
Abstract
Tanshinone IIA is a compound purified from the Chinese herb Danshen (Radix Salviae Miltiorrhiza Bge). The neuroprotective effect of tanshinone IIA was investigated in a neonatal rat model of hypoxia-ischemia brain damage. Hypoxia-ischemia encephalopathy was induced in rats at day 7 of postnatal age by ligation of the right common carotid artery, followed by 2 h of hypoxia. Tanshinone IIA (10 mg/kg, i.p.) was injected daily from day 2 before surgery for 9 or 16 d. Our results demonstrated significant and sustained brain damage in the hypoxia-ischemia– and vehicle-treated groups at 1 and 3 wk after surgery. Treatment with tanshinone IIA significantly reduced the severity of brain injury, as indicated by the increase in ipsilateral brain weight and neuron density, compared with those of sham-operated animals. The recovery of sensorimotor function and histology was observed in animals that received tanshinone IIA. The plasma of tanshinone IIA–treated rats exhibited higher antioxidant activities, as reflected by the oxygen radical absorbance capacity assay, compared with the vehicle-treated rats. In the neural progenitor cell line C17.2 that was subjected to 2,2′-azobis (2-amidino propane hydrochloride)–induced oxidative stress, tanshinone IIA increased cell viability and protected against mitochondrial damage (JC-1 assay). Our results suggest that tanshinone IIA has antioxidative activities and that treatment that is started before a hypoxic-ischemic insult is partially neuroprotective. Further studies are required to elucidate whether rescue treatment with tanshinone IIA is effective and to determine whether its protective effect is also associated with secondary cooling of the brain.
Similar content being viewed by others


Main
Hypoxia-ischemia encephalopathy (HIE) in the newborn infant is a major cause of acute mortality and chronic neurologic disability in survivors. During hypoxic-ischemic brain injury, mitochondrial functions are compromised and the lack of electron acceptors leads to free radical leakage from the mitochondria into the cytoplasm, causing lipid peroxidation of polyunsaturated fatty acid–rich neuronal membranes (1,2).
Danshen is derived from the dried root or rhizome of Salviae Miltiorrhizae Bge. In recent decades, Danshen has been commonly used in China for the treatment of angina pectoris and cerebrovascular disorders such as ischemic stroke (3). Tanshinone IIA, a derivative of phenanthrenequinone, is one of the key components of Danshen (Fig. 1). It has antioxidant properties and has been demonstrated to protect against lipid peroxidation in in vitro and in vivo studies (4,5). Tanshinone IIA can prevent the neuroblastoma cell line PC12 from apoptosis induced by serum-free culture conditions (6). It has the potential to penetrate the blood–brain barrier and acts as an inhibitor of acetylcholinesterase (AchE) (7). These observations prompted us to speculate that tanshinone IIA may protect against HIE in neonates. In the present study, we investigated the neuroprotective effect of tanshinone IIA on a validated neonatal rat model of hypoxia-ischemia brain injury. We examined changes in neuropathology and sensorimotor functions in response to tanshinone IIA in this model. We also studied the antioxidant capacity in the plasma of these animals and in cultures of a mouse neural progenitor cell line C17.2. Our results suggest that tanshinone IIA has antioxidative activities and provides neuroprotection against hypoxia-ischemia–induced brain injury in neonatal rats.

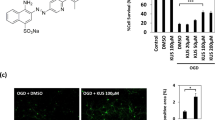
Structural formula of tanshinone IIA.
METHODS
Animal protocols.
All procedures were carried out in accordance with guidelines approved by the Animal Ethics Committee of The Chinese University of Hong Kong. Sprague-Dawley rat pups were kept with their dams in the Laboratory Animal Services Center with a light:dark cycle of 12:12 h and allowed food and water ad libitum.
Induction of hypoxia-ischemia in neonatal rats.
Hypoxic-ischemic brain injury was induced in rat pups (12–15 g) on day 7 of postnatal age. They were anesthetized by ether. The right common carotid artery was exposed and ligated with size 4-0 surgical sutures to stop the blood flow. The entire procedure was completed in <10 min. After carotid ligation, the pups were returned to their dams and allowed to recover for 2 h. Hypoxia then was induced by exposing the animals to a humidified gas mixture that contained 8% oxygen in nitrogen at 37°C for 2 h. The daily injection of tanshinone IIA was administered to the pups before they were returned to their dams after hypoxic exposure. Sham-operated pups underwent the same surgical procedure but did not receive carotid ligation and exposure to hypoxia.
Administration of tanshinone IIA.
