
Abstract
Age-related macular degeneration (AMD) is characterized by pathological changes in the retinal pigment epithelium (RPE) and loss of photoreceptors. Growing evidence has demonstrated that reactive microglial cells trigger RPE dysfunction and loss of photoreceptors, and inflammasome pathways and complement activation contribute to AMD pathogenesis. We and others have previously shown that adenosine A2A receptor (A2AR) blockade prevents microglia-mediated neuroinflammatory processes and mediates protection to the retina. However, it is still unknown whether blocking A2AR in microglia protects against the pathological features of AMD. Herein, we show that an A2AR antagonist, SCH58261, prevents the upregulation of the expression of pro-inflammatory mediators and the alterations in the complement system triggered by an inflammatory challenge in human microglial cells. Furthermore, blockade of A2AR in microglia decreases the inflammatory response, as well as complement and inflammasome activation, in ARPE-19 cells exposed to conditioned medium of activated microglia. Finally, we also show that blocking A2AR in human microglia increases the clearance of apoptotic photoreceptors. This study opens the possibility of using selective A2AR antagonists in therapy for AMD, by modulating the interplay between microglia, RPE and photoreceptors.
Similar content being viewed by others


Introduction
Age-related macular degeneration (AMD) is a major cause of vision loss worldwide and the leading cause of blindness in the elderly in developed countries1. The accumulation of cellular debris as drusen in the subretinal space, beneath the retinal pigment epithelium (RPE), is a main feature of early and intermediate AMD, which can progress to photoreceptor loss, representing the so-called dry AMD. Wet AMD is characterized by choroidal neovascularization, with new blood vessels arising from the choroid through the RPE layer into the outer retina, leading to photoreceptor dysfunction2.
Growing evidence supports a critical role of the immune system in AMD3,4. Recruitment of microglial cells, the immunocompetent cells of the central nervous system (CNS), has been associated with the progression and severity of AMD5,6. Indeed, microglia reactivity is involved in the recruitment and activation of complement system7,8 and inflammasome pathways9, which may contribute to photoreceptor degeneration10.
The complement system and inflammasome pathways, key innate immune defenses against inflammation, orchestrate critical responses in the CNS. Several studies reported the involvement of the complement cascades in both acute and chronic disease conditions11,12. Genetic variations of several complement-related genes are associated with AMD pathogenesis13,14,15,16. Similarly, elevated levels of complement factors have been detected in the plasma and aqueous humor of AMD patients17,18. Moreover, complement proteins are present in drusen. Notably, A2E, a RPE lipofuscin and drusen component, triggers microglia reactivity and complement activation by increasing complement factor B (CFB) and decreasing complement factor H (CFH) expression, leading to photoreceptor degeneration8.
Inflammasome is a large multiprotein complex that takes part in the innate immune response, promoting interleukin 1β (IL-1β) and interleukin 18 (IL-18) maturation, through caspase-1-mediated cell death, known as pyroptosis19. AMD-associated inflammasome activation has been widely described. There is evidence showing the ability of drusen to induce the activation of inflammasome, namely NLRP3 inflammasome, in retinal mononuclear cells20. Inflammasome has been detected in the RPE, in both dry and wet AMD, and might contribute to RPE degeneration21,22. Interestingly, factors released from reactive microglia can also induce the activation of NLRP3 inflammasome in ARPE-19 cells9.
Adenosine is a crucial neuromodulator in the CNS, and blockade of adenosine A2A receptor (A2AR) affords robust neuroprotection against different neurodegenerative conditions23. We have recently reported that A2AR blockade prevents retinal microglia reactivity and the associated neuroinflammatory response, conferring protection to retinal ganglion cells, in experimental models of glaucoma24,25,26. Still, the impact of A2AR modulation on the complement system and its potential beneficial effects on AMD remain to be elucidated.
Hence, the main aim of this work was to evaluate the outcome of A2AR blockade in the microglial complement system. Moreover, we evaluated the potential beneficial effects of blocking A2AR in human microglial cells on the inflammatory response of ARPE-19 cells, inflammasome activation and photoreceptor loss.
Results
Inflammatory stimuli increase the release of adenosine and the density of A2AR in human microglial cells
It is well known that A2AR is up-regulated under noxious conditions24,25,27. Therefore, we firstly investigated whether the incubation of human microglial cells with Zymosan and phorbol myristate acetate (PMA), two potent pro-inflammatory stimuli28, would be able to increase the levels of extracellular adenosine, which then could boost A2AR activation, and affect A2AR expression.
In control conditions, the concentration of extracellular adenosine was 308 ± 152 pmol/μL (n = 7). The incubation with Zymosan + PMA for 6 h or 24 h increased the concentration of adenosine to 593.7 ± 205.4 or 941.1 ± 205 ρmol/μL, respectively (n = 4–7; Fig. 1A). Also, A2AR mRNA expression was significantly up-regulated (1.3-fold increase, n = 5) after 6 h incubation with Zymosan + PMA when compared with the control condition (Fig. 1B). Moreover, the incubation of human microglia with Zymosan + PMA for 6 h or 24 h increased the immunoreactivity of A2AR (159 ± 12.1 and 128.6 ± 8.7% of control, respectively; n = 4; Fig. 1C and D). These results demonstrate that pro-inflammatory conditions can trigger an increase in the release of adenosine from human microglia that may then signal through the upregulated A2AR.

Inflammatory stimulus increases the release of adenosine and the expression and density of A2AR in human microglia. Human immortalized microglial cells were challenged with Zymosan + PMA for 6 h or 24 h. (A) Adenosine concentration in culture supernatants. Data are expressed as percentage of control and represent the mean ± SEM of 3–7 independent experiments (presented with the scatterplot with the individual data points). (B) The mRNA levels of A2AR were assessed by qPCR relatively to hprt (housekeeping gene). Results are normalized to control from 3–7 independent experiments. Mann-Whitney test. (C) Microglial cells (red; phalloidin) were immunostained for A2AR (green). Nuclei were stained with DAPI (bue). Images are representative of 4–7 independent experiments. (D) Densitometric analysis of A2AR immunoreactivity. Data represent the mean ± SEM, with the scatterplot with the individual data points. Results are expressed as percentage of control and were obtained from 4–7 independent experiments. Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
Treatment with an A2AR antagonist reduces the inflammatory response in human microglia
Considering that A2AR expression and density increased in human microglia exposed to Zymosan + PMA and that a selective antagonist of A2AR prevented the neuroinflammatory response24, we next investigated whether A2AR blockade could influence human microglia reactivity.
