P < 0.001–0.05). This shows that incubation with Aβ42 oligomers is accompanied by hyperactivity as early as 24 h. This was also true when examining all neurons regardless of spontaneous activity levels and when only included neurons recorded at both time points (see Supplementary Figures 2A–C).
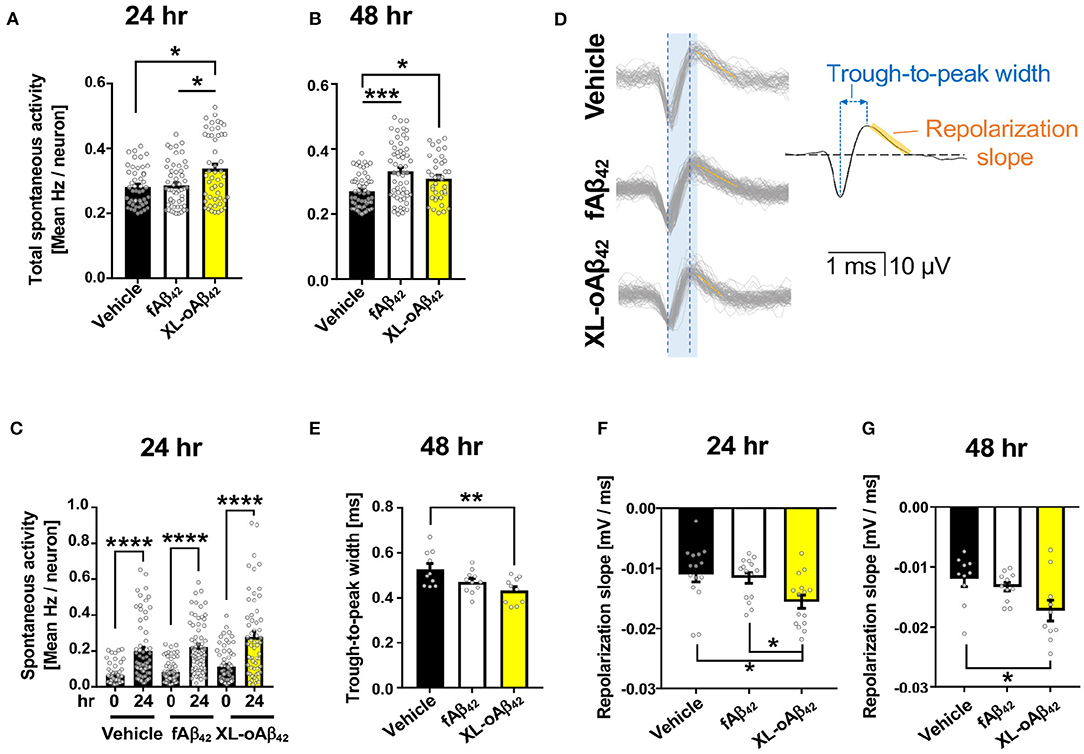
Figure 2. Oligomeric Aβ42 assemblies cause neuronal hyperactivity and altered extracellular spike waveforms. Microelectrode array recordings of spontaneous activity in P1 primary cortical neurons measured 24 and 48 h after addition of vehicle, 100 nM of fAβ42, or of XL-oAβ42. (A,B) Mean neuronal frequency of active neurons showing spontaneous activity during 5 min recordings. Neurons analyzed met a minimum threshold of 0.2 Hz firing rate. 24 h, *P < 0.05, vs. Vehicle or fAβ42; 48 h, *P < 0.05, ***P < 0.001, vs. Vehicle; Kruskal-Wallis analysis by Dunn's test. (C) Comparison of spontaneous activity measured from the same individual neuron populations over time, from untreated (time 0) to 24 h incubation with vehicle, fAβ42, or XL-oAβ42. ****P < 0.0001, vs. time 0; Kruskal-Wallis analysis by Dunn's test. (D) Representative extracellular waveforms from each condition (gray = overlays of multiple waveforms from a single neuron; black = fitted mean waveform for analysis), identifying the trough-to-peak width (blue) and repolarization slope (orange). Shaded blue box indicates trough-to-peak width observed in control wave forms, dotted blue lines indicate measurement in XL-oAβ42. Orange hashed lines indicate mean repolarization slope of each wave form. (E) Quantification of trough-to-peak width in P1 cortical neurons incubated with fAβ42 or XL-oAβ42 for 48 h. **P < 0.01 vs. Vehicle 48 h. (F–G) Quantification of repolarization slope in P1 neurons in each condition. *P < 0.05 vs. Vehicle 24 and 48 h or fAβ42 24 h, respectively. one-way ANOVA and Tukey's post-test. Data expressed as mean ± s.e.m., with individual data point from 5 independent experiments.
Did the presence of oligomers change the biophysical properties of neurons? To address this question, we next assessed whether the extracellular waveforms of the recorded action potentials differed between neurons exposed to fibrillar and oligomeric forms (Figures 2D–G and Supplementary Figure 2D). We found that two critical features of the extracellular waveform changed after incubation with oligomeric Aβ42. First, the trough-to-peak width of waveforms (Figure 2D) recorded from neurons incubated with oligomeric Aβ42 was significantly decreased relative to controls after 48 h (Figure 2E; P < 0.01); this effect was not apparent after 24 h (Supplementary Figure 1D). Second, the repolarization slope significantly decreased for neurons incubated with Aβ42 oligomers, but not fibrils, relative to controls after 24 h (Figure 2F; P = 0.0103), an effect which persisted through 48 h (Figure 2G; P < 0.05). Together, this shows that incubation with Aβ42 oligomers was accompanied by spontaneous neuronal hyperactivity and altered extracellular waveforms.
Activated Macrophages Effectively Prevent Synaptic Loss and Neuritic Arborization Retraction Caused by Aβ42 Oligomers
To determine whether naïve BM-derived (MΦBM) or GA-activated macrophages (GA-MΦ) can confer neuroprotection, P1 CN were co-cultured with MΦBM or GA-MΦ in the presence of fAβ42 or XL-oAβ42 (Figure 3; experimental scheme and neurite tracing quantification method are shown in Figures 3A,B). Substantial synaptic loss and neuritic retraction were observed, especially after exposure to Aβ42 oligomers (Figures 3C,D,F,G vs. Figure 3E; P < 0.01; 32 and 65% VGluT1/PSD95-synaptic loss in response to fibrils and oligomers, respectively). Importantly, reductions in pre- and postsynaptic puncta number (and area) after exposure to fAβ42 were fully restored when co-cultured with MΦBM or GA-MΦ (Figure 3C and Supplementary Figures 3A, B; P < 0.01), suggesting that MΦ have the capacity to effectively protect synapses against Aβ42 fibril toxicity within this time frame. However, untreated MΦ had limited ability to protect synapses against damage induced by Aβ42 oligomers (Figure 3C; P < 0.05), although importantly, GA activation of MΦ promoted full protection also against these highly synaptotoxic oligomers (Figure 3C and Supplementary Figures 3A,B; P < 0.01). While minimal to no protection against neuritic arborization loss was observed following co-culturing cortical neurons with naïve MΦ in the presence of either fAβ42 or XL-oAβ42 (Figure 3D), significant protection to neuronal dendritic structure was obtained with GA-activated MΦ, as measured by neurite process lengths (Figure 3D; P < 0.05). Interestingly, even in the absence of Aβ, co-cultures of cortical neurons with GA-MΦ vs. naïve MΦ had a significant increase in synaptic density (Figure 3C; P < 0.05). Thus, in primary cortical neurons, GA-activated MΦ effectively prevented loss of synapses and defects in arborization of neurite processes induced by both Aβ42 fibrils and oligomers.