Tanshinone IIA was ethanol extracted and purified from Radix Salviae Miltiorrhiza Bge (Sichuan, China) with a purity of 99.9% determined by HPLC (System Gold 508/125/168; Beckman Coulter Inc., Fullerton, CA). The extract was dissolved in 0.1% DMSO (Sigma Chemical Co., St. Louis, MO) and made up to a concentration of 1 mg/mL in PBS (pH 7.4). The rat pups were randomly divided into three groups: sham-operated group (n = 50), vehicle-treated group (PBS that contained 0.1% DMSO; n = 59), and tanshinone IIA–treated group (n = 59). Tanshinone IIA was administered by i.p. injection at a dose of 10 mg · kg−1 · d−1 from 2 d before surgery (postnatal day 5) for 9 or 16 d. The injection of tanshinone IIA or vehicle on the day of the surgery was administered to the pups immediately after hypoxia exposure, and they then were returned to their dams. The animals were killed at 1, 2, or 3 wk after surgery by using ketamine (0.05 mL/kg) and xylazine (0.01 mg/kg; Alfasan, Woerden, Holland).
Brain weight.
The cerebral hemispheres, brain stem, and cerebellum were removed from the skull. The hemispheres were separated by a longitudinal midline incision, and each hemisphere was weighed on a high-precision, digital balance (sensitivity ±0.001 g). The difference in weights between the ipsilateral (right) and contralateral (left) brain was calculated using the formula % Damage = (C − I)/C × 100, where C and I denote weights of the contralateral and ipsilateral hemispheres, respectively.
Histology.
Rat brains at 3 wk after surgery were fixed in 10% neutral formaldehyde and kept at 4°C. Two-millimeter-thick coronal blocks were cut from the brain, with the most frontal cut being 2 mm from the frontal pole of the intact hemisphere. The tissue blocks were dehydrated in 70% ethanol, embedded in paraffin wax, and sectioned. Five-micrometer sections were stained with hematoxylin and eosin and examined under light microscopy
Immunostaining with neuron-specific enolase.
At 3 wk after surgery, three rats from each group were anesthetized with ketamine (0.05 mL/kg) and xylazine (0.01 mg/kg) and then transcardially infused with 0.9% saline followed by 10% ice-cold neutral formaldehyde (Sigma Chemical Co.). Their brains were removed, placed in 10% neutral formaldehyde overnight at room temperature, and processed for paraffin histology. Five-micrometer sections were cut and deparaffinized in xylene and graded alcohol before they were immersed in citrate buffer (pH 7.6) for antigen retrieval in a microwave oven. The sections were placed in 3% hydrogen peroxide for 20 min to block endogenous peroxidase and then incubated in 5% rabbit serum (Dako, Glostrup, Demark) for 10 min. The primary MAb against neuron-specific enolase (NSE; 1:400 dilution; Calbiochem, Darmstadt, Germany) (8) was added onto the sections, which then were incubated overnight at room temperature. The sections were treated further with a biotinylated rabbit anti-mouse antibody (1:1000 dilution; Dako) for 40 min before incubation with horseradish peroxidase (HRP; Zymed, San Francisco, CA) for 45 min. Color development was performed in 3,3′-diaminobenzidine tetrahydrochloride (Sigma Chemical Co.) solution for 10 min. After staining, the sections were washed and coverslipped with Permount (Fisher Scientific, Loughborough, UK).
Counting of cortical neurons in the sensorimotor area of the forelimb.
The number of cortical neurons in the sensorimotor area of the forelimb (9) were counted in five randomly selected frontal sections by an investigator who was blinded to the allocation of treatment groups. These neurons were identified by their location, size, and NSE staining. The neuron density of each group (three rats) was expressed as the mean number of neurons per 10,000 μm2.
Functional test.
A standard postural reflex test was performed to evaluate the extent of neural recovery in rat pups 3 wk after surgery (10). The investigator had no previous information on the treatment group of the rats. The pup (sham-operated group n = 23; vehicle-treated group n = 26; tanshinone IIA–treated group n = 27) was held by the tail 50 cm above a table. Normal rat pups extended both forelimbs toward the table (score 0). Pups with brain damage flexed the forelimb contralateral to the damaged hemisphere (score 1). The pup then was put onto the table, and a lateral pressure was applied behind the shoulder until the forelimbs slid. A reduced resistance to this lateral force toward the left side (contralateral to the brain damage) was considered abnormal (score 2). Results are presented as the percentage of rats in each of the functional groups.
Body temperature measurement.
In an independent experiment, daily rectal temperature was measured in the three treatment groups of neonatal rats, using a Bioprober temperature sensor (0.7 mm diameter) connected directly to the Powerlab/16S system (AD Instruments Pty Ltd, Victoria, Australia), and data were analyzed by the Powerlab Chart V4.2 software. The unrestrained pups were acclimatized inside an infant incubator (Air-shields-C100; Narco Scientific, Hatboro) at 32°C for 30 min before their rectal temperatures were recorded (11).