The incubation of human microglia with Zymosan + PMA for 6 h up-regulated the mRNA expression of the inflammatory markers CCL2, TNF and IL-8 (n = 7). Pre-treatment with 50 nM SCH58261, the A2AR antagonist, reduced the stimulatory effect of Zymosan + PMA (n = 7; Fig. 2A–C). SCH58261 by itself did not alter the expression of these pro-inflammatory markers.

A2AR blockade reduces the inflammatory response in human microglia. Human microglial cells were challenged with Zymosan + PMA in the absence or presence of 50 nM SCH58261. The mRNA levels of CCL2 (A), TNF (B) and IL-8 (C) were assessed by qPCR. Results are normalized to control from 5–7 independent experiments. (D) The release of NO was assessed by the Griess reaction assay in culture supernatants. Results are expressed in percentage of control and presented as the mean ± SEM of 4–7 independent experiments (the individual data points for each condition are also presented). (E) Human microglial cells were incubated with Zymosan + PMA for 2 h in the absence or presence of SCH58261, before adding polystyrene beads for 6 h (white arrows). Images were acquired and the phagocytic efficiency was determined (F). Results are expressed as percentage of control and presented as the mean ± SEM of 5 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
The incubation of microglia with Zymosan + PMA for 6 h or 24 h also increased the release of NO by human microglia, as determined by the nitrite concentration in culture supernatants (223.9 ± 37.1 and 298.3 ± 63.3% of control, respectively, n = 6–8). The blockade of A2AR prevented the increase in NO induced by Zymosan + PMA (118.7 ± 12.3 and 116.8 ± 20.8% of control for 6 h and 24 h, respectively) (Fig. 2D).
Since reactive microglia are characterized by the ability to phagocytose foreign material, we determined the phagocytic efficiency of human microglia following exposure to Zymosan + PMA, in the absence or presence of the A2AR antagonist. The incubation of human microglia with Zymosan + PMA significantly increased the number of phagocytosed polystryrene microparticles (206.9 ± 36% of control, n = 5). This effect was partially reduced by the pre-treatment with SCH58261 (138 ± 20.6% of control, n = 5). Taken together, these results indicate that A2AR blockade prevents the shift of human microglia into a reactive phenotype.
Blockade of A2AR prevents the upregulation of complement components in human microglia induced by Zymosan + PMA
Complement system modulation is an emerging therapeutic strategy for AMD29. Although A2AR blockade controls microglia reactivity23, the direct effects of A2AR blockade on the complement system of microglia are unknown. Hence, we addressed this question in human immortalized microglial cells to evaluate whether the A2AR antagonist could modulate the expression of complement factor in microglial cells.
Incubation of human microglia with Zymosan + PMA for 6 h increased the expression of complement components associated with the activation of the cascade: complement C3 (Fig. 3A), CFB (Fig. 3B), CD91 (also known as C1qr; Fig. 3C), and complement C5a receptor 1 (C5AR1) (Fig. 3D). When the cells were pre-treated with the A2AR selective antagonist, the upregulation of these components was prevented. Concerning the inhibitory complement components, Zymosan + PMA decreased CFH (Fig. 3E), CFI (Fig. 3F), CD46 (also known as MCP; Fig. 3G), and CD55 (also known as DAF; Fig. 3H), and these inhibitory effects were absent in the presence of the A2AR antagonist. No alterations were observed in the mRNA levels of C1s (Fig. 3I). Treatment of human microglia with SCH58261 did not change the expression of complement components.

Blockade of A2AR prevents the alterations in complement cascade components in reactive human microglial cells. Human microglial cells were challenged with Zymosan + PMA in the presence or absence of 50 nM SCH58261. The expression levels of complement cascade components C3 (A), CFB (B), CFH (C), CFI (D), CD93 (E), C1s (F), C5AR1 (G), CD46 (H) and CD55 (I) were assessed by qPCR. Results are normalized to control and presented as the mean ± SEM of 5–7 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
A2AR blockade prevents microglia reactivity induced by exposure to ARPE-19 debris
Drusen components can induce microglia reactivity and complement activation8. Therefore, we evaluated whether A2AR blockade could prevent the reactivity of human microglia induced by RPE debris, which were used to mimic drusen components.
We first evaluated the ability of different concentrations and times of exposure of ARPE-19 debris to induce an inflammatory response in microglial cells (Sup. Figure 1). All challenges with ARPE-19 cell debris induced up-regulation of CCL2 and IL-8 mRNA expression, but not TNF. We choose 5 μg/μL ARPE-19 cell debris since it was the shorter time point and lower concentration.
Then, human microglial cells were challenged with RPE debris (5 μg/μL of ARPE-19 cell debris for 6 h) 45 minutes after incubation with A2AR antagonist. The A2AR antagonist partially prevented the increase in the mRNA expression of CCL2 (Fig. 4A) and IL-8 (Fig. 4B). The mRNA expression levels of TNF were not altered by ARPE-19 cell debris in the absence or presence of SCH58261 (Fig. 4C).

A2AR blockade prevents ARPE-19 debris-induced microglia inflammatory response. Human microglia were exposed to 5 µg ARPE-19 apoptotic cell debris for 6 h in the absence or presence of the A2AR antagonist. The mRNA expression levels of CCL2 (A), TNF (B) and IL-8 (C) were assessed by qPCR. Results are normalized to control and presented as the mean ± SEM of 5 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
A2AR blockade prevents changes in complement components in microglia induced by exposure to ARPE-19 debris
We then evaluated whether the ARPE-19 cell debris were able to induce alterations in the expression of complement components of human microglia. When human microglia were incubated for 6 h with ARPE-19 cell debris the mRNA expression of C3 (Fig. 5A; n = 5), CFB (Fig. 5B; n = 5) and C5AR1 (Fig. 5C; n = 5) increased and this effect was prevented by the blockade of A2AR. The transcript levels of the inhibitory components CFH (Fig. 5D), CFI (Fig. 5E), CD46 (Fig. 5F) and CD55 (Fig. 5G) in human microglia presented a tendency to decrease upon exposure to ARPE-19 cell debris (n = 5). Pre-treatment with SCH58261 partially prevented these alterations (n = 5).