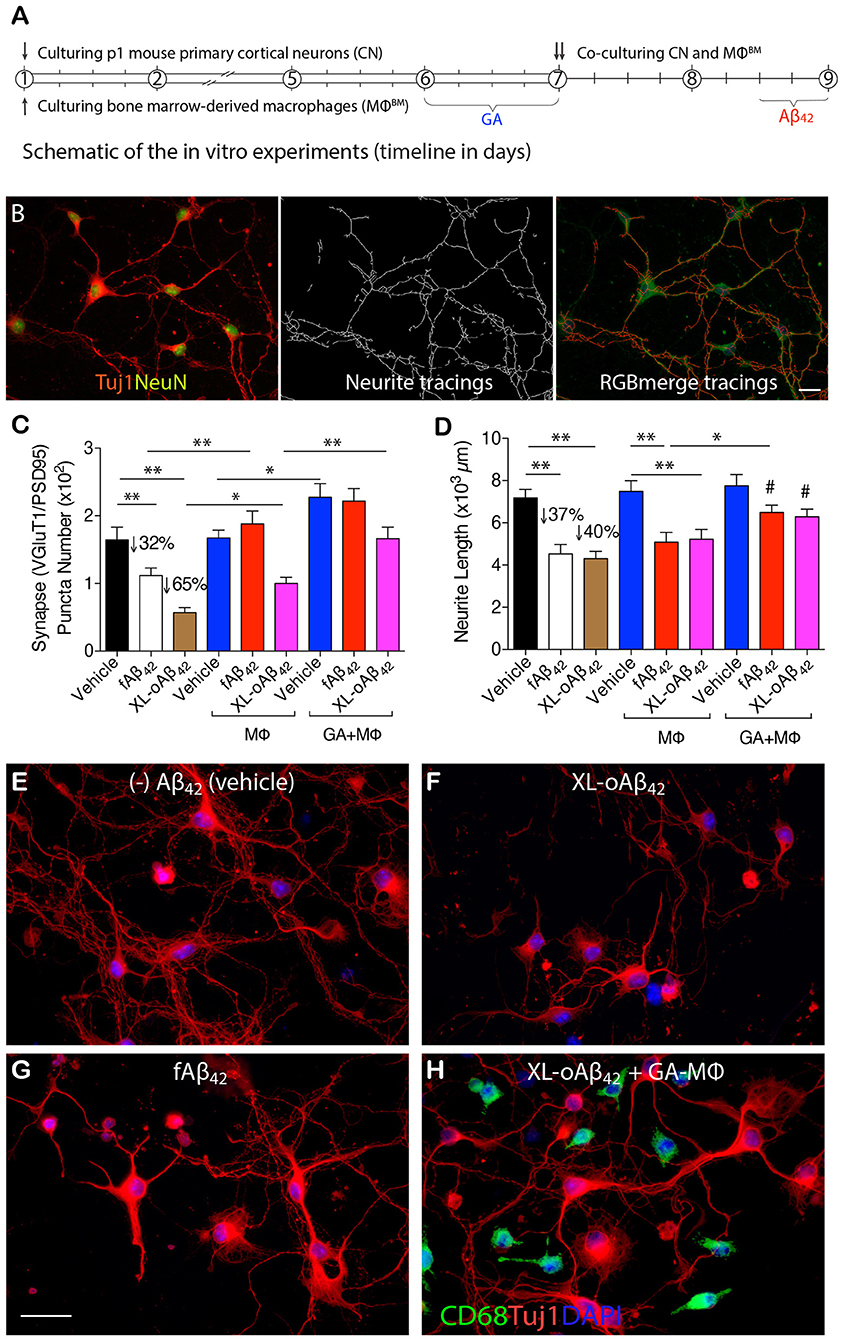
Figure 3. Activated MΦ effectively protect against oligomeric Aβ42-induced synaptic and neuritic arborization loss in primary cortical neurons. (A) Schematic of the in vitro experiments (timeline in days). P1 cortical neurons (treated with 100 nM XL-oAβ42, fAβ42, or vehicle for 12 h, respectively), bone marrow-derived MΦ (MΦBM), and GA-activated MΦBM (GA-MΦ) were cultured for 9 d. (B) Representative microphotographs of P1 neurons labeled with anti-Tuj1 and -NeuN serum (left), neuritic tracings with NeuriteTracer (83) (middle), and RGB merge tracings (right). Scale bar represents 20 μm. (C) Quantification of colocalized VGluT1/PSD95 synaptic puncta number in P1 neurons incubated with fAβ42, XL-oAβ42, or vehicle, and P1 neurons co-cultured with MΦ or with GA-MΦ. Note that fAβ42 and XL-oAβ42 both reduced the VGluT1/PSD95 synaptic density that was significantly preserved by co-culturing with MΦ. This effect was enhanced by co-culturing with GA-MΦ. (D) Quantification of neuritic length of P1 neurons incubated with fAβ42, XL-oAβ42, or vehicle, and P1 neurons co-cultured with MΦ or with GA-MΦ. Note that co-culturing with GA-MΦ significantly prevented decreases in neuritic length from fAβ42 or XL-oAβ42. Data expressed as mean ± s.e.m.; n = 48 fields analyzed from 3 independent experiments; *P < 0.05, **P < 0.01, comparisons as indicated by lines; #P < 0.05, vs. fAβ42 or XL-oAβ42 alone (no MΦ), by one-way ANOVA and Tukey's post-test. (E-H) Representative microphotographs of primary P1 neurons incubated with (E) vehicle, (F) XL-oAβ42, (G) fAβ42, and (H) co-cultured with GA-MΦ + XL-oAβ42. Scale bar = 20 μm.
MMP-9/Extracellular Degradation Is Predominant for Macrophage Clearance of Aβ42 Oligomers
To investigate how macrophages, including GA-activated macrophages, facilitate clearance of Aβ42 oligomers vs. fibrils and potentially increase synaptic protection, we characterized MΦ phenotypes, with and without GA stimulation, in response to the same quantities of XL-oAβ42 and pre-formed fAβ42 (100 nM; Figure 4). First, to assess surface recognition and intracellular uptake of Aβ42 assemblies, MΦ were incubated with XL-oAβ42 or fAβ42 for 30 min and labeled for Aβ (6E10), early endosome antigen 1 (EEA1), and class B scavenger receptor CD36 or class A type 1 scavenger receptor SCARA1 (encoded by macrophage scavenger receptor 1 – MSR1 gene). There was a notable increase in CD36 expression, intracellular o/fAβ42, and o/fAβ42 colocalized within EEA1 endosomes in GA-MΦ vs. naïve MΦ (Figures 4A, B). A quantitative analysis of scavenger receptors indicated a significant increase in CD36 expression (Figure 4C; P < 0.01; Supplementary Figure 4A; P < 0.05) in GA-MΦ compared to untreated MΦ, but not in SCARA1 for XL-oAβ42 (Supplementary Figure 4B). Regardless of GA, exposure to Aβ42 oligomers substantially upregulated surface expression of CD36 in MΦ (Figure 4C; P < 0.001). GA-MΦ exhibited significantly more intracellular uptake of both fAβ42 and XL-oAβ42 vs. naïve MΦ (Figures 4D,E; P < 0.0001 and P < 0.001, respectively). Both naïve and GA-MΦ were on average six times less effective in intracellular uptake of oligomeric vs. fibrillar Aβ42 (Figure 4D vs. Figure 4E). Strikingly, as compared with fibrils, Aβ42 oligomers were approximately 2,500 times less targeted into early endosomes (Figure 4F vs. Figure 4G), as measured by 6E10+-Aβ puncta number colocalized in EEA1+ vesicles after 30 min phagocytosis assay. Hence, these data suggest that while intracellular Aβ42 fibrils are predominantly targeted to the endosomal-lysosomal proteolysis pathway, intracellular oligomers mostly escape these proteolytic vesicles in MΦ. Nonetheless, GA-MΦ significantly increased the presence of both Aβ42 fibrils and oligomers in EEA1+ endosomes (Figures 4F,G; P < 0.01–0.0001).