Oxygen radical absorbance capacity assay.
With the use of the same peroxyl radical generator for both lipophilic and hydrophilic antioxidants, ORAC levels were measured in the plasma of rat pups 2 wk after surgery (12). Ten pups were randomly selected from each treatment group. Peripheral blood samples were collected from the left heart ventricle. They were centrifuged at 10,000 × g for 10 min at 4°C. Total ORAC in plasma was measured as described by Wang et al. Ten μL 2,2′-azobis (2-amidinopropane) hydrochloride (160 mM; AAPH; Sigma Chemical Co.) was added to a reaction mixture of 300 μL of β-phycoerythrin (3.8 mg/L; Sigma Chemical Co.) and 20 μL of Trolox (range 0–100 μM; Sigma Chemical Co.) or whole plasma (1:200 dilution) and incubated in a dual-scanning spectrofluorometer (Gemini SpectraMAX; Molecular Devices, Tokyo, Japan) at 37°C. The phycoerythrin fluorescence decay was monitored at 2-min intervals for 70 min at 546 nm excitation and 565 nm emission. ORAC values were calculated according to the area under the curve using the SOFTmax PRO software (Molecular Devices, Tokyo, Japan) and expressed as the equivalence of 1 μM Trolox.
Effects of tanshinone IIA on C17.2 cell viability.
The mouse neural progenitor cell line C17.2 was cultured in IMDM (GIBCO, CA) supplemented with 10% (vol/vol) FCS (GIBCO) in an atmosphere of 5% CO2/95% humidified air at 37°C. AAPH was prepared as a stock solution (100 mg/mL) in IMDM and diluted to a 4-mg/mL working solution immediately before use. C17.2 cells (5 × 103/well) were seeded in a 96-well plate. After being cultured in IMDM with 10% FCS overnight, the cells were washed twice with PBS and then incubated with AAPH at 4 mg/mL in serum-free IMDM. In the vehicle-treated and tanshinone IIA–treated cultures, the vehicle and tanshinone IIA at different doses (0, 0.02, 0.05, 0.1, and 0.2 μg/mL) were added. After 8 h of incubation, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution (5 mg/mL, 20 μL per well) was added. After 4 h, the supernatant was discarded and DMSO (200 μL) was added to each well. The suspension was placed on a micro-vibrator for 5 min, and the absorbance (A) was measured at 570 nm with a microplate spectrophotometer (μ-Quant Microplate Spectrophotometer; Bio-tek, Instruments Inc., Winooski, VT). Cell viability relative to the control culture (without AAPH) was calculated by the formula (absorbance of treated sample/absorbance of control sample) × 100%.
Effects of tanshinone IIA on mitochondrial integrity.
Mitochondrial integrity was assessed by monitoring the mitochondrial membrane potential changes using the ApoAlert Mitochondrial Membrane Sensor Kit (BD Clontech, Palo Alto, CA) according to the manufacturer's instructions. JC-1 is a membrane-permeable lipophilic cation fluorescent indicator that is specifically accumulated in the mitochondria as a result of a negative interior potential. The loss of this membrane potential causes shifts in fluorescence emission from red (aggregates) to green (monomers). C17.2 cells in IMDM cultures that contained 10% FCS were subjected to oxidative stress (AAPH, 4 mg/mL) and treated with 0 or 0.1 μg/mL tanshinone IIA. After 8 h, 1 × 106 cells were incubated with 1 mL of diluted JC-1 for 15 min at 37°C in a 5% CO2 incubator. The cells then were washed with 1 mL of incubation buffer and resuspended in 1 mL of buffer. The monomers and aggregates of JC-1 were detected at the FL1 and FL2 channels, respectively.
Statistical analysis.
The data are expressed as means ± SD. Results from in vivo studies were compared by one-way ANOVA. The χ2 test was performed for the mortality assay and functional test. The functional scorings were also compared by the Mann Whitney U test. The paired t test was used to analyze in vitro results. A p < 0.05 was considered to be statistically significant.
RESULTS
Mortality.
The mortality rates of pups in the vehicle-treated and tanshinone IIA–treated groups were 11.0 and 12%, respectively. These pups died during either surgery or hypoxia. All surviving pups in the three treatment groups did not display differences in their total body weights, with mean range of 23.3–24.4 g at 1 wk and 96.4–100 g at 3 wk after surgery. No discernible physiologic and behavioral changes as a result of intoxication were observed.
Brain weight.