A2AR blockade prevents microglia complement cascade alterations induced by ARPE-19 debris. Human microglia were exposed to 5 µg/mL ARPE-19 apoptotic cell debris for 6 h in the presence or absence of A2AR antagonist. The mRNA expression levels of complement cascade components C3 (A), CFB (B), CFH (C), CFI (D), C5AR1 (E), CD46 (F) and CD55 (G) were assessed by qPCR. Results are normalized to control and presented as the mean ± SEM of 5 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
The blockade of A2AR in microglia prevents an inflammatory response and complement activation in ARPE-19 cells induced by exposure to microglia conditioned medium
We have previously reported that the exposure of ARPE-19 cells to conditioned medium from reactive microglia increases the expression of pro-inflammatory markers9. In addition, we demonstrated that the blockade of A2AR prevents retinal inflammation24,25. Therefore, we hypothesized that if A2AR blockade halts microglia reactivity, this would be sufficient to limit RPE responses to microglia-conditioned medium (MCM).
The mRNA expression of CCL2 and IL-8 (Fig. 6A and B) was up-regulated in ARPE-19 cells (2.98- and 11.7-fold increase comparing with the control MCM; n = 6) after exposure to MCM from Zymosan + PMA-treated cells. The incubation of ARPE-19 cells with cell culture supernatants from microglia pre-treated with A2AR antagonist before Zymosan + PMA exposure did not change the expression of CCL2 and IL-8, compared with the control.

Blockade of A2AR in microglia prevents the inflammatory response and complement cascade alterations in ARPE-19 cells. Microglia were pretreated with 50 nM SCH58261 and then challenged with Zymosan + PMA for 6 h. MCM was added to ARPE-19 cells for 48 h. The mRNA expression of CCL2 (A) IL-8 (B), CFB (C), CFH (D), and CD55 (E) was assessed by qPCR. Results are normalized to control and presented as the mean ± SEM of 6 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
The exposure of ARPE-19 cells to MCM from cells exposed to Zymosan + PMA increased the mRNA expression of CFB (2.15-fold increase relative to control MCM; n = 6; Fig. 6C) and downregulated CFH and CD55 mRNA expression (0.71- and 0.9-fold change, respectively; Fig. 6D and E), suggesting the activation of the complement cascade in ARPE-19 cells. The A2AR blockade in microglial cells prevented the alterations in CFB, CFH, and CD55 detected in RPE cells. Also, MCM obtained from microglial cells exposed to SCH58261 did not alter the expression of these markers. In order to exclude a potential carryover effect, ARPE-19 cells were directly incubated with Zymosan + PMA, but no changes in the mRNA expression of these markers were detected (Supplementary Fig. 2). Importantly, ARPE-19 cells are not immunoreactive for A2AR (Supplementary Fig. 3) and do not express A2AR mRNA (data not shown).
A2AR blockade in microglia prevents inflammasome activation in RPE cells
We have recently described a role for reactive microglia in the activation of RPE inflammasome9. Therefore, we investigated whether blocking A2AR in microglia could prevent the activation of inflammasome in ARPE-19 cells triggered by exposure to MCM collected from microglia cultures exposed to pro-inflammatory conditions.
When ARPE-19 cells were exposed to MCM from non-stimulated microglial cell cultures, no significant alterations were detected in the expression of several components (caspase-1, IL-1β and IL-18) of the inflammasome pathway components of ARPE-19 cells. However, in ARPE-19 cells exposed to MCM from microglial cell cultures exposed to Zymosan + PMA there was an up-regulation of the mRNA expression levels of caspase 1 (CASP1; Fig. 7A), IL-1β (Fig. 7B) and IL-18 (Fig. 7C). Importantly, pre-treatment of human microglia with the A2AR antagonist prevented this effect (Fig. 7). MCM obtained from microglial cells exposed to SCH58261 did not alter the expression of these markers.

A2AR blockade in reactive human microglia prevents inflammasome activation in ARPE-19 cells. Microglia were pretreated with 50 nM SCH58261 and then challenged with Zymosan + PMA for 6 h. MCM was added to ARPE-19 cells for 48 h. The mRNA expression of caspase-1 (A) IL-1β (B), and IL-18 (C) was assessed by qPCR. Results are normalized to control and presented as the mean ± SEM of 6 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
Blockade of A2AR increases microglia clearance of photoreceptor apoptotic debris
An effective phagocytic clearance of photoreceptor apoptotic debris by microglia is fundamental for the maintenance of retinal integrity30,31. Hence, we next investigated the impact of A2AR blockade on the clearance of photoreceptor debris by microglia. For this purpose, control or Zymosan + PMA-treated human microglia, in the absence or presence of SCH58261, were exposed to the 661 W photoreceptor cell line apoptotic debris and the phagocytic efficiency was determined (Fig. 8A). Blockade of A2AR significantly increased the phagocytic efficiency of microglia (137.8 ± 6.1% of non-treated, n = 4; Fig. 8B), suggesting an increase in the clearance of apoptotic material from photoreceptors.

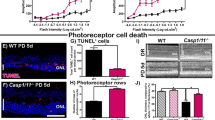
Blockade of A2AR increases the clearance of apoptotic photoreceptors debris by human microglial cells and prevents microglia-mediated increase of photoreceptor apoptosis. Human microglial cells (green) were exposed to CellTracker CM-DiI-labelled photoreceptors (red) in the absence or presence of SCH58261 for 6 h. Nuclei were counterstained with DAPI (blue). Representative images are depicted in (A) and the phagocytic efficiency was determined (B). Results are expressed as percentage of control and presented as the mean ± SEM of 4 independent experiments (the individual data points for each condition are also presented). (C) Photoreceptors were exposed to microglia conditioned medium for 48 h and the activity of caspase 3/7 was determined. Results are expressed as percentage of control and presented as the mean ± SEM of 3 independent experiments (the individual data points for each condition are also presented). Kruskall-Wallis test, followed by Dunn’s multiple comparison test.
Control of microglia reactivity by the A2AR antagonist prevents 661 W photoreceptor cell death
In order to further explore the influence of blocking A2AR on microglial-induced neurotoxicity, 661 W photoreceptor-like cells were incubated for 48 h with untreated or Zymosan + PMA-treated MCM, in the absence or presence of SCH58261. Thereafter, the activity of caspases 3/7 was determined to estimate the apoptotic activity in 661 W cells (Fig. 8C). 661 W photoreceptors cultured with MCM from reactive cells showed increased caspase 3/7 activity (289.5 ± 54% of the control, n = 3). The activity of caspase 3/7 was significantly reduced when microglia were pre-treated with SCH58261 (115.6 ± 6% of control; n = 3), suggesting that the control of microglia reactivity due to A2AR blockade protects against the neurotoxic effects of microglia.