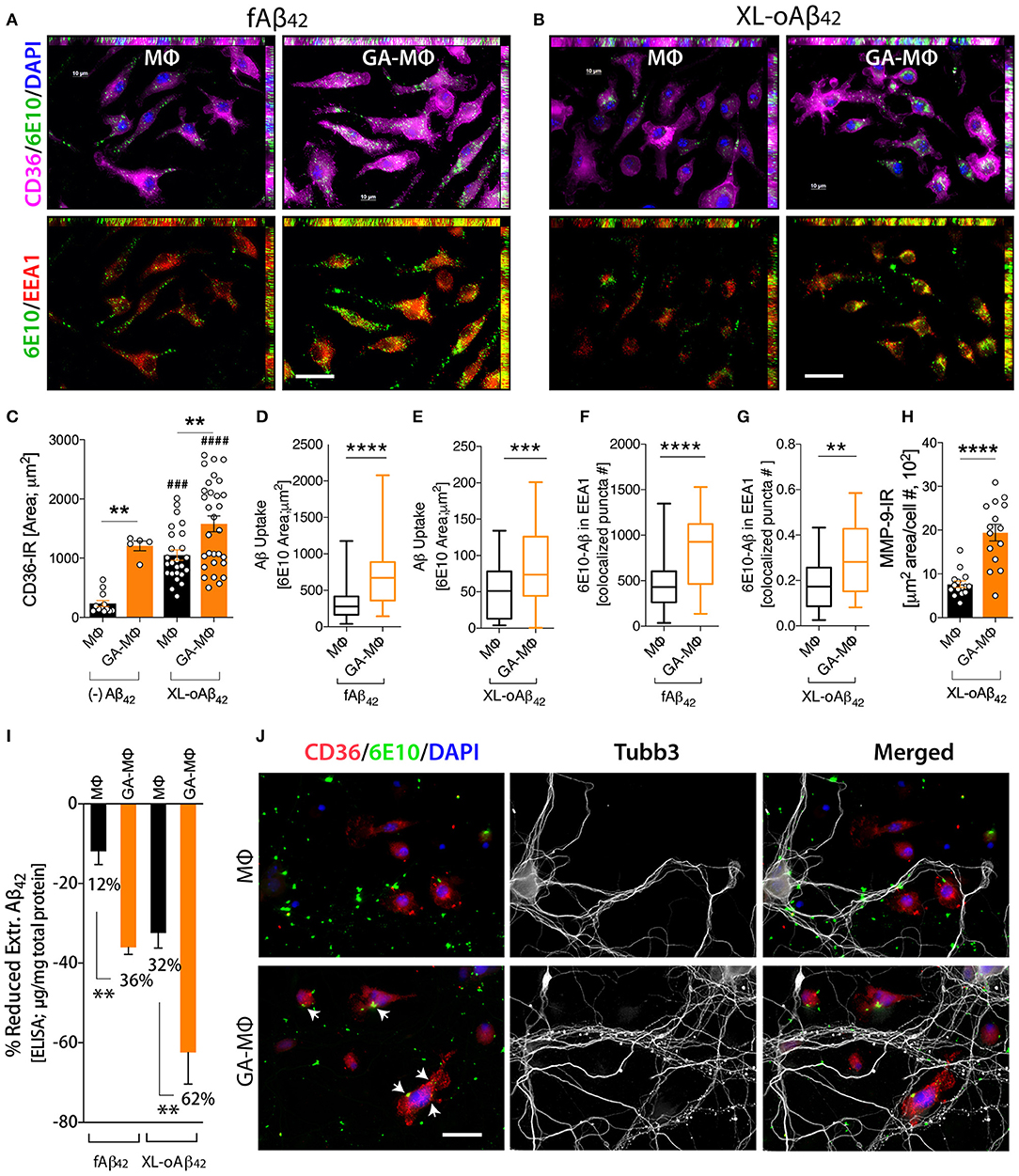
Figure 4. Induced oligomeric Aβ42 clearance via GA activation of CD36/EEA1-intra- and MMP-9-extracellular mechanisms in MΦ, leading to neuronal preservation. (A–G) GA-activated (24 h) or naïve MΦ were exposed to 100 nM Aβ42 assemblies for 30 min, and co-labeled for Aβ (6E10, green), type B scavenger receptor CD36 (magenta), and early endosomal marker (EEA1, red). Cells were counterstained with DAPI (blue). Representative micrographs demonstrate elevated surface CD36 expression, intracellular Aβ, and Aβ colocalized within EEA1 endosomes in GA-MΦ vs. control MΦ, 30 min fAβ42 (A) and XL-oAβ42 (B) uptake assays. (C) Quantitative immunoreactive analysis of CD36 signals showed a substantial increase in CD36 in GA-MΦ. Basal levels of CD36 expression without Aβ42 were higher in GA-MΦ vs. control MΦ. This increase maintained its significance after exposure to XL-oAβ42. Regardless of GA effects, the presence of oligomeric Aβ42 substantially increased CD36 levels in both control MΦ and GA-MΦ. (D,E) Quantitative analysis of intracellular Aβ in MΦ following 30 min fAβ42 (D) and XL-oAβ42 (E). (F,G) Quantitative analysis of fAβ42 (F) and XL-oAβ42 (G) within EEA1-positive vesicles in early endosomes in GA-MΦ vs. control MΦ. Co-labeled 6E10/EEA1- immunoreactive puncta number per MΦ is displayed. (H) Increased MMP-9 signal in GA-MΦ after exposure to XL-oAβ42. (I) ELISA quantification of extracellular Aβ42 after 24 h incubation with fAβ42 or XL-oAβ42 compared to 30 min basal peptide levels. (J) Representative microphotographs of co-cultured P1 neurons and MΦ exposed to XL-oAβ42 for 24 h, stained with CD36, 6E10, Tubb3, and DAPI. P1 cortical neurons co-cultured with GA-MΦ vs. control MΦ exhibit increased neuritic outgrowth density with increased CD36-mediated Aβ uptake (arrows) and reduced extracellular Aβ42. Data expressed as mean ± s.e.m., with individual data point; n = 48 fields analyzed from 3 independent experiments; immunoreactive areas are normalized by cell number; **P < 0.01, ***P < 0.001, ****P < 0.0001, GA-MΦ vs. MΦ with fAβ42 or XL-oAβ42, and ###P < 0.001, ####P < 0.0001 for comparisons to MΦ without Aβ [(–) Aβ42], by one-way ANOVA with Tukey's test or two-tailed student t-test. Scale bar = 20 μm.
Next, we evaluated the extent to which MΦBM degrade Aβ42 oligomers as compared to fibrillar forms in the extracellular space. To this end, we first assessed the expression of matrix metallopeptidase 9 (MMP-9)—an Aβ-degrading enzyme—by analysis of MMP-9+-immunoreactive area in MΦ (Figure 4H). Then, we determined Aβ1−42 concentrations in supernatant of MΦ following incubation with 100nM of either fAβ42 or XL-oAβ42, using a highly sensitive ELISA assay (Figure 4I). We previously showed that after exposure to Aβ42 fibrils, GA-MΦ exhibited a significant increase in MMP-9 levels (63, 70). Yet, the response to oligomers has not been reported. Here, we found that MMP-9 expression in GA-MΦ was markedly increased after incubation with XL-oAβ42 (Figure 4H; P < 0.0001). Examination of extracellular degradation as a function of Aβ42 conformation indicated significant decreases in concentration of Aβ42 in the media of GA-MΦ vs. naïve MΦ after 24 h incubation with either fAβ42 or XL-oAβ42 (Figure 4I; 36 or 62% reduction, respectively, P < 0.01). Likewise, the substantial reduction in fAβ42 and XL-oAβ42 concentrations in the extracellular media of GA-treated vs. untreated MΦ can be seen after 30 min of incubation with these Aβ species (Supplementary Figures 4C,D; P < 0.001). Notably, at the two incubation time points (30 min and 24 h incubation), Aβ42 oligomers were primarily degraded in the extracellular rather than the intracellular space of macrophages and were considerably more accessible for extracellular degradation than their fibril counterparts (Figure 4I; 32% vs. 12 and 62% vs. 36% for MΦ and GA-MΦ, respectively; Supplementary Figures 4C,D; P < 0.001). Representative microscopic images in Figure 4J panel illustrate the increase in oAβ42 uptake and CD36 expression by GA-activated MΦ, along with a decrease in extracellular or synaptic-bound oAβ42, which resulted in synaptic and neuritic outgrowth preservation in primary cortical neurons. Overall, these data suggest that GA induces CD36/EEA1-mediated macrophage clearance, and moreover, MMP-9/extracellular degradation of synaptotoxic Aβ42 oligomers, ultimately protecting synapses, their dendritic structure, and neural function.
Severe Loss of Excitatory Pre- and Postsynaptic Terminals in the Entorhinal Cortex and Hippocampus of ADtg Mice
Given the crucial role of entorhinal cortex layers 2 and 3 in connecting brain cortical regions to the hippocampus, we sought to analyze pre- and postsynaptic VGluT1 and PSD95 biomarkers in substructures of the entorhinal cortex (ENT) and hippocampus (HIPPO) of 10- and 13-month-old symptomatic ADtg vs. wild type (WT) mice (Figures 5A–E; Supplementary Figures 5–8). Due to lack of differences between the lateral (ML-L) and the medial (ML-M) blade molecular layers in pre- and postsynaptic areas across all experimental groups (Supplementary Figure 5), both regions were combined in our analyses (Figures 5D,E). Ten-month-old ADtg mice already exhibited significant pre- and postsynaptic losses in the molecular layers (ML) of the dentate gyrus (DG) and the stratum lacunosum-moleculare (SLM), but not in the stratum oriens (SO) nor in the stratum radiatum (SR) of the cornu ammonis 1 (CA1; Supplementary Figure 6). At 13 months, ADtg mice exhibited 50-62% presynaptic VGluT1-immunoreactive (IR) area loss in all hippocampal regions as compared to WT mice (Figure 5D; P < 0.05–0.001). Similarly, analyses of postsynaptic PSD95-IR areas in 13-month-old ADtg mice vs. WT littermates indicated significant reductions by 28–42% in all hippocampal regions (Figure 5E; P < 0.05). In ENT layers 2 and 3, 68 and 53% of presynaptic VGluT1-positive areas were lost (Figure 5D; P < 0.001), while 38 and 51% of postsynaptic PSD95 signals were reduced (Figure 5E; P < 0.05 and P < 0.01) in ADtg mice compared to WT mice, respectively. Overall, our data demonstrate a progressive and considerable loss of VGLUT/PSD95 synaptic density, while the presynaptic VGluT1 terminals appear more vulnerable to Aβ accumulation in the 13-month ADtg animals.