At both assessment time points of 1 and 3 wk after hypoxia-ischemia treatment, pups in the vehicle group had decreased weights of the ipsilateral hemisphere (hypoxia-ischemia side), compared with those in the sham-operated group (p < 0.05; Table 1). Pups that were treated with tanshinone IIA for 9 or 16 d had significantly heavier right brain weights and less brain damage (p < 0.05) in comparison with those of the vehicle group. Similar effects were observed in the total brain weight at 3 wk after surgery (p < 0.05). The contralateral brain weights of all groups were similar at both time points. The recovery, however, remained incomplete as demonstrated by the 13.2 and 11.5% reduction of the relative brain weights at 1 and 3 wk, respectively (p < 0.05, tanshinone IIA versus sham-operated).
Cerebral cortical atrophy.
Examination of the external morphology of the brain at 3 wk after the hypoxia-ischemia treatment demonstrated that whereas the sham-operated pups had normal brain size and hemispheric morphology (Fig. 2A), severe atrophy of the right brain occurred in the vehicle-treated animals (Fig. 2B). Tanshinone IIA treatment reduced the extent of atrophy, and the right hemisphere almost returned to its normal size and morphology (Fig. 2C). Coronal sections through the striatum showed that sham-operated pups had normal brain histology (Fig. 2D), whereas vehicle-treated pups that were subjected to hypoxia-ischemia had brain damage at varying extents, ranging from slight neuronal loss to a marked degeneration of the ipsilateral hemisphere (Fig. 2E). Cell death and necrotic nuclei were found scattered in the cerebral cortex, and the degenerative change was especially obvious in the outer layer of the cerebral cortex. Treatment with tanshinone IIA resulted in a remarkable reduction in the severity of injury (Fig. 2F). The neuronal damage was moderate, compared with those of the vehicle group. The hemispheric structure seemed intact, with the only abnormality being the slight reduction in size.

Effects of tanshinone IIA on brain morphology and histology 3 wk after surgery. (A) Normal external brain morphology of a sham-operated pup. (B) Atrophy (arrow) of the ipsilateral hemisphere in a vehicle-treated pup. (C) Reduced injury (arrow) in a tanshinone IIA–treated brain. Histology of the coronal sections through the striatum (hematoxylin and eosin staining) demonstrates the normal brain hemisphere of a sham-operated pup (D), an extensive degenerative area (arrow) in the cortex of the ipsilateral side of a vehicle-treated pup (E), and near-complete recovery (arrow) in a tanshinone IIA–treated animal (F). When the sections were stained with NSE, numerous positive cells (brown staining) were found throughout the cortex, including the sensorimotor area of the forelimb of the sham-operated pup (G). In the same cortical area of the vehicle-treated pups, the number of positive cells was dramatically decreased (H). Treatment with tanshinone IIA increased the number of positive cells (I). Magnification: ×8 in D–F; ×200 in G–I.
Cortical neurons by NSE staining.
At 3 wk after surgery, the number of cortical neurons in the sensorimotor area of the forelimb in the ipsilateral hemisphere of the vehicle-treated animals was significantly decreased in comparison with that of the sham-operated group (p < 0.05; Figs. 2G and H and Fig. 3). Treatment with tanshinone IIA significantly increased the number of neurons (p < 0.05; Figs. 2H and I and Fig. 3), although it remained significantly less than that of the sham-operated pups (p < 0.05; Fig. 3). The numbers of cortical neurons in the contralateral hemispheres were similar in the three treatment groups.

NSE-positive cortical neurons in the sensorimotor area of the forelimb at 3 wk after surgery. The number of the cortical neurons was significantly decreased in the vehicle-treated pups compared with the sham-operated control (*p < 0.05). Tanshinone IIA treatment significantly increased the neuron number, compared with the vehicle-treated group (§p < 0.05; n= 3 for each group).
Postural reflex test.
All sham-operated animals obtained score 0, and none of them had abnormal postural responses of score 1 and score 2 (Fig. 4). The vehicle-treated pups demonstrated abnormal postural responses to the reflex test, and significant numbers of them were classified in scores 1 and 2 (p < 0.05). Some animals in this group displayed normal postural responses. Rat pups in the tanshinone IIA group had improved postural reflex as demonstrated by the increased proportion of those that got a score 0 and reduced numbers with scores 1 and 2 (p < 0.05). Despite the improved functions, the performance of tanshinone IIA–treated pups remained inferior to that of sham-operated pups (p < 0.05). Similar results were observed when these scores were compared using the Mann Whitney U test.

Scores of the postural reflex test at 3 wk after surgery. Sham-operated (▪; n = 23) pups exhibited normal response. Vehicle-treated animals (□; n = 26) had defective response as shown by the low distribution in score 0 and high distribution in scores 1 and 2 (*p < 0.05). Tanshinone-treated pups (□; n = 27) performed better than the vehicle-treated animals in all three scores (§p < 0.05), but their reflex functions remained inferior to the sham-operated animals (**p < 0.05).
Body temperature.