Discussion
In the present work, using in vitro models, we show for the first time that the blockade of A2AR in microglia impacts the expression of the complement system components in microglial cells. Importantly, the blockade of A2AR in microglia also reduces the inflammatory response in RPE cells, as well as the activation of the complement cascade and inflammasome. Likewise, blocking A2AR in microglia reduces photoreceptor cell death elicited by exposure to the supernatants of reactive microglia and increases the clearance of photoreceptor apoptotic material (Fig. 9).

The blockade of A2AR in microglial cells prevent the loss of photoreceptors and reduced RPE dysfunction. Changes in homeostasis, trigger microglia reactivity that up-regulate inflammatory mediators and activate the complement cascade. This induce an inflammatory response is accompanied by complement and inflammasome activation on RPE cells and increased photoreceptor cell death. When microglial cells are treated with a selective antagonist of A2AR, the microglial immune response is reduced (effects indicated with red symbols). Importantly, the blockade of A2AR in microglia reduces the inflammatory response in RPE cells, as well as the activation of the complement cascade and inflammasome. Likewise, blocking A2AR in microglia reduces photoreceptor cell death elicited by exposure to the supernatants of reactive microglia and increases the clearance of photoreceptor apoptotic material.
Increased expression of A2AR, as occurs in retinal microglia24, has been associated with several CNS noxious conditions23,32. It has been shown that activation of A2AR promotes microglia-mediated inflammatory responses, by potentiating NO release by activated microglia33. Here, by exposing human microglial cells to Zymosan and PMA, known to induce microglia activation by mimicking pathogenic infection28,34, we report an increase in the concentration of extracellular adenosine as well as in the expression of A2AR in these cells, suggesting that A2AR signaling might be boosted in microglia thus promoting inflammation.
Parallel to what we have previously described for rat microglia24, blockade of A2AR in human microglial cells prevents the reactive microglia phenotype, as indicated by the decrease in the expression of inflammatory mediators (CCL2, TNF and IL-8) and the reduced release of NO and phagocytic efficiency. Importantly, the exposure of human microglia to the apoptotic human ARPE-19 cell debris that resemble drusen material35 also up-regulated the expression of inflammatory mediators (CCL2 and IL-8), which was also prevented by the blockade of A2AR. Interestingly TNF expression was increased in microglia incubated with Zymosan + PMA but not with ARPE-19 debris, which suggests that different signaling pathways are elicited by the different noxious conditions. Several signaling pathways have been demonstrated to regulate TNF expression, such as nuclear factor kappa B (NF-κB), as well as members of the mitogen activated protein kinase family, including ERK1/2 (extracellular receptor activated kinases 1/2), p38 and c-jun N-terminal kinase (JNK)36. Future studies can focus on the signaling pathways activated in each condition.
Exposure of human microglia to an inflammatory stimulus or to ARPE-19 cell debris also induced the activation of the complement cascade by up-regulating the expression of C3, CFB, C1q and C5AR1 and down-regulating CFH, CFI, CD46 and CD93. Previous studies reported that the up-regulation of C3 in microglia is associated with the pathogenesis of age-related retinal diseases7,37,38. Furthermore, when exposed to A2E, a drusen component, microglial cells up-regulate the expression of CFB and down-regulate the levels of CFH, a process that has been associated with photoreceptor loss8. Likewise, increased plasma levels and aqueous humor concentrations of complement components in AMD patients have been associated with low responsiveness to current therapies17,18. Therefore, supplemental therapies targeting complement system activation have been suggested as novel strategies for AMD management17,29,39. The selective antagonist of A2AR was able to prevent the alterations occurring in the complement system components in reactive human microglia, suggesting that A2AR not only modulates microglia pro-inflammatory responses, but can also modulate the activation of complement cascade.
Several studies have suggested a crosstalk between signals arising from reactive microglial cells and RPE that regulate the expression and secretion of pro-inflammatory, chemotactic, and pro-angiogenic molecules9,40,41. This crosstalk may be also associated with drusen formation as a product of local inflammatory responses42. Here we show that reactive microglial cells may also trigger the activation of the complement system in ARPE-19 cells, further corroborating the role of microglia on RPE dysfunction. Again, the blockade of A2AR in microglia had the ability to prevent the activation of the complement system in RPE, suggesting that the control of microglia reactivity is sufficient to prevent microglia-induced effects on RPE.
NLPR3 inflammasome activation in the RPE has been described in AMD models21,43,44 and has been proposed to be a causal factor for RPE dysfunction and degeneration45. In accordance with our recent report9, we detected increased expression of caspase-1, IL-1β and IL-8 in ARPE-19 cells, supporting the idea that reactive microglial cells release factors that induce the activation of NLRP3 inflammasome in ARPE-19 cells. Remarkably, when microglial cells were treated with SCH58261, the activation of inflammasome in ARPE-19 was abolished, suggesting that the blockade of A2AR in microglia modulates inflammasome activation in RPE. In macrophages, adenosine modulates the inflammasome activation through A2AR46, which is in accordance with our data, where we found that blocking this receptor prevents the up-regulation of NLRP3 and IL-18 in reactive microglial cells (Supplementary Figure 4). However, there were no previous reports about the modulation of RPE inflammasome by A2AR. Also, we have previously described that reactive microglia-released material might induce activation of autophagy in RPE9. Although we have not evaluated the role of microglial A2AR blockade on the ARPE-19 autophagic pathways, one can speculate that autophagy might also be impacted, since autophagy regulates NLRP3 inflammasome activation47,48. Indeed, future studies should focus on understanding a possible role of A2AR modulation in autophagy, which has been shown to have a fundamental role on the onset of AMD, as it modulates RPE inflammasome activation49,50,51 and RPE cell death52,53.