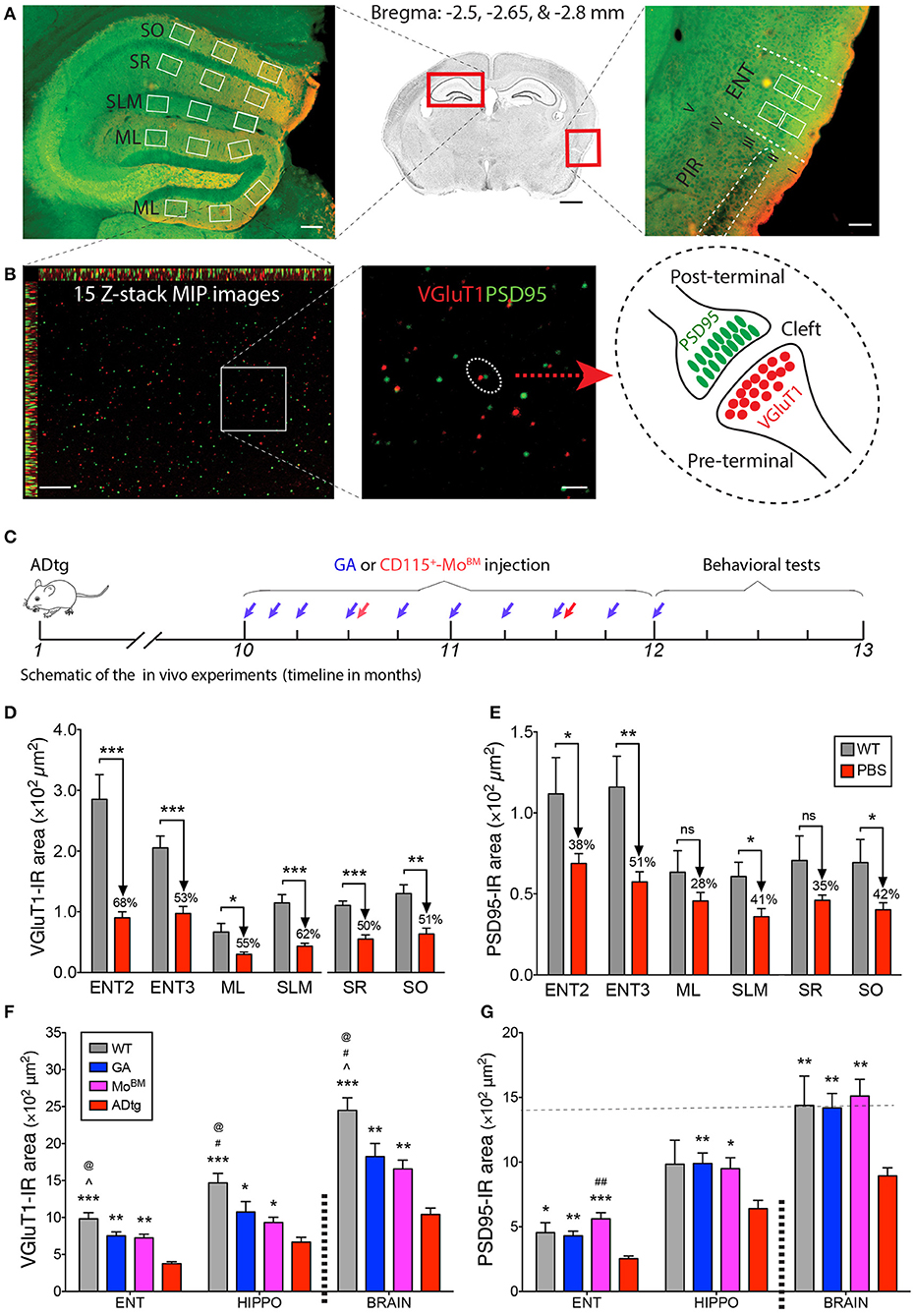
Figure 5. Cortical and hippocampal synaptic rescue by CD115+-monocyte blood enrichment and GA immunization in ADtg mice. (A,B) VGluT1/PSD95 synaptic quantification scheme in entorhinal cortex (ENT) and hippocampus (HIPPO). (A) (Middle) Representative crystal violet stained image of a coronal brain section at Bregma −2.65 mm. Scale bar = 1 mm. (Left) Representative microimage of HIPPO stained with the presynaptic (VGluT1, red) and postsynaptic (PSD95, green) markers. Note that 3 fields are chosen in each of the lateral and medial blade molecular layers (ML) of dentate gyrus (DG), as well as in the stratum lacunosum-moleculare (SLM), stratum radiatum (SR), and stratum oriens (SO) of CA1. (Right) Representative microphotograph of ENT immunolabled with the same pre- and postsynaptic markers. Note that ENT adjacent to piriform cortex (PIR) and 2 same fields are precisely selected from layers 2 and 3. Scale bar = 100 μm. (B) Representative microphotographs from the lateral blade ML immunostained with the same synaptic markers. Each high-magnification maximum intensity projection (MIP) image contains 15 Z-stack optical scanning (left). Scale bar = 10 μm. (Middle) Magnification for visualizing the proximity of pre- and postsynaptic signals. Scale bar = 2 μm. (Right) Schematic representation of pre- and post-terminal synapse. (C) Schematic of the in vivo experiments, where 10-month-old ADtg mice (all males) were injected with weekly s.c. GA immunization or monthly i.v. CD115+-MoBM injections for a duration of 2 months. Control groups were either naïve WT mice or monthly i.v. PBS injected ADtg mice. At the completion of the experiment, mice underwent behavioral testing, were euthanized, and brains were collected for analyses. (D,E) Quantification of presynaptic VGluT1 (D) and postsynaptic PSD95 (E) areas in ENT2/3 and hippocampal substructures of 13-month-old ADtg mice vs. age-matched WT littermates. (F,G) Quantification of VGluT1 (F) and PSD95 (G) immunoreactive areas in the entorhinal cortex, hippocampus, and whole brain of WT, GA-immunized, MoBM-injected, and PBS-control mice. Data expressed as mean ± s.e.m.; n = 6 mice per group; *P < 0.05, **P < 0.01, ***P < 0.001, and #P < 0.001, ##P < 0.0001, compared to ADtg control; @P < 0.05 compared to MoBM, ∧P < 0.05 compared to GA, one-way ANOVA with Tukey's test.
Synaptic Rescue Following Enhanced Cerebral Recruitment of Macrophages via Immunomodulation Therapy in ADtg Mice
To better understand the role of peripheral Mo/MΦ in preservation of excitatory synapses within AD-relevant cortical and hippocampal regions, we analyzed both pre- and postsynaptic density in substructures of ENT and HIPPO following weekly s.c. injections of GA immunization or monthly i.v. injections of CD115+ MoBM into the peripheral blood (schematic of in vivo experiment in Figure 5C). Rescue pre- and postsynaptic density was noted in ENT2/3 by GA immunization or blood enrichment with MoBM in ADtg mice (Figures 5F,G, P < 0.05–0.001; Supplementary Figure 7, P < 0.05–0.01). Similarly, GA led to rescue of pre-VGluT1 and post-PSD95 synapses in HIPPO across various ML, SLM, SR, and SO subregions (Figures 5F,G, P < 0.05–0.01; Supplementary Figure 8). Notably, throughout the analyzed brain regions, both immunomodulation approaches rescued postsynaptic density (i.e., PSD95-IR area) in symptomatic ADtg mice to levels similar or higher than those observed in WT mice (Figure 5G; Supplementary Figures 7B, 8B). No cumulative effect was noted between a group of ADtg mice treated with both GA and MoBM (combined treatment) and groups of mice treated with GA or MoBM alone (data not shown). Taken together, our in vitro and in vivo data suggest that immunomodulation approaches that deliver macrophages to reduce pathogenic Aβ42 burden at synaptic clefts substantially preserve pre- and postsynaptic terminals.
To identify confounders that potentially predict excitatory synaptic loss and contribute to immunotherapy-based synaptic preservation, we carried out multiple Pearson's coefficient (r) correlation analyses in the 13-month-old mouse cohort (Supplementary Figure 9). Specifically, we explored possible relationships between synaptic integrity and parameters of disease stage (cognitive function in Barnes maze test, astrogliosis, and amyloidosis) and therapeutic response (cerebral MΦ recruitment and phenotype). A strong linear association was observed between cerebral astrocytosis (GFAP+ reactive astrocyte area) and impaired cognitive function, as measured by error count on day 7 (long-term memory retention) of Barnes maze test (Supplementary Figure 9A; P = 0.0187, Pearson's r = 0.60). Similar correlations were found between hippocampal and cortical astrogliosis and other parameters of cognitive deficit in the Barnes maze test (not shown), suggesting that astrogliosis is a strong predictor of cognitive decline. Comparable to these observations, tight correlations were detected between cortical astrocytosis and postsynaptic loss (Supplementary Figures 9B–D; P = 0.028–0.007 and Pearson's r = −0.55 to −0.61). These data show an association between astrogliosis and cognitive outcome.