At 4 h after hypoxia, the rectal temperatures of the sham-operated, vehicle-treated, and tanshinone IIA–treated animals were not different (35.7 ± 0.30, 35.6 ± 0.12, and 35.5 ± 0.25°C, respectively; p > 0.05). Increase in daily body temperatures from 2 d before surgery to 7 d after surgery (mean values 35.2°C to 36.3°C) was observed, in parallel with progressing postnatal age of the animals. There were no differences in the daily rectal temperatures between the three groups at all time points measured.
Plasma ORAC.
The ORAC was significantly higher in the plasma of animals that were treated with tanshinone IIA (p < 0.05) when compared with that of the vehicle-treated animals (Fig. 5).

The ORAC in rat plasma. Tanshinone IIA increased the antioxidant capacity of rat plasma when compared with the vehicle-treated group (*p < 0.05; n = 10 for each group).
Effects of tanshinone IIA on viability of C17.2 neural progenitor cells.
After seeding overnight, C17.2 cells grew to a monolayer, and most of them exhibited a flattened, elongated, fibroblast-like morphology (Fig. 6A). After treatment with AAPH for 8 h, which induced oxidative damages to these cells, most of the cells lost their normal fibroblast-like morphology and appeared round in shape. Many of them were detached from their neighboring cells (Fig. 6B). MTT assay showed that the viability of the C17.2 cells was dropped to 38 ± 0.09% of the normal control (Fig. 6D). Tanshinone IIA increased the viability of AAPH-treated cells in a dose-dependent manner at the range of 0.02–0.1 μg/mL (Fig. 6D), with its effect leveling off at the concentration of 0.2 μg/mL. The cell morphology partially recovered in the presence of tanshinone IIA, and many cells appeared normal in shape with flat and extended cytoplasm (Fig. 6C).

In vitro effect of tanshinone IIA on neural progenitor cell line C17.2. C17.2 cells were cultured in the presence of AAPH (4 mg/mL) for 8 h with or without tanshinone IIA (0.1 μg/mL). (A) Without AAPH and tanshinone IIA, cells had normal morphology. (B) After treatment with AAPH, many cells rounded up and were detached from their neighboring cells. (C) Co-incubation with tanshinone IIA effectively protected the cells from AAPH-induced damages with improved morphology. (D) In the MTT assay, tanshinone IIA significantly increased cell viability in a dose-dependent manner (0.02–0.2 μg/mL; * p < 0.05, eight replicated wells in three independent experiments, n = 3). Magnification: ×20 in A–C.
Effects of tanshinone IIA on mitochondrial integrity.
In control untreated C17.2 cells (Fig. 7), the majority of the cell population had intact mitochondria (R1, red fluorescence aggregates), with a low proportion of early apoptotic (R2) and apoptotic (R3, green fluorescence monomers) cells. The treatment with AAPH increased the total apoptotic cells (p = 0.02), and similar trends were demonstrated in the R2 (p = 0.16) and R3 (p = 0.06) populations, compared with the control cultures. The presence of tanshinone IIA significantly decreased early apoptosis (R2, p = 0.03), and a trend was observed in the proportion of total apoptotic cells (R2 + R3, p = 0.09).

The protective effect of tanshinone IIA on mitochondrial damage induced by AAPH. C17.2 cells were cultured in IMDM with 10% FCS for 8 h (A), with AAPH (B) or AAPH and tanshinone IIA (C). After staining with JC-1, dot-blot analysis showed decreased intact cells (R1) and increased early apoptotic (R2) and apoptotic (R3) cells in cultures that contained AAPH (B). The treatment with tanshinone IIA (D) significantly decreased the proportion of early apoptotic cells (R2) in culture (n = 4). *AAPH vs AAPH + tanshinone IIA group, p < 0.05; §AAPH vs control, p < 0.05.
DISCUSSION
The rat model of hypoxia-ischemia brain injury induced at day 7 of postnatal age was established by Levine (13) and modified by Rice et al. (14). At this stage of development, the rat brain is histologically similar to that of a 32- to 34-wk-gestation human infant. The neuropathologic damages observed in these animals are in good agreement with their behavioral deficits (2). Various drugs, such as 21-aminosteroid Tirilazad mesylate (U-74006F) (15), platelet-activating factor antagonist WEB (15), erythropoietin (16), and a herbal compound huperzine A (17), have been used in this model with some degree of success in reducing brain damage. A recent trial has demonstrated that moderate hypothermia treatment improved the most serious outcomes (death and severe motor scores) in human neonatal encephalopathy (18).