Photoreceptor cell loss by apoptosis is a major feature of AMD54, and an effective microglia-mediated clearance of the apoptotic cells is considered to be associated with an anti-inflammatory phenotype55. Indeed, it has been shown that photoreceptor proteins are able to induce microglia phagocytic activity and promote the inflammatory response56. Notably, the selective A2AR antagonist was able to increase the phagocytosis of photoreceptor debris by microglia, suggesting that blocking A2AR appears to have additional beneficial effects, such as increased clearance of the apoptotic material. Also, a more efficient clearance of the apoptotic material might contribute to a reduction of the inflammatory response triggered by the exposure to photoreceptor proteins. In this work we demonstrated that the A2AR antagonist reduces the phagocytosis of polystyrene beads by microglia, while it increased the phagocytosis of apoptotic cells. These apparent contradictory results operated by A2AR may imply molecular signals that induce distinct responses in microglia: it promotes the clearance of dead cells probably to halt the inflammatory response but when microglia face inert material, such as polystyrene beads, microglia phagocytose less when treated with SCH58261.
In fact, the crosstalk between microglial cells and photoreceptors is indubitable, since factors present in the culture medium of reactive microglial cells are able to induce photoreceptor cell apoptosis57,58,59. Importantly, blockade of microglial A2AR is also able to reduce this outcome, suggesting that modulation of microglial cells reactivity by A2AR blockade can afford protective effects against photoreceptor cell death.
The role of inflammation and particularly the role of retinal microglia reactivity in the pathophysiology of AMD is unquestionable. Moreover, microglia-RPE crosstalk have long been associated with AMD pathogenesis, being speculated that this crosstalk can act as a positive feedback perpetuating the inflammatory response and sub-retinal microglia accumulation, fostering the proper mechanisms that promote AMD-related retinal dysfunction40. Complement proteins8,37, IL-1β60 and NF-κB10,57 have been already proposed as microglial modulatory factors that can in turn modulate the RPE inflammatory response being involved in photoreceptor loss. Still, novel therapeutic strategies aiming diminishing the effects of these mechanisms are required to reduce immune response in the context of AMD. Noteworthy, the control of the complement system has been identified as a possible therapeutic strategy for managing AMD29, but some concerns might be raised relatively to its efficacy. Recently, ROCHE announced that the clinical trial (phase IIB) for Lampalizumab (NCT01602120), an antibody-binding fragment of a humanized monoclonal antibody that binds to complement factor D, which has been implicated in the pathogenesis of geographic atrophy61, did not reduced the geographic atrophy lesion area at 1 year of treatment62.
In this study, the different cell lines allowed us to dissect the role of A2AR blockade in microglial cells, the immune cells of the CNS, without the contribution of infiltrating immune cells, and the possible crosstalk between microglia, RPE cells and photoreceptors. Although it is important to refer that despite having some advantages over primary cells, cell lines have some limitations because they do not always accurately replicate the features of native cells in the tissue. Indeed, ARPE-19 cell line is one of the most commonly used cell lines for the study of RPE. However, these cells exhibit particularities different from RPE cells in their native environment, such as reduced transepithelial resistance, reduced levels of secreted cytokines, and different morphology63,64,65. Despite the limitations of the cell lines, the present work paves the way to a better understanding on the role of microglial A2AR as a putative target for the treatment of AMD. Nevertheless, more studies are needed, using primary cultures and animal models of AMD to ascertain the therapeutic potential of A2AR antagonists.
In summary, in previous studies, we have suggested that selective A2AR blockade might be seen as a possible therapeutic strategy for glaucoma24,25,26,66. In this work, using in vitro models, we show that an adequate modulation of microglial cell reactivity by blocking A2AR also presents beneficial effects on the modulation of the complement system, and might impact the microglia crosstalk with RPE and photoreceptors.
Methods
Cell lines and pharmacological compounds
The immortalized human microglia-SV40 cell line derived from primary human microglia was purchased from Applied Biological Materials, Inc. (ABM, Inc.). 661 W photoreceptor-like cells were a kind gift from Prof. Muayyad Al-Ubaidi (Department of Cell Biology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA). ARPE-19 cells were obtained from ATCC (CRL-2302). Zymosan A from Saccharomyces cerevisiae and Phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich. The A2AR antagonist SCH58261 was purchased from Merck Millipore.
Cell cultures and treatments
Human microglial cells were grown in collagen-coated T-75 cm2 flasks and cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, at 37 °C in a humidified atmosphere of 5% CO2. Microglia were pretreated for 45 min with 50 nM SCH58261 (A2AR antagonist) and then challenged with Zymosan A (50 µg/mL) and PMA (100 nM) for 6 h. For immunocytochemistry and assessment of mRNA expression, microglial cells were plated at a density of 1 × 104 cells and 1 × 105 cells/cm2, respectively, in collagen-coated cover-slips on 12-well or 6-well plates (Fig. 10A).

Schematic overview of the experimental design. (A) Human immortalized microglial cells were pretreated for 45 min with 50 nM SCH58261 (A2AR antagonist) and then challenged with Zymosan A (50 µg/mL) and PMA (100 nM) for 6 h. (B) Human immortalized microglial cells were challenged with 5 μg/μL ARPE-19 cell debris for 6 h, previously untreated or treated with 50 nM SCH58261. (C) ARPE-19 cell line was cultured with MCM for 48 h. (D) 661 W photoreceptor cell line was cultured with MCM for 48 h.
Since drusen may result from cellular remnants and debris derived from degenerated RPE cells, debris obtained by serum-starved ARPE-19 cells were used to model the composition of drusen in vitro, as we reported previously9. To obtain RPE debris, ARPE‐19 cells were starved by serum deprivation for 2 weeks, harvested, centrifuged, and washed three times with PBS, and the pellet was frozen at −20 °C. Microglial cells were incubated with 5 μg/μL ARPE-19 cell debris for 6 h, previously treated or not with 50 nM SCH58261 (Fig. 10B).
ARPE-19 cell line was cultured in DMEM/F12 supplemented with 10% FBS and 1% penicillin/streptomycin. One day after seeding, the MCM was added to ARPE-19 cells for 48 h (Fig. 10C).
661 W photoreceptor cells were cultured in high glucose DMEM with L-glutamine, supplemented with 10% FBS and 1% penicillin/streptomycin, and maintained at 37 °C in a humidified atmosphere of 5% CO2. One day after seeding, the MCM was added to 661 W cells for 48 h (Fig. 10D).
Adenosine quantification
The extracellular concentration of adenosine was quantified in human microglial cells supernatant using a fluorometric kit (BioVision), according to the instructions provided by the manufacturer.