In relation to Aβ-plaque pathology, both cortical and hippocampal regions further revealed close inverse associations with postsynaptic density (Supplementary Figures 9E–H; P = 0.011–0.002 and Pearson's r = −0.57 to −0.70). Similar correlations were also observed for presynaptic areas (not shown). These data suggest that cerebral Aβ-plaque burden are important indicators of synaptic loss in this murine model. We then established the relationship between excitatory synaptic integrity and cognition, showing significant associations between hippocampal and cortical synaptic density and cognitive function (Supplementary Figures 9I,J; P = 0.015–0.009 and Pearson's r = −0.45 to −0.53). The latter cognitive performance was determined by escape latency times in the learning and memory reversal phase on day 9 of the Barnes maze test. These data suggest that synaptic integrity is tightly associated with cognitive function, and that both astrogliosis and amyloidosis adversely affect it.
We previously showed that effective immunomodulation approaches in ADtg mice, such as GA immunization or blood enrichment with MoBM, reduced Aβ-plaque pathology and preserved cognition (50, 52, 63, 70). These therapeutic effects were achieved via cerebral recruitment of Iba-1+/CD115+/CD45hi Mo/MΦ that expressed high levels of osteopontin (OPN; encoded by the gene secreted phosphoprotein 1 or SPP1) (63, 70). OPN/SPP1 is an immunoregulatory cytokine shown to both promote synaptogenesis (93–95) and be crucial for MΦ-mediated Aβ42 clearance (70). Significant and strong associations were observed between cerebral and hippocampal Iba-1+/CD45hi Mo/MΦ (Mo/MΦ cell count) and cognitive function, as measured by latency times and error counts on day 9 of the Barnes maze test (Supplementary Figures 9K,L; P = 0.0472–0.011 and Pearson's r = −0.64 – −0.76). We further found a significant and robust association between OPN and postsynaptic but not presynaptic density areas (Supplementary Figures 9M,N; P = 0.0133 and Pearson's r = 0.69). These analyses suggest that increased Mo/MΦ-mediated OPN/SPP1 presence in the brain is linked with synaptic integrity and cognitive preservation.
Discussion
This study supports the conclusion that activated MΦBM can reduce Aβ42 oligomers and protect synaptic integrity and neuronal structure. Initially, we determined the impact of defined and stabilized populations of soluble Aβ42 oligomers on excitatory synapses, neuritic arborization, and neuronal function as compared to pure fibrils. We indicated that synaptotoxicity was not elicited by non-aggregating sAβ42 peptides but was dependent on their conformation state; cortical neurons were far more vulnerable to Aβ42 oligomers than to fibrils. Aβ42 oligomers caused severe synaptic loss, neurite arborization retraction, and hyperactivity, as well as changes in two critical features of the extracellular waveform in cortical neurons. We further found that MΦBM effectively protect against synaptic loss and neurite retraction caused by Aβ42 fibrils yet provide insufficient protection against oligomers. Importantly, MΦ activation with glatiramer acetate (GA, an FDA-approved drug) fully protected neurons from detrimental effects triggered by both oligomers and fibrils. We identified distinct MΦ-mediated mechanisms for clearance of oligomers as compared to fibrils leading to synaptic protection. While eradication of Aβ42 oligomers predominately involves increased extracellular/MMP-9 degradation, fibrils are primarily eliminated via CD36/EEA1+-endosomal intracellular proteolysis. Strikingly, both pathways are boosted in MΦ stimulated with GA. Finally, our in vivo studies corroborated the in vitro findings, demonstrating that loss of cortical and hippocampal excitatory synapses in transgenic mouse models of AD is reversed by increased cerebral recruitment of Mo-derived MΦ, achieved via GA immunomodulation or peripheral-blood enrichment of BM-derived monocytes. Overall, these findings should guide future therapeutic interventions for AD to boost MΦ-mediated clearance of synaptotoxic Aβ42 oligomers and preserve synapses and cognition.
Our data in mouse primary cortical neurons provide evidence to support the notion that Aβ42 oligomers pose a greater threat to the neuronal network than do fibrils. These results align with previous studies reporting that Aβ42 oligomers impair neuronal plasticity and activation of excitatory mGluR hippocampal neurons, as well as exhibit direct synaptotoxic effects leading to cognitive impairments (8, 13, 15, 23–29, 96). As compared to postsynaptic terminals, presynaptic terminals displayed greater vulnerability to Aβ42 assemblies in both in vivo and in vitro models. We found that compared to fibrils, stabilized Aβ42 oligomers caused greater synaptic loss and neuritic retraction along with hyperactivity and altered action potential shape in primary cortical neurons. Due to the massive reduction in synapses (~70%) observed in response to Aβ42 oligomers as early as 12 h after incubation, a corresponding reduction in neuronal activity was anticipated. To the contrary, however, we found elevated levels of spontaneous activity after incubation with oligomers. These observations are in accordance with previous studies in murine models of AD that showed increased neuronal excitability near Aβ plaques or in Aβ-depositing mouse brains, using Ca2+ imaging and in vivo intracellular recording (97, 98). Combined with the modified action potential shape we documented, this suggests that oligomeric Aβ42 directly affects the biophysical (intrinsic) properties of neurons, possibly via altered ion channel densities, in addition to its effects on synapses. Because oligomeric Aβ42 species are highly metastable and exist in dynamically changing mixtures, the conclusions of previous reports on Aβ42 oligomers may have been confounded by the existence of heterogeneous Aβ populations. By using defined and stabilized Aβ42 oligomers, this study establishes the precise neuropathological consequences caused by oligomers vs. fibrils and supports therapeutic avenues that clear cerebral Aβ42 oligomers to prevent such neuronal and synaptic damage.
One powerful method to clear Aβ42 oligomers and protect synapses was revealed in this study. Cortical neurons that were severely impaired by Aβ42 oligomers showed partial recovery of pre- and postsynaptic density and neurite arborization when co-cultured with MΦBM, and full recovery with GA-MΦ. While naïve MΦ were able to effectively protect against synaptic and neurite length loss caused by the fibrils, they exhibited poor capacity to prevent oligomeric-induced synaptotoxicity. Yet, GA-MΦ successfully eliminated both fibrils and oligomers, essentially protecting neuronal network integrity despite these adverse conditions. Postsynaptic terminals, which were less vulnerable to Aβ42 species, were fully rescued by the presence of GA-MΦ. However, it would be interesting to look at the alteration of inhibitory gephyrin immunosignal expression following the administration of GA or GA-MΦ. We discovered that the EEA1+-early endosomal/lysosomal pathway was far more involved in the MΦ-mediated engulfment of fibrils than of oligomers. In contrast, reduction of Aβ42 in the extracellular space of MΦ was substantially more efficient for oligomers than for fibrils, potentially through enhanced release and activity of proteolytic enzymes (i.e., MMP-9). These in vitro data corroborate our in vivo studies in ADtg mice that demonstrated enhanced MMP-9 expression by infiltrating macrophages surrounding cerebral Aβ plaques in GA-immunized animals (63, 70). Notably, GA enhanced both internalization and extracellular pathways in MΦ to break down synaptotoxic Aβ42 forms. Recent studies indicated that depleting microglia with colony-stimulating factor 1 receptor (CSF1R) signaling inhibitors could impair formation of the plaques in an AD model (99, 100). Microglial depletion was further found not only to lead to a complete rescue of deficits in proliferation, differentiation and survival of adult hippocampal neural progenitor cells (101), but also significantly reduce ApoE, which is a major constituent of amyloid plaques and promotes their aggregation and deposition (102). It would be interesting to look at changes of synapses, Aβ deposition and cognition when microglial depletion plus GA immunotherapy is applied to this ADtg model. Additional studies are needed to further establish direct structure-function connection between these and other Aβ42 forms (e.g., dimers, trimers, etc.), synaptotoxicity, and macrophage-mediated synaptic protection. A limitation of this study is that it does not allow for conclusions regarding potential rescue of neuronal function by macrophages due to lack of electrophysiological studies in macrophages and neurons co-cultures. Yet, since this study clearly demonstrates neuroprotection by morphological (neurite length) and synaptic biomarker measurements, future studies should evaluate whether these are accompanied with functional protection.
In this study, we demonstrated that progressive cortical and hippocampal pre- and postsynaptic loss tightly correlated with increased cerebral Aβ burden in the double-transgenic APPSWE/PS1ΔE9 murine model. The direct association between pre- and postsynaptic biomarkers and cognitive scores further emphasizes a potential link between Aβ42, synaptotoxicity, and cognitive decline. Moreover, the unexpected strong association between synaptic density and GFAP+ astrogliosis suggested that beyond direct Aβ toxicity, chronically reactive astrocytes not only fail to protect synapses, but also detrimentally impair them and lead to cognitive decline. Future investigations are warranted to better understand the impact of astrocytes and their phenotype on synaptic and cognitive decline in AD.