In the present study, rat pups were subjected to right common artery occlusion combined with systemic hypoxia. We observed a reproducible pattern of brain injury, in terms of brain weight, histopathology, and number of sensorimotor neurons, in the ipsilateral but not the contralateral hemispheres. The neural damage was persistent at 3 wk after surgery and was translated to a compromised response to the postural reflex challenge. To investigate the protective effects of tanshinone IIA on this animal model, we adopted a pretreatment protocol as reported by several research teams (19–21). Our data showed that rat pups that were treated with tanshinone IIA 2 d before the induction of ischemia hypoxia for 16 d had marked recoveries in the ipsilateral hemispheres in all studied parameters. More significant, these animals scored better in the functional test, compared with the vehicle-treated group. To our knowledge, these data provided the first evidence that tanshinone IIA could confer marked histopathologic and behavioral protection against neonatal hypoxia-ischemia brain injury in vivo. Our data are in line with those of Lam et al. (22), who demonstrated that tanshinone IIA at 16 mg/kg had the ability to reduce neuronal damage in an adult mouse model subjected to transient focal cerebral ischemia. One limitation of our model is that pretreating hypoxia-ischemia patients is unlikely to be possible in a clinical setting. Another concern is the lack of information on the pharmacokinetics of tanshinone IIA and its metabolites on neonatal rats. Lam et al. (22) reported that in brain tissues of adult mice, the level of tanshinone IIA peaked at 60 min and slowly declined to 50% after 5–6 h. Considering the effectiveness of tanshinone IIA on neuroprotection, issues on the timing of administration and therapeutic dose deserve further investigation.
Our findings raise the question as to how tanshinone IIA protects the brain from hypoxia-ischemia brain injury. Several protective mechanisms have been proposed. Studies in the hypoxia-ischemia model showed that some cholinesterase inhibitors such as HupA and donepezil had neuroprotective effects (17,23). Tanshinone IIA is an AchE inhibitor that has the potential to penetrate the blood–brain barrier (7). Lam et al. (22) reported that tanshinone IIA can readily penetrate the brain, reaching a peak concentration 60 min after i.p. injection to adult mice. In neonatal rats, excessive glutamate release during and after a hypoxia-ischemia insult might activate N-methyl-d-aspartate channel receptors, leading to further influx of calcium into cells, thus inducing neuronal death (24,25). Tanshinone IIA has been demonstrated to reduce glutamate-induced cytotoxicity by antagonizing cerebral NMDA receptors (6,26). Changes in body temperature are known to modulate neuronal damage in animal experiments and in clinical trials (27). Hypothermia might suppress the release of excitotoxins, nitric oxide, and free radicals during reperfusion (28,29). In line with another report (11), we demonstrated changes in body temperature with increasing postnatal age of the animals. We did not detect significant changes in the rectal temperatures of the three groups at 4 and 24 h after hypoxia, indicating that the neuroprotective effect of tanshinone IIA was possibly not mediated by hypothermia. However, one has to take into consideration that rectal temperature measurements, although correlating well with brain temperatures (30,31), might not be sensitive enough to detect small differences of the latter (29). In addition, the animals were actively warmed in the incubator before rectal temperature measurement, and there is the possibility that this procedure might have masked a mild decrease in body temperature induced by the treatment. Oxygen radicals are markedly produced after a hypoxic-ischemic insult. Several lines of evidence have shown that tanshinone IIA can reverse the abnormalities induced by free radical injury as previously described by our laboratory and other researchers (26,32). It could also protect the mitochondrial membrane from ischemia-reperfusion injuries and lipid peroxidation (33). In this study, we demonstrated that tanshinone IIA significantly improved the antioxidant capacity in rat plasma and protected the C17.2 progenitors against AAPH-induced cell death. We further confirmed that tanshinone IIA exerted a protective effect on mitochondrial membrane potential and decreased cell death secondary to oxidative stress.
The contribution of tanshinone IIA–induced antioxidant capacity to the protection against brain damage in our model remains to be investigated further. Our data and other evidence have shown an effective neuroprotective action by tanshinone IIA, possibly mediated by multiple pathways in hypoxic-ischemic neonatal animals. To date, the applications of neuroprotective agents, including tanshinone IIA, have not achieved total recovery of the damaged brain in various animal models. It thus is important to investigate the mechanism of each agent and the possibility of an integrated treatment strategy in maximizing the outcome. The present study suggests that pretreatment with tanshinone IIA is partially protective for hypoxia-ischemia in the immature rat. As the treatment protocol spanned a very wide period before and after the hypoxic-ischemic insult, it is uncertain which treatment times were optimal for protection or whether rescue treatment is equally effective. We have confirmed that tanshinone IIA has antioxidant effects that may be beneficial, although other potentially relevant pathways might be involved. This study, however, did not purposefully evaluate whether brain cooling secondary to the treatment may have contributed to neuroprotection. Further studies are required to address these issues to verify the clinical potential of tanshinone IIA in helping to alleviate brain damage.