Immunocytochemistry and image analysis
Cells were fixed with 4% formaldehyde, washed with PBS, and incubated in blocking buffer containing 10% goat serum and 0.3% Triton X-100. Subsequently, cells were incubated with the antibodies rabbit anti-Iba1 (1:150; Wako) or rabbit anti-A2AR (1:200; Thermo Scientific) in a solution containing 2.5% goat serum and 0.1% Triton X-100 for 1 h at room temperature. Cells were incubated with goat anti-rabbit Alexa 488 (A-11012, Life Technologies) or with 0.1 μg/ml Phalloidin-TRITC (Sigma-Aldrich) (in the case of A2AR labeling). Cells were then washed with PBS and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Cover slips were mounted with fluorescent mounting medium (Dako), and fluorescence photomicrographs were taken with an AxioImager.M2 microscope (Carl Zeiss).
Densitometric analysis of A2AR staining was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and mean grey value was calculated as previously described25, with the formula:
Real-time quantitative Polymerase Chain Reaction (PCR)
Total RNA was extracted from human microglial cells or ARPE-19 using the NucleoSpin® RNA Mini Kit (Macherey-Nagel), according to the manufacturer’s instructions. RNA was quantified spectrophotometrically using a NanoDrop 2000 (Thermo Scientific) and then stored at −80 °C. Amplification of cDNA was with RevertAid™ H Minus First strand cDNA Synthesis Kit (Thermo Scientific), according to the instructions provided by the manufacturer, using 1 µg of total RNA.
Amplification of 50 ng cDNA was conducted in an ABI7900HT machine (Applied Biosystems) in 10 μl reaction mixtures containing 1 × TaqMan Universal PCR Master Mix (Applied Biosystems), 200 nM of primers, and 0.125 μL of dual-labeled UPL probe (Roche Applied Science). The reaction parameters were as follows: 2 min 50 °C hold, 30 min 60 °C hold, and 5 min 95 °C hold, followed by 45 cycles of 20 s 94 °C melt and 1 min 60 °C anneal/extension. Primer sequences and UPL probe numbers are listed on Table 1. Ct values were converted to “relative quantification” using the 2∆∆Ct method, using the method described by Livak67. Hprt was used as housekeeping gene.
Nitrite measurement
Nitric oxide concentration was estimated in the supernatant of human microglial cells by quantifying the concentration of nitrites using the Griess reaction method (Promega), as previously described and according to the manufacturer’s instructions. The results are presented as percentage of control.
Microparticles phagocytosis assay
After platting and overnight seeding, microglial cells were treated as described above and 2 h after incubated with 2 µL of micro-particles based on polystyrene solutions (Sigma-Aldrich) for 6 h. After washing 3 times with PBS, 5 images were randomly acquired for each condition under an inverted microscope (Carl Zeiss). Phagocytic efficiency was calculated as described previously24. Results are presented as percentage of control.
661 W apoptotic material phagocytosis assay
661 W photoreceptor cells were starved by serum deprivation and harvested and fluorescently labeled using CellTracker CM-DiI (Invitrogen, Carlsbad, CA, USA). Human microglial phagocytic efficiency was assessed as previously described59. Briefly, microglial cells were treated with 150 µL of apoptotic 661W-labeled solution for 6 h in the presence or absence of SCH58261 (50 nM) (Fig. 10B). Cells were then fixed with 4% formaldehyde and immunolabeled with an antibody rabbit anti-Iba1 (1:150; Wako)68, as described above. Preparations were observed with an inverted fluorescence microscope AxioImager.M2 microscope (Carl Zeiss) and from each experimental condition, at least 5 fields were randomly acquired. Phagocytic efficiency was determined using the formula previously described24. Results are presented as percentage of control.
661 W photoreceptor apoptosis assay
Microglia-induced neurotoxicity was investigated as previously described59. Briefly, 661 W photoreceptor cells were incubated for 48 h with culture supernatants from control-, Zymosan + PMA-, Zymosan + PMA + SCH58261-, or SCH58261-treated microglial cells. Apoptotic cell death was determined using the Caspase-Glo® 3/7 Assay (Promega), according to the manufacturer’s instructions. Relative luciferase units (RLUs) reflect the level of apoptotic cell death. Results are presented as percentage of control.
Statistical analysis
Normality of the data was assessed with Shapiro-Wilk normality test. Since data did not follow a Gaussian distribution, all data was analyzed using non-parametric tests: Mann-Whitney test (for comparison of two groups) or Kruskall-Wallis test (when comparing more than two groups), followed by Dunn’s multiple comparison correction test, as indicated in the figure legends. The values of P are presented above the bars for each comparison. The results are presented as mean ± standard error of the mean (SEM) and the individual data points are also shown. Values of P < 0.05 were considered statistically significant. The statistical analysis was performed in Prism 7.0 Software for Mac OS X (GraphPad Software, Inc).
References
Wong, W. L. et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health 2, e106–116, https://doi.org/10.1016/S2214-109X(13)70145-1 (2014).
Wong, T. Y. et al. The natural history and prognosis of neovascular age-related macular degeneration: a systematic review of the literature and meta-analysis. Ophthalmology 115, 116–126, https://doi.org/10.1016/j.ophtha.2007.03.008 (2008).
Kauppinen, A., Paterno, J. J., Blasiak, J., Salminen, A. & Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci 73, 1765–1786, https://doi.org/10.1007/s00018-016-2147-8 (2016).
Chen, M. & Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukoc Biol 98, 713–725, https://doi.org/10.1189/jlb.3RI0615-239R (2015).
Madeira, M. H., Boia, R., Santos, P. F., Ambrosio, A. F. & Santiago, A. R. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediators Inflamm 2015, 673090, https://doi.org/10.1155/2015/673090 (2015).
Karlstetter, M. et al. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res 45, 30–57, https://doi.org/10.1016/j.preteyeres.2014.11.004 (2015).
Rutar, M. et al. Analysis of complement expression in light-induced retinal degeneration: synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Invest Ophthalmol Vis Sci 52, 5347–5358, https://doi.org/10.1167/iovs.10-7119 (2011).
Ma, W., Coon, S., Zhao, L., Fariss, R. N. & Wong, W. T. A2E accumulation influences retinal microglial activation and complement regulation. Neurobiol Aging 34, 943–960, https://doi.org/10.1016/j.neurobiolaging.2012.06.010 (2013).
Nebel, C., Aslanidis, A., Rashid, K. & Langmann, T. Activated microglia trigger inflammasome activation and lysosomal destabilization in human RPE cells. Biochem Biophys Res Commun 484, 681–686, https://doi.org/10.1016/j.bbrc.2017.01.176 (2017).