Here, we showed that both immunotherapies—GA immunization or blood-enrichment with CD115+ monocytes—restored presynaptic and fully rescued postsynaptic density in ADtg mice exhibiting significant synaptic loss. In our previous studies, these immunomodulation approaches have led to increased cerebral infiltration of monocyte-derived macrophages directly involved in Aβ clearance, leading to improved hippocampal-based memory and learning (52, 63). In this study, cognitive function, which directly correlated with synaptic integrity, also strongly associated with cerebral recruitment of (GFP+) Iba1+/CD45high monocytes and macrophages. These data suggest that MΦ activity in the brain associated with decreased synaptotoxic Aβ levels can enhance synaptic protection and regeneration. One interesting question would be what happens if microglia as residential macrophages are treated with GA. Immunophenotypes of microglia expressed differently in dementia with AD pathology. For example, Iba1 was negatively associated with AD while levels of CD68, MSR-A, and CD64 expression were positively related to the presence of dementia (103). In a previous paper we determined a significant reduction of GFAP expression in the cortex of AD mice following GA or/and GA-MΦ immunotherapy (63), and found GFAP expression in this model was negatively associated with pre-synapse VGluT1. We now further demonstrated that the relationship of GFAP levels vs. post-synapse PSD95 in the cortex of AD mice exhibits the same behavior pattern as that of GFAP vs. VGluT1.
Cerebral clearance of Aβ by innate immune cells has been shown to take place via multiple mechanisms, including phagocytosis and enzymatic degradation (48, 49, 51, 54, 56, 57, 61, 104–106). Increasing evidence over the past decade has established the notion that peripheral MΦ contribute to the phagocytosis of cerebral Aβ plaque, an idea that is further supported by reports of defective Aβ phagocytosis by MΦ isolated from AD patients (52, 53, 58, 60, 63, 104, 107). Several studies corroborating this idea have shown the involvement of scavenger receptors such as CD36 and SCARA-1 in facilitating fibrillar Aβ engulfment by innate immune cells. Rare variants in the TREM2 gene were also associated with diminished MΦ phagocytosis and increased risk for AD (108–110). Indeed, parallel to the aforementioned investigations, this study's findings indicated elevated levels of surface scavenger receptors (e.g., CD36) in response to not only fibrillar but also oligomeric Aβ42, with increased uptake by GA-MΦ. Importantly, the role of MΦ and, moreover, GA-activated MΦ in the clearance of non-fibrillar oligomeric Aβ forms has not been previously studied (111, 112). Enhanced colocalization of oligomers and fibrils in EEA1+-vesicles in GA-MΦ imply that these Aβ assemblies are redirected to lysosomal degradation rather than for perinuclear or endoplasmic reticulum pathologic accumulation (113). Therefore, this FDA-approved drug emerges again as a promising modality to safely stimulate phagocytic MΦ and improve ability to resist synaptotoxic Aβ forms associated with AD.
In the absence of Aβ, cortical neurons supplemented with GA-MΦ exhibited elevated levels of synaptic density and neuritic length that were higher than that of the baseline levels detected in cortical neurons alone. These results suggest that increased synaptic density in the presence of GA-MΦ is a result of both Aβ42 clearance and upregulation of neurotrophic factors stimulating synaptogenesis, such as insulin-like growth factor 1 (IGF-1), transforming growth factor beta 1 (TGFβ1), and/or OPN/SPP1 (50, 66, 70). In our previous study, we observed that OPN/SPP1 was markedly elevated in MΦ following GA treatment in vitro and in vivo (70). Here, a strong correlation (Pearson's r = 0.7) was found between cerebral OPN/SPP1 levels and postsynaptic density, which can explain increased synaptogenesis in the presence of MΦBM overexpressing OPN in the brains of immunized ADtg mice (70). In support of these findings, we reported that harnessing monocyte-derived macrophages by GA- or MOG-45D-immunization in ADtg mice enhanced cerebral expression of neurotrophic factors such as IGF-1, TGFβ1, and nuclear transcription factor early growth response 1 (Egr-1) (50, 58, 66), concomitant with recovered hippocampal neurogenesis. Hence, this immunomodulation approach holds great promise to positively affect neural regeneration as well as synaptic and cognitive preservation through combined MΦ activity of eradicating pathogenic Aβ42 forms and increasing neurotrophic support.
In conclusion, therapeutic approaches designed to prevent synaptotoxicity or even rescue synapses are becoming more prominent. Our extended data from both in vitro and in vivo studies may signify that GA-stimulated macrophages confer a potential synaptoprotective phenotype, at least in part through augmented capacity to eliminate highly toxic Aβ42 oligomers and induce synaptic preservation and regeneration. Hence, this study encourages the development of a macrophage-based approach as therapeutic intervention for AD.
Data Availability Statement
The raw data and protocols used to generate datasets for this study are presented in the article text, figures and tables. Supplementary data are also available online. Extended data can be made available upon reasonable request from the corresponding author to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by Cedars-Sinai Medical Center Institutional Animal Care and Use Committee (IACUC).
Author Contributions
MK-H: study conception, design, study supervision, and final approval of the manuscript. SL: major experimental contributor, data acquisition and design. VG, YK, D–TF, JS, AR, and TT: experimental contributors with assistance from MK-H and SL. EH, JS, and DT: preparation of Aβ42 assemblies. DD: correlational analysis with guidance from MK-H. SL, VG, and MK-H: data analysis, interpretation, and presentation. MK-H, SL, EH, D–TF, TT, YK, UR, KB, and DT: discussion on intellectual content and manuscript editing. MK-H, SL, EH, VG, D–TF, UR, and DT: manuscript drafting, editing, and revisions.
Funding
This study was supported by the BrightFocus Foundation A2013328S00 grant (formerly AHAF; MK-H), The Coins for Alzheimer's Research Trust (C.A.R.T; MK-H) Fund, NIH/NIA R01AG056478 and R01AG055865 (MK-H), The Cheryl and Haim Saban Foundation (MK-H), The Marciano Family Foundation (MK-H), National Natural Science Foundation of China 81671229, 81871032 (SL) and the National Center for Advancing Translational Sciences through CTSI (UL1TR000124; MK-H).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mia Oviatt for assistance with editing this manuscript and Prof. Jevgenij Raskatov for his helpful comments. This paper is dedicated to Dr. Salomon Moni Hamaoui and Lillian Jones Black, both of whom died from Alzheimer's disease.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00049/full#supplementary-material
References
1. James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K, Bennett DA. Contribution of Alzheimer disease to mortality in the United States. Neurology. (2014) 82:1045–50. doi: 10.1212/WNL.0000000000000240
2. Alzheimer's Association. 2018 Alzheimer's disease facts and figures. Alzheimer's Dementia. (2018) 14:367–429. doi: 10.1016/j.jalz.2018.02.001
3. Butterfield DA. Phosphoproteomics of Alzheimer disease brain: insights into altered brain protein regulation of critical neuronal functions and their contributions to subsequent cognitive loss. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2031–9. doi: 10.1016/j.bbadis.2018.08.035
4. Cardozo PL, De Lima IBQ, Maciel EMA, Silva NC, Dobransky T, Ribeiro FM. Synaptic Elimination in Neurological Disorders. Curr Neuropharmacol. (2019) 17:1071–95. doi: 10.2174/1570159X17666190603170511
5. Selkoe DJ. Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. (2001) 3:75–80. doi: 10.3233/JAD-2001-3111
6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. (2002) 297:353–6. doi: 10.1126/science.1072994
7. Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. (2008) 192:106–13. doi: 10.1016/j.bbr.2008.02.016
8. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. (2008) 14:837–42. doi: 10.1038/nm1782
9. Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. (2011) 54(Suppl. 1):S204–17. doi: 10.1016/j.neuroimage.2010.06.020
10. La Morgia C, Ross-Cisneros FN, Koronyo Y, Hannibal J, Gallassi R, Cantalupo G, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. (2016) 79:90–109. doi: 10.1002/ana.24548
11. Koronyo YB, Biggs D, Barron E, Boyer DS, Pearlman JA, Au WJ. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer's disease. JCI Insight. (2017) 2:e93621. doi: 10.1172/jci.insight.93621
12. Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front Cell Neurosci. (2015) 9:191. doi: 10.3389/fncel.2015.00191
13. Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. (2007) 27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007
14. Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. (2007) 101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x
15. Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. (2009) 62:788–801. doi: 10.1016/j.neuron.2009.05.012
16. Hayden EY, Conovaloff JL, Mason A, Bitan G, Teplow DB. Preparation of pure populations of covalently stabilized amyloid beta-protein oligomers of specific sizes. Anal Biochem. (2017) 518:78–85. doi: 10.1016/j.ab.2016.10.026
17. Li S, Jin M, Liu L, Dang Y, Ostaszewski BL, Selkoe DJ. Decoding the synaptic dysfunction of bioactive human AD brain soluble Abeta to inspire novel therapeutic avenues for Alzheimer's disease. Acta Neuropathol Commun. (2018) 6:121. doi: 10.1186/s40478-018-0626-x
18. Brinkmalm G, Hong W, Wang Z, Liu W, O'malley TT, Sun X, et al. Identification of neurotoxic cross-linked amyloid-beta dimers in the Alzheimer's brain. Brain. (2019) 142:1441–57. doi: 10.1093/brain/awz066
19. Raskatov JA. What is the “Relevant” Amyloid beta42 concentration? Chembiochem. (2019) 20:1725–6. doi: 10.1002/cbic.201900097
20. Yasumoto T, Takamura Y, Tsuji M, Watanabe-Nakayama T, Imamura K, Inoue H, et al. High molecular weight amyloid beta1-42 oligomers induce neurotoxicity via plasma membrane damage. Faseb j. (2019) 33:9220–34. doi: 10.1096/fj.201900604R