Abbreviations
- AAPH:
-
2,2′-azobis (2-amidino propane hydrochloride)
- AchE:
-
acetylcholinesterase
- HIE:
-
hypoxia-ischemia encephalopathy
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NSE:
-
neuron-specific enolase
- ORAC:
-
oxygen radical absorbance capacity
References
Buonocore G, Perrone S, Bracci R 2001 Free radicals and brain damage in the newborn. Biol Neonate 79: 180–186
Feng Y, Shi W, Huang M, LeBlanc MH 2003 Oxypurinol administration fails to prevent hypoxic-ischemic brain injury in neonatal rats. Brain Res Bull 59: 453–457
Zhu XP 1998 Chinese Material Medica: Chemistry, Pharmacology and Applications. Harwood Academic Publisher, Amsterdam, pp 459–463
Niu XL, Ichimori K, Yang X, Hirota Y, Hoshiai K, Li M, Nakazawa H 2000 Tanshinone II-A inhibits low density lipoprotein oxidation in vitro. Free Radic Res 33: 305–312
Zhou GY, Zhao BL, Hou JW, Ma GE, Xin WJ 1999 Protective effects of sodium tanshinone IIA sulphonate against adriamycin-induced lipid peroxidation in mice hearts in vivo and in vitro. Pharmacol Res 40: 487–491
Ji ZN, Liu GQ 2001 Inhibition of serum deprivation-induced PC12 cell apoptosis by tanshinone II A. Acta Pharmacol Sin 22: 459–462
Ren Y, Houghton PJ, Hider RC, Howes MJ 2004 Novel diterpenoid acetylcholinesterase inhibitors from Salvia miltiorrhiza. Planta Med 70: 201–204
Riederer BM, Berbel P, Innocenti GM 2004 Neurons in the corpus callosum of the cat during postnatal development. Eur J Neurosci 19: 2039–2046
Paxinox GWC 1986 The Rat Brain in Stereotaxic Coordination. Academic Press Inc, San Diego, pp 15–23
Calvert JW, Yin W, Patel M, Badr A, Mychaskiw G, Parent AD, Zhang JH 2002 Hyperbaric oxygenation prevented brain injury induced by hypoxia-ischemia in a neonatal rat model. Brain Res 951: 1–8
Colman AS, Miller JH 2002 Lack of involvement of mu(1) opioid receptors in dermorphin-induced inhibition of hypoxic and hypercapnic ventilation in rat pups. Respir Physiol Neurobiol 131: 199–212
Wang CC, Chu CY, Chu KO, Choy KW, Khaw KS, Rogers MS, Pang CP 2004 Trolox-equivalent antioxidant capacity assay versus oxygen radical absorbance capacity assay in plasma. Clin Chem 50: 952–954
Levine S 1960 Anoxic-ischemic encephalopathy in rats. Am J Pathol 36: 1–17
Rice JE 3rd, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141
Viswanath M, Palmer C, Roberts RL 2000 Reduction of hypoxic-ischemic brain swelling in the neonatal rat with PAF antagonist WEB 2170: lack of long-term protection. Pediatr Res 48: 109–113
Kumral A, Ozer E, Yilmaz O, Akhisaroglu M, Gokmen N, Duman N, Ulukus C, Genc S, Ozkan H 2003 Neuroprotective effect of erythropoietin on hypoxic-ischemic brain injury in neonatal rats. Biol Neonate 83: 224–228
Wang LS, Zhou J, Shao XM, Tang XC 2002 Huperzine A attenuates cognitive deficits and brain injury in neonatal rats after hypoxia-ischemia. Brain Res 949: 162–170
Eicher DJ, Wagner CL, Katikaneni LP, Hulsey TC, Bass WT, Kaufman DA, Horgan MJ, Languani S, Bhatia JJ, Givelichian LM, Sankaran K, Yager JY 2005 Moderate hypothermia in neonatal encephalopathy: efficacy outcomes. Pediatr Neurol 32: 11–17
Ekert P, MacLusky N, Luo XP, Lehotay DC, Smith B, Post M, Tanswell AK 1997 Dexamethasone prevents apoptosis in a neonatal rat model of hypoxic-ischemic encephalopathy (HIE) by a reactive oxygen species-independent mechanism. Brain Res 747: 9–17
Galvin KA, Oorschot DE 2003 Continuous low-dose treatment with brain-derived neurotrophic factor or neurotrophin-3 protects striatal medium spiny neurons from mild neonatal hypoxia/ischemia: a stereological study. Neuroscience 118: 1023–1032
Wang X, Zhu C, Wang X, Gerwien JG, Schrattenholz A, Sandberg M, Leist M, Blomgren K 2004 The nonerythropoietic asialoerythropoietin protects against neonatal hypoxia-ischemia as potently as erythropoietin. J Neurochem 91: 900–910
Lam BY, Lo AC, Sun X, Luo HW, Chung SK, Sucher NJ 2003 Neuroprotective effects of tanshinones in transient focal cerebral ischemia in mice. Phytomedicine 10: 286–291