Zhang, C. et al. Activation of microglia and chemokines in light-induced retinal degeneration. Mol Vis 11, 887–895 (2005).
Wagner, E. & Frank, M. M. Therapeutic potential of complement modulation. Nat Rev Drug Discov 9, 43–56, https://doi.org/10.1038/nrd3011 (2010).
Orsini, F., De Blasio, D., Zangari, R., Zanier, E. R. & De Simoni, M. G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci 8, 380, https://doi.org/10.3389/fncel.2014.00380 (2014).
Cipriani, V. et al. Genetic variation in complement regulators and susceptibility to age-related macular degeneration. Immunobiology 217, 158–161, https://doi.org/10.1016/j.imbio.2011.09.002 (2012).
Fritsche, L. G. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48, 134–143, https://doi.org/10.1038/ng.3448 (2016).
Wu, L. et al. Association between polymorphisms of complement pathway genes and age-related macular degeneration in a Chinese population. Invest Ophthalmol Vis Sci 54, 170–174, https://doi.org/10.1167/iovs.12-10453 (2013).
Black, J. R. & Clark, S. J. Age-related macular degeneration: genome-wide association studies to translation. Genet Med 18, 283–289, https://doi.org/10.1038/gim.2015.70 (2016).
Lechner, J. et al. Higher plasma levels of complement C3a, C4a and C5a increase the risk of subretinal fibrosis in neovascular age-related macular degeneration: Complement activation in AMD. Immun Ageing 13, 4, https://doi.org/10.1186/s12979-016-0060-5 (2016).
Schick, T. et al. Local complement activation in aqueous humor in patients with age-related macular degeneration. Eye (Lond) 31, 810–813, https://doi.org/10.1038/eye.2016.328 (2017).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10, 241–247, https://doi.org/10.1038/ni.1703 (2009).
Celkova, L. & Doyle, S. L. & Campbell, M. NLRP3 Inflammasome and Pathobiology in AMD. J Clin Med 4, 172–192, https://doi.org/10.3390/jcm4010172 (2015).
Tseng, W. A. et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci 54, 110–120, https://doi.org/10.1167/iovs.12-10655 (2013).
Brandstetter, C., Patt, J., Holz, F. G. & Krohne, T. U. Inflammasome priming increases retinal pigment epithelial cell susceptibility to lipofuscin phototoxicity by changing the cell death mechanism from apoptosis to pyroptosis. J Photochem Photobiol B 161, 177–183, https://doi.org/10.1016/j.jphotobiol.2016.05.018 (2016).
Santiago, A. R. et al. Role of microglia adenosine A(2A) receptors in retinal and brain neurodegenerative diseases. Mediators Inflamm 2014, 465694, https://doi.org/10.1155/2014/465694 (2014).
Madeira, M. H. et al. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure-induced transient ischemic injury. Transl Res 169, 112–128, https://doi.org/10.1016/j.trsl.2015.11.005 (2016).
Madeira, M. H. et al. Adenosine A2AR blockade prevents neuroinflammation-induced death of retinal ganglion cells caused by elevated pressure. J Neuroinflammation 12, 115, https://doi.org/10.1186/s12974-015-0333-5 (2015).
Boia, R. et al. Treatment with A2A receptor antagonist KW6002 and caffeine intake regulate microglia reactivity and protect retina against transient ischemic damage. Cell Death Dis 8, e3065, https://doi.org/10.1038/cddis.2017.451 (2017).
Cunha, R. A. How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem 139, 1019–1055, https://doi.org/10.1111/jnc.13724 (2016).
Harrigan, T. J., Abdullaev, I. F., Jourd’heuil, D. & Mongin, A. A. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem 106, 2449–2462, https://doi.org/10.1111/j.1471-4159.2008.05553.x (2008).
Xu, H. & Chen, M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur J Pharmacol 787, 94–104, https://doi.org/10.1016/j.ejphar.2016.03.001 (2016).
Wolf, S. A., Boddeke, H. W. & Kettenmann, H. Microglia in Physiology and Disease. Annu Rev Physiol 79, 619–643, https://doi.org/10.1146/annurev-physiol-022516-034406 (2017).
Kim, S. Y. Retinal phagocytes in age-related macular degeneration. Macrophage (Houst) 2, https://doi.org/10.14800/macrophage.698 (2015).
Yu, L. et al. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol 63, 338–346, https://doi.org/10.1002/ana.21313 (2008).
Saura, J. et al. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem 95, 919–929, https://doi.org/10.1111/j.1471-4159.2005.03395.x (2005).
Smith, M. E., van der Maesen, K., Somera, F. P. & Sobel, R. A. Effects of phorbol myristate acetate (PMA) on functions of macrophages and microglia in vitro. Neurochem Res 23, 427–434 (1998).
Karlstetter, M. et al. Polysialic acid blocks mononuclear phagocyte reactivity, inhibits complement activation, and protects from vascular damage in the retina. EMBO Mol Med 9, 154–166, https://doi.org/10.15252/emmm.201606627 (2017).
Sweet, M. J. & Hume, D. A. Endotoxin signal transduction in macrophages. J Leukoc Biol 60, 8–26 (1996).
Natoli, R. et al. Retinal Macrophages Synthesize C3 and Activate Complement in AMD and in Models of Focal Retinal Degeneration. Invest Ophthalmol Vis Sci 58, 2977–2990, https://doi.org/10.1167/iovs.17-21672 (2017).
Rutar, M., Valter, K., Natoli, R. & Provis, J. M. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS One 9, e93343, https://doi.org/10.1371/journal.pone.0093343 (2014).
Nozaki, M. et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA 103, 2328–2333, https://doi.org/10.1073/pnas.0408835103 (2006).
Ma, W., Zhao, L., Fontainhas, A. M., Fariss, R. N. & Wong, W. T. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS One 4, e7945, https://doi.org/10.1371/journal.pone.0007945 (2009).
Anderson, D. H. et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res 29, 95–112, https://doi.org/10.1016/j.preteyeres.2009.11.003 (2010).
Zhou, J., Jang, Y. P., Kim, S. R. & Sparrow, J. R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA 103, 16182–16187, https://doi.org/10.1073/pnas.0604255103 (2006).
Wang, Y. et al. NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration. Int J Mol Sci 17, https://doi.org/10.3390/ijms17010073 (2016).
Brandstetter, C., Mohr, L. K., Latz, E., Holz, F. G. & Krohne, T. U. Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. J Mol Med (Berl) 93, 905–916, https://doi.org/10.1007/s00109-015-1275-1 (2015).