21. Shankar GM, Walsh DM. Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener. (2009) 4:48. doi: 10.1186/1750-1326-4-48
22. Yu L, PetyukVA, Tasaki S, Boyle PA, Gaiteri C, Schneider JA, et al. Association of cortical beta-amyloid protein in the absence of insoluble deposits with Alzheimer disease. JAMA Neurol. (2019) 76:818–26. doi: 10.1001/jamaneurol.2019.0834
23. Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. (2007) 27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007
24. Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, et al. Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci. (2007) 27:10578–87. doi: 10.1523/JNEUROSCI.2444-07.2007
25. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. (2010) 330:198. doi: 10.1126/science.1194653
26. Tang Y, Scott DA, Das U, Edland SD, Radomski K, Koo EH, et al. Early and selective impairments in axonal transport kinetics of synaptic cargoes induced by soluble amyloid beta-protein oligomers. Traffic. (2012) 13:681–93. doi: 10.1111/j.1600-0854.2012.01340.x
27. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. (2002) 416:535–9. doi: 10.1038/416535a
28. Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener. (2014) 9:48. doi: 10.1186/1750-1326-9-48
29. Umeda T, Ramser EM, Yamashita M, Nakajima K, Mori H, Silverman MA, et al. Intracellular amyloid beta oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol Commun. (2015) 3:51. doi: 10.1186/s40478-015-0230-2
30. Selkoe DJ. Developing preventive therapies for chronic diseases: lessons learned from Alzheimer's disease. Nutr Rev. (2007) 65:S239–43. doi: 10.1111/j.1753-4887.2007.tb00370.x
31. Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. (2006) 281:4292–9. doi: 10.1074/jbc.M511018200
32. Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. (2008) 28:4231–7. doi: 10.1523/JNEUROSCI.5161-07.2008
33. Nitsch RM, Hock C. Targeting beta-amyloid pathology in Alzheimer's disease with Abeta immunotherapy. Neurotherapeutics. (2008) 5:415–20. doi: 10.1016/j.nurt.2008.05.013
34. Cho JE, Kim JR. Recent approaches targeting beta-amyloid for therapeutic intervention of Alzheimer's disease. Recent Pat CNS Drug Discov. (2011) 6:222–33. doi: 10.2174/157488911796958002
35. Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M. Clearance of cerebral Abeta in Alzheimer's disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci. (2017) 74:2167–201. doi: 10.1007/s00018-017-2463-7
36. Guedes JR, Lao T, Cardoso AL, El Khoury J. Roles of microglial and monocyte chemokines and their receptors in regulating Alzheimer's disease-associated amyloid-β and tau pathologies. Front neurol. (2018) 9:549. doi: 10.3389/fneur.2018.00549
37. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
38. Tejera D, Heneka MT. Microglia in Alzheimer's disease: the good, the bad and the ugly. Curr Alzheimer Res. (2016) 13:370–80. doi: 10.2174/1567205013666151116125012
39. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
40. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
41. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique microglia type associated with restricting development of Alzheimer's Disease. Cell. (2017) 169:1276–90 e1217. doi: 10.1016/j.cell.2017.05.018
42. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81 e569. doi: 10.1016/j.immuni.2017.08.008
43. Song WM, Colonna M. The identity and function of microglia in neurodegeneration. Nat Immunol. (2018) 19:1048–58. doi: 10.1038/s41590-018-0212-1
44. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
45. Ziegler-Waldkirch S, D'errico P, Sauer JF, Erny D, Savanthrapadian S, Loreth D, et al. Seed-induced Abeta deposition is modulated by microglia under environmental enrichment in a mouse model of Alzheimer's disease. Embo J. (2018) 37:167–82. doi: 10.15252/embj.201797021
46. Wang S, Colonna M. Microglia in Alzheimer's disease: a target for immunotherapy. J Leukoc Biol. (2019) 106:219–27. doi: 10.1002/JLB.MR0818-319R
47. Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta-amyloid. Am J Pathol. (1992) 140:1389–99.
48. Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. (1996) 17:553–65. doi: 10.1016/S0896-6273(00)80187-7
49. Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, Mcconlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. (2001) 7:612–8. doi: 10.1038/87945
50. Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, et al. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. (2006) 103:11784–9. doi: 10.1073/pnas.0604681103
51. Simard AR, Rivest S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer's disease. Mol Psychiatry. (2006) 11:327–35. doi: 10.1038/sj.mp.4001809
52. Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur J Neurosci. (2007) 26:413–6. doi: 10.1111/j.1460-9568.2007.05652.x
53. El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. (2007) 13:432–8. doi: 10.1038/nm1555
54. Takata K, Kitamura Y, Yanagisawa D, Morikawa S, Morita M, Inubushi T, et al. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS Lett. (2007) 581:475–8. doi: 10.1016/j.febslet.2007.01.009
55. Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, et al. Degradation of fibrillar forms of Alzheimer's amyloid beta-peptide by macrophages. Neurobiol Aging. (2008) 29:707–15. doi: 10.1016/j.neurobiolaging.2006.12.001
56. Richard KL, Filali M, Prefontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. (2008) 28:5784–93. doi: 10.1523/JNEUROSCI.1146-08.2008
57. Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, et al. Clearance mechanisms of Alzheimer's amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatry. (2009) 14:469–86. doi: 10.1038/mp.2008.96
58. Koronyo-Hamaoui M, Ko MK, Koronyo Y, Azoulay D, Seksenyan A, Kunis G, et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: local elevation of IL-10 and MMP-9. J Neurochem. (2009) 111:1409–24. doi: 10.1111/j.1471-4159.2009.06402.x
59. Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer's disease. CNS Neurol Disord Drug Targets. (2010) 9:168–73. doi: 10.2174/187152710791011982
60. Lebson L, Nash K, Kamath S, Herber D, Carty N, Lee DC, et al. Trafficking CD11b-positive blood cells deliver therapeutic genes to the brain of amyloid-depositing transgenic mice. J Neurosci. (2010) 30:9651–8. doi: 10.1523/JNEUROSCI.0329-10.2010
61. Lai AY, Mclaurin J. Clearance of amyloid-beta peptides by microglia and macrophages: the issue of what, when and where. Future Neurol. (2012) 7:165–76. doi: 10.2217/fnl.12.6
62. Bernstein KE, Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Lopes DH, et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer's-like cognitive decline. J Clin Invest. (2014) 124:1000–12. doi: 10.1172/JCI66541
63. Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V, et al. Therapeutic effects of glatiramer acetate and grafted CD115(+) monocytes in a mouse model of Alzheimer's disease. Brain. (2015) 138:2399–422. doi: 10.1093/brain/awv150
64. He H, Altomare D, Ozer U, Xu H, Creek K, Chen H, et al. Cancer cell-selective killing polymer/copper combination. Biomater Sci. (2016) 4:115–20. doi: 10.1039/C5BM00325C
65. Frenkel D, Maron R, Burt DS, Weiner HL. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J Clin Invest. (2005) 115:2423–33. doi: 10.1172/JCI23241
66. Bakalash S, Pham M, Koronyo Y, Salumbides BC, Kramerov A, Seidenberg H, et al. Egr1 expression is induced following glatiramer acetate immunotherapy in rodent models of glaucoma and Alzheimer's disease. Invest Ophthalmol Vis Sci. (2011) 52:9033–46. doi: 10.1167/iovs.11-7498
67. Koronyo Y, Salumbides BC, Black KL, Koronyo-Hamaoui M. Alzheimer's disease in the retina: imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener Dis. (2012) 10:285–93. doi: 10.1159/000335154
68. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. (2015) 6:7967. doi: 10.1038/ncomms8967
69. Kunis G, Baruch K, Miller O, Schwartz M. Immunization with a myelin-derived antigen activates the brain's choroid plexus for recruitment of immunoregulatory cells to the CNS and attenuates disease progression in a mouse model of ALS. J Neurosci. (2015) 35:6381–93. doi: 10.1523/JNEUROSCI.3644-14.2015
70. Rentsendorj A, Sheyn J, Fuchs DT, Daley D, Salumbides BC, Schubloom HE, et al. A novel role for osteopontin in macrophage-mediated amyloid-beta clearance in Alzheimer's models. Brain Behav Immun. (2018) 67:163–80. doi: 10.1016/j.bbi.2017.08.019
71. Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci. (2014) 17:304–11. doi: 10.1038/nn.3606
72. Criscuolo C, Fontebasso V, Middei S, Stazi M, Ammassari-Teule M, Yan SS, et al. Entorhinal Cortex dysfunction can be rescued by inhibition of microglial RAGE in an Alzheimer's disease mouse model. Sci Rep. (2017) 7:42370. doi: 10.1038/srep42370
73. Frontinan-Rubio J, Sancho-Bielsa FJ, Peinado JR, Laferla FM, Gimenez-Llort L, Duran-Prado M, et al. Sex-dependent co-occurrence of hypoxia and beta-amyloid plaques in hippocampus and entorhinal cortex is reversed by long-term treatment with ubiquinol and ascorbic acid in the 3xTg-AD mouse model of Alzheimer's disease. Mol Cell Neurosci. (2018) 92:67–81. doi: 10.1016/j.mcn.2018.06.005
74. Marzi SJ, Leung SK, Ribarska T, Hannon E, Smith AR, Pishva E, et al. A histone acetylome-wide association study of Alzheimer's disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat Neurosci. (2018) 21:1618–27. doi: 10.1038/s41593-018-0253-7
75. Nakazono T, Jun H, Blurton-Jones M, Green KN, Igarashi KM. Gamma oscillations in the entorhinal-hippocampal circuit underlying memory and dementia. Neurosci Res. (2018) 129:40–6. doi: 10.1016/j.neures.2018.02.002
76. Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. (2003) 100:330–5. doi: 10.1073/pnas.222681699
77. Rahimi F, Maiti P, Bitan G. Photo-induced cross-linking of unmodified proteins (PICUP) applied to amyloidogenic peptides. J Vis Exp. (2009) 12:1071. doi: 10.3791/1071
78. Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. (2012) 7:1741–54. doi: 10.1038/nprot.2012.099
79. Li S, Nie EH, Yin Y, Benowitz LI, Tung S, Vinters HV, et al. GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat Neurosci. (2015) 18:1737–45. doi: 10.1038/nn.4146