Zhang HY, Tang XC 2000 Huperzine B, a novel acetylcholinesterase inhibitor, attenuates hydrogen peroxide induced injury in PC12 cells. Neurosci Lett 292: 41–44
Abdel-Hamid KM, Tymianski M 1997 Mechanisms and effects of intracellular calcium buffering on neuronal survival in organotypic hippocampal cultures exposed to anoxia/aglycemia or to excitotoxins. J Neurosci 17: 3538–3553
Egawa-Tsuzuki T, Ohno M, Tanaka N, Takeuchi Y, Uramoto H, Faigle R, Funa K, Ishii Y, Sasahara M 2004 The PDGF B-chain is involved in the ontogenic susceptibility of the developing rat brain to NMDA toxicity. Exp Neurol 186: 89–98
Wang AM, Sha SH, Lesniak W, Schacht J 2003 Tanshinone (Salviae miltiorrhizae extract) preparations attenuate aminoglycoside-induced free radical formation in vitro and ototoxicity in vivo. Antimicrob Agents Chemother 47: 1836–1841
Asai S, Zhao H, Kohno T, Takahashi Y, Nagata T, Ishikawa K 2000 Quantitative evaluation of extracellular glutamate concentration in postischemic glutamate re-uptake, dependent on brain temperature, in the rat following severe global brain ischemia. Brain Res 864: 60–68
Battin MR, Dezoete JA, Gunn TR, Gluckman PD, Gunn AJ 2001 Neurodevelopmental outcome of infants treated with head cooling and mild hypothermia after perinatal asphyxia. Pediatrics 107: 480–484
DeBow SB, Clark DL, MacLellan CL, Colbourne F 2003 Incomplete assessment of experimental cytoprotectants in rodent ischemia studies. Can J Neurol Sci 30: 368–374
Bona E, Hagberg H, Loberg EM, Bagenholm R, Thoresen M 1998 Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: short- and long-term outcome. Pediatr Res 43: 738–745
Thoresen M, Bagenholm R, Loberg EM, Apricena F, Kjellmer I 1996 Posthypoxic cooling of neonatal rats provides protection against brain injury. Arch Dis Child Fetal Neonatal Ed 74: F3–F9
Wu TW, Zeng LH, Fung KP, Wu J, Pang H, Grey AA, Weisel RD, Wang JY 1993 Effect of sodium tanshinone IIA sulfonate in the rabbit myocardium and on human cardiomyocytes and vascular endothelial cells. Biochem Pharmacol 46: 2327–2332
Zhao BL, Jiang W, Zhao Y, Hou JW, Xin WJ 1996 Scavenging effects of salvia miltiorrhiza on free radicals and its protection for myocardial mitochondrial membranes from ischemia-reperfusion injury. Biochem Mol Biol Int 38: 1171–1182
Acknowledgements
We thank David Lo, Department of Anatomical and Cellular Pathology, The Chinese University of Hong Kong, for providing practical advice and support. We also thank Dr. David Walsh, Department of Anatomy, The University of New South Wales, Australia, for providing the C17.2 cell line.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by an Area of Excellence grant from the University Grants Committee of the Hong Kong SAR for the project “Chinese Medicine Research and Further Development” (Ref no. AoE/B-10/01).
Rights and permissions
About this article
Cite this article
Xia, W., Yang, M., Fok, T. et al. Partial Neuroprotective Effect of Pretreatment with Tanshinone IIA on Neonatal Hypoxia-Ischemia Brain Damage. Pediatr Res 58, 784–790 (2005). https://doi.org/10.1203/01.PDR.0000180550.99162.BC
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000180550.99162.BC
This article is cited by
-
Tanshinone I alleviates motor and cognitive impairments via suppressing oxidative stress in the neonatal rats after hypoxic-ischemic brain damage
Molecular Brain (2017)
-
Tanshinone IIA Rescued the Impairments of Primary Hippocampal Neurons Induced by BV2 Microglial Over-Activation
Neurochemical Research (2015)
-
Protective Effect of Tanshinone IIA Against Infarct Size and Increased HMGB1, NFκB, GFAP and Apoptosis Consequent to Transient Middle Cerebral Artery Occlusion
Neurochemical Research (2014)
-
Tanshinone IIA activates calcium-dependent apoptosis signaling pathway in human hepatoma cells
Journal of Natural Medicines (2012)
-
Anti-inflammatory effects of Chinese medicinal herbs on cerebral ischemia
Chinese Medicine (2011)