Gao, J. et al. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediators Inflamm 2015, 690243, https://doi.org/10.1155/2015/690243 (2015).
Ouyang, X. et al. Adenosine is required for sustained inflammasome activation via the A(2)A receptor and the HIF-1alpha pathway. Nat Commun 4, 2909, https://doi.org/10.1038/ncomms3909 (2013).
Zhong, Z., Sanchez-Lopez, E. & Karin, M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol 34, 12–16 (2016).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230, https://doi.org/10.1038/ni.1980 (2011).
Piippo, N. et al. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochim Biophys Acta 1843, 3038–3046, https://doi.org/10.1016/j.bbamcr.2014.09.015 (2014).
Iannaccone, A. et al. Circulating Autoantibodies in Age-Related Macular Degeneration Recognize Human Macular Tissue Antigens Implicated in Autophagy, Immunomodulation, and Protection from Oxidative Stress and Apoptosis. PLoS One 10, e0145323, https://doi.org/10.1371/journal.pone.0145323 (2015).
Iannaccone, A. et al. Retinal pigment epithelium and microglia express the CD5 antigen-like protein, a novel autoantigen in age-related macular degeneration. Exp Eye Res 155, 64–74, https://doi.org/10.1016/j.exer.2016.12.006 (2017).
Kaarniranta, K., Tokarz, P., Koskela, A., Paterno, J. & Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol Toxicol 33, 113–128, https://doi.org/10.1007/s10565-016-9371-8 (2017).
Yao, J. et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 11, 939–953, https://doi.org/10.1080/15548627.2015.1041699 (2015).
Dunaief, J. L., Dentchev, T., Ying, G. S. & Milam, A. H. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol 120, 1435–1442 (2002).
Sierra, A., Abiega, O., Shahraz, A. & Neumann, H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 7, 6, https://doi.org/10.3389/fncel.2013.00006 (2013).
Kohno, H. et al. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem 288, 15326–15341, https://doi.org/10.1074/jbc.M112.448712 (2013).
Yang, L. P., Zhu, X. A. & Tso, M. O. A possible mechanism of microglia-photoreceptor crosstalk. Mol Vis 13, 2048–2057 (2007).
Karlstetter, M. et al. Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J Neuroinflammation 11, 3, https://doi.org/10.1186/1742-2094-11-3 (2014).
Aslanidis, A. et al. Activated microglia/macrophage whey acidic protein (AMWAP) inhibits NFkappaB signaling and induces a neuroprotective phenotype in microglia. J Neuroinflammation 12, 77, https://doi.org/10.1186/s12974-015-0296-6 (2015).
Natoli, R. et al. Microglia-derived IL-1beta promotes chemokine expression by Muller cells and RPE in focal retinal degeneration. Mol Neurodegener 12, 31, https://doi.org/10.1186/s13024-017-0175-y (2017).
Katschke, K. J. Jr. et al. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J Biol Chem 287, 12886–12892, https://doi.org/10.1074/jbc.M112.345082 (2012).
Roche, F. H.-L. ROCHE - Investor update - media release (Acessed October 25, 2017), https://www.roche.com/dam/jcr:a3969247-3a45-4501-8940-306fbd1dc97f/en/med-cor-2017-09-08b.pdf (2017).
Ablonczy, Z. et al. Human retinal pigment epithelium cells as functional models for the RPE in vivo. Invest Ophthalmol Vis Sci. 52, 8614–8620, https://doi.org/10.1167/iovs.11-8021 (2011).
Dunn, K. C. et al. Use of the ARPE-19 cell line as a model of RPE polarity: basolateral secretion of FGF5. Invest Ophthalmol Vis Sci 39, 2744–2749 (1998).
Fronk, A. H. & Vargis, E. Methods for culturing retinal pigment epithelial cells: a review of current protocols and future recommendations. J Tissue Eng 7, 2041731416650838, https://doi.org/10.1177/2041731416650838 (2016).
Madeira, M. H. et al. Caffeine administration prevents retinal neuroinflammation and loss of retinal ganglion cells in an animal model of glaucoma. Sci Rep 6, 27532, https://doi.org/10.1038/srep27532 (2016).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Luckoff, A., Scholz, R., Sennlaub, F., Xu, H. & Langmann, T. Comprehensive analysis of mouse retinal mononuclear phagocytes. Nat Protoc 12, 1136–1150, https://doi.org/10.1038/nprot.2017.032 (2017).
Acknowledgements
M.H. Madeira was a recipient of a short-term fellowship from the Federation of European Biochemical Societies (FEBS). The work was supported by DFG research unit FOR2240 (Lymph) Angiogenesis and cellular immunity in inflammatory diseases of the eye, Foundation for Science and Technology (PEst UID/NEU/04539/2013), COMPETE-FEDER (POCI-01-0145-FEDER-007440), and Centro 2020 Regional Operational Program (CENTRO-01-0145-FEDER-000008: BrainHealth 2020).
Author information
Authors and Affiliations
Contributions
M.H.M., K.R., A.R.S. and T.L. conceived and designed the experiments. M.H.M. performed the experiments. M.H.M., K.R., A.F.A., A.R.S. and T.L. analyzed the data. A.F.A., A.R.S. and T.L. contributed with reagents/materials/analysis tools. M.H.M., K.R., A.F.A., A.R.S. and T.L. wrote the paper. All contributing authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Madeira, M.H., Rashid, K., Ambrósio, A.F. et al. Blockade of microglial adenosine A2A receptor impacts inflammatory mechanisms, reduces ARPE-19 cell dysfunction and prevents photoreceptor loss in vitro. Sci Rep 8, 2272 (2018). https://doi.org/10.1038/s41598-018-20733-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20733-2
This article is cited by
-
E3 ubiquitin ligase Herc3 deficiency leads to accumulation of subretinal microglia and retinal neurodegeneration
Scientific Reports (2024)
-
Targeting adenosine A2A receptors for early intervention of retinopathy of prematurity
Purinergic Signalling (2024)
-
Identification of immune associated potential molecular targets in proliferative diabetic retinopathy
BMC Ophthalmology (2023)
-
Forward genetic screening using fundus spot scale identifies an essential role for Lipe in murine retinal homeostasis
Communications Biology (2023)
-
Gene regulation in activated microglia by adenosine A3 receptor agonists: a transcriptomics study
Purinergic Signalling (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