80. Paxinos G, Franklin KB. The Mouse Brain in Stereotaxic Coordinates. 2nd ed. San Diego, CA: Academic Press (2001).
81. Eroglu C, Allen NJ, Susman MW, O'rourke NA, Park CY, Ozkan E, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. (2009) 139:380–92. doi: 10.1016/j.cell.2009.09.025
82. Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp. (2010) 19:2270. doi: 10.3791/2270
83. Pool M, Thiemann J, Bar-Or A, Fournier AE. NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J Neurosci Methods. (2008) 168:134–9. doi: 10.1016/j.jneumeth.2007.08.029
84. Bitan G, Teplow DB. Preparation of aggregate-free, low molecular weight amyloid-beta for assembly and toxicity assays. Methods Mol Biol. (2005) 299:3–9. doi: 10.1385/1-59259-874-9:003
85. Bitan G, Lomakin A, Teplow DB. Amyloid beta-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem. (2001) 276:35176–84. doi: 10.1074/jbc.M102223200
86. Bitan G, Teplow DB. Rapid photochemical cross-linking–a new tool for studies of metastable, amyloidogenic protein assemblies. Acc Chem Res. (2004) 37:357–64. doi: 10.1021/ar000214l
87. Bitan G. Structural study of metastable amyloidogenic protein oligomers by photo-induced cross-linking of unmodified proteins. Methods Enzymol. (2006) 413:217–36. doi: 10.1016/S0076-6879(06)13012-8
88. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. (1982) 157:105–32. doi: 10.1016/0022-2836(82)90515-0
89. Borreguero JM, Urbanc B, Lazo ND, Buldyrev SV, Teplow DB, Stanley HE. Folding events in the 21-30 region of amyloid beta-protein (Abeta) studied in silico. Proc Natl Acad Sci USA. (2005) 102:6015–20. doi: 10.1073/pnas.0502006102
90. Yamin G, Coppola G, Teplow DB. Design, characterization, and use of a novel amyloid beta-protein control for assembly, neurotoxicity, and gene expression studies. Biochemistry. (2016) 55:5049–60. doi: 10.1021/acs.biochem.6b00579
91. Fradinger EA, Maji S, Lazo ND, Teplow DB. Studying amyloid β-protein assembly. In: Xia W, Xu H, editors. Amyloid Precursor Protein, A Practical Approach. Boca Raton, FL: CRC Press (2005). p. 83–110.
92. Finkelstein A, Kunis G, Seksenyan A, Ronen A, Berkutzki T, Azoulay D, et al. Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS. PLoS ONE. (2011) 6:e22374. doi: 10.1371/journal.pone.0022374
93. Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal. (2009) 3:311–22. doi: 10.1007/s12079-009-0068-0
94. Rittling SR. Osteopontin in macrophage function. Expert Rev Mol Med. (2011) 13:e15. doi: 10.1017/S1462399411001839
95. Chan JL, Reeves TM, Phillips LL. Osteopontin expression in acute immune response mediates hippocampal synaptogenesis and adaptive outcome following cortical brain injury. Exp Neurol. (2014) 261:757–71. doi: 10.1016/j.expneurol.2014.08.015
96. Zhao J, Li A, Rajsombath M, Dang Y, Selkoe DJ, Li S. Soluble abeta oligomers impair dipolar heterodendritic plasticity by activation of mGluR in the hippocampal CA1 region. iScience. (2018) 6:138–50. doi: 10.1016/j.isci.2018.07.018
97. Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. (2008) 321:1686–9. doi: 10.1126/science.1162844
98. Kellner V, Menkes-Caspi N, Beker S, Stern EA. Amyloid-beta alters ongoing neuronal activity and excitability in the frontal cortex. Neurobiol Aging. (2014) 35:1982–91. doi: 10.1016/j.neurobiolaging.2014.04.001
99. Spangenberg EE, Green KN. Inflammation in Alzheimer's disease: lessons learned from microglia-depletion models. Brain Behav Immun. (2017) 61:1–11. doi: 10.1016/j.bbi.2016.07.003
100. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
101. Ortega-Martinez S, Palla N, Zhang X, Lipman E, Sisodia SS. Deficits in enrichment-dependent neurogenesis and enhanced anxiety behaviors mediated by expression of Alzheimer's disease-linked Ps1 variants are rescued by microglial depletion. J Neurosci. (2019) 39:6766–80. doi: 10.1523/JNEUROSCI.0884-19.2019
102. Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. (2019) 22:191–204. doi: 10.1038/s41593-018-0296-9
103. Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, Brayne C, et al. Microglial immunophenotype in dementia with Alzheimer's pathology. J Neuroinflammation. (2016) 13:135. doi: 10.1186/s12974-016-0601-z
104. Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. (2006) 49:489–502. doi: 10.1016/j.neuron.2006.01.022
105. Yamada K, Hashimoto T, Yabuki C, Nagae Y, Tachikawa M, Strickland DK, et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J Biol Chem. (2008) 283:34554–62. doi: 10.1074/jbc.M801487200
106. Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid beta-peptide elimination from the brain. J Neurochem. (2010) 115:1077–89. doi: 10.1111/j.1471-4159.2010.07002.x
107. Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. (2005) 18:134–42. doi: 10.1016/j.nbd.2004.09.009
108. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
109. Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, et al. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet. (2014) 23:5838–46. doi: 10.1093/hmg/ddu277
110. Finelli D, Rollinson S, Harris J, Jones M, Richardson A, Gerhard A, et al. TREM2 analysis and increased risk of Alzheimer's disease. Neurobiol Aging. (2015) 36:546. e549–13. doi: 10.1016/j.neurobiolaging.2014.08.001
111. Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, et al. Impact of peripheral myeloid cells on amyloid-beta pathology in Alzheimer's disease-like mice. J Exp Med. (2015) 212:1811–8. doi: 10.1084/jem.20150479
112. Varvel NH, Grathwohl SA, Degenhardt K, Resch C, Bosch A, Jucker M, et al. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-beta deposition in mouse models of Alzheimer's disease. J Exp Med. (2015) 212:1803–9. doi: 10.1084/jem.20150478
Keywords: Alzheimer's disease, neurodegeneration, immunomodulation therapy, amyloid-beta, regeneration, synaptogenesis
Citation: Li S, Hayden EY, Garcia VJ, Fuchs D-T, Sheyn J, Daley DA, Rentsendorj A, Torbati T, Black KL, Rutishauser U, Teplow DB, Koronyo Y and Koronyo-Hamaoui M (2020) Activated Bone Marrow-Derived Macrophages Eradicate Alzheimer's-Related Aβ42 Oligomers and Protect Synapses. Front. Immunol. 11:49. doi: 10.3389/fimmu.2020.00049
Received: 15 October 2019; Accepted: 09 January 2020;
Published: 31 January 2020.
Edited by:
Tanuja Chitnis, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Enrico Castroflorio, Medical Research Council Harwell (MRC), United KingdomRobert Adam Harris, Karolinska Institutet (KI), Sweden
Copyright © 2020 Li, Hayden, Garcia, Fuchs, Sheyn, Daley, Rentsendorj, Torbati, Black, Rutishauser, Teplow, Koronyo and Koronyo-Hamaoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya Koronyo-Hamaoui, maya.koronyo@cshs.org