
Abstract
Cancer immunotherapy has revolutionised cancer treatment, with immune checkpoint blockade (ICB) therapy and adoptive cell therapy (ACT) increasingly becoming standard of care across a growing number of cancer indications. While the majority of cancer immunotherapies focus on harnessing the anti-tumour CD8+ cytotoxic T cell response, the potential role of CD4+ ‘helper’ T cells has largely remained in the background. In this review, we give an overview of the multifaceted role of CD4+ T cells in the anti-tumour immune response, with an emphasis on recent evidence that CD4+ T cells play a bigger role than previously thought. We illustrate their direct anti-tumour potency and their role in directing a sustained immune response against tumours. We further highlight the emerging observation that CD4+ T cell responses against tumours tend to be against self-derived epitopes. These recent trends raise vital questions and considerations that will profoundly affect the rational design of immunotherapies to leverage on the full potential of the immune system against cancer.
Similar content being viewed by others

Introduction
Cancer immunotherapy has advanced rapidly in the clinic in recent years because of two main therapeutic drivers: immune checkpoint blockade (ICB) therapy using antibodies blocking inhibitory receptors of the immune system across tumours [1,2,3,4], and adoptive cell therapy (ACT) using T cells engineered to express chimaeric antigen receptors (CAR T cells) targeting blood malignancies [5, 6]. These therapeutic modalities have largely focused on boosting the quantity and quality of anti-tumour CD8+ cytotoxic T lymphocyte (CTL) responses to generate therapeutic benefits. However, despite ongoing efforts to extend the therapeutic reach and increase the safety of ICBs and ACT [3, 4, 7,8,9], typically by investigating the therapeutic potential of rationally designed combination therapies (e.g. tumour vaccines with ICBs, radio- and chemotherapy with ICBs etc.) [10,11,12,13], there remain significant limitations in the clinical efficacies of both these treatment modalities.
Recently, it has become increasingly clear that CD4+ T cells play a critical role in developing and sustaining effective anti-tumour immunity, even in cancer immunotherapies specifically designed to activate a CD8+ CTL response. In this review, we discuss new developments detailing the multifaceted involvement of CD4+ T cells in the anti-tumour immune response and revisit older paradigms on the roles of CD4+ T cells in tumour immunity. Finally, we will highlight some novel emergent aspects of anti-tumour CD4+ T cells and offer our perspective on future directions to accelerate translation of this knowledge into clinical therapies.
A brief history of trends in cancer immunotherapy targeting CD4+ T cells
CD4+ T cells are highly versatile, polyfunctional cells that constitute the second arm of adaptive T cell immunity alongside their sister lineage of CD8+ cytotoxic T cells. CD4+ T cells can differentiate into one of several diverse functional subtypes in response to context-dependent signals (Fig. 1), which in turn allows them to provide ‘help’ to appropriate effector immune cells in their primary role as central co-ordinators of the immune response. CD4+ T cells primarily mediate anti-tumour immunity by providing help for CD8+ CTL and antibody responses, as well as via secretion of effector cytokines such as interferon-γ (IFNγ) and tumour necrosis factor-α (TNFα), and, under specific contexts, via direct cytotoxicity against tumour cells (Fig. 2). The earliest efforts to induce CD4+ T cell responses against tumours were attempts to generate TH1-polarised CD4+ T cells by vaccination with peptide epitopes (Table 1). These peptides were typically derived from highly immunogenic tumour-associated antigens [14, 15] including members of the cancer testis antigen family such as NY-ESO1 [16, 17] or melanoma-associated antigens such as MAGE-A3 [18]. In particular, these studies focused on boosting CD4+ T cell-derived secretion of TH1-characteristic tumoricidal cytokines (e.g. IFNγ) as a readout of increased anti-tumour CD4+ responses. Other variations of this strategy were attempts to isolate and expand tumour-reactive CD4+ T cells from patient tumour-infiltrating lymphocytes (TILs) using tumour-derived antigens with major histocompatibility complex (MHC) II-restricted epitopes and then re-infusing them as a form of ACT [19, 20], or more recently, by engineering autologous CD4+ T cells from cancer patients to express synthetic chimaeric antigen receptors that recognise antigenic epitopes on tumour cells.

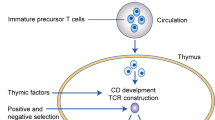
CD4+ T cells are T lymphocytes that express T cell receptors (TCRs) recognising peptide antigens presented in the context of Class II major histocompatibility complex (MHC II) molecules. CD4+ T cells express the TCR co-receptor CD4, which binds to the β2 domain of MHC II and facilitates TCR engagement with peptide-MHC II complexes on antigen-presenting cells [111]. During thymic development, the cell fate of developing thymocytes is decided by their TCR affinity for self-peptide-MHC complexes presented by thymic epithelial cells. Thymocytes that have little to no affinity for self-peptide do not initiate activating signals from their TCR complexes and thus die by neglect. Conversely, thymocytes with high self-reactivity are negatively selected and deleted by apoptosis. Thymocytes with intermediate TCR affinities below the negative selection threshold receive positive selection via activating TCR signals and complete thymic maturation as naïve conventional T cells (TH0). Some thymocytes with moderately high affinities to self-antigen are redirected into the regulatory T cell (Treg) developmental pathway, where they acquire immunosuppressive function to regulate tissue homoeostasis and resolution of immune responses [53, 112]. Upon receiving cues from the cytokine milieu together with TCR activation, naive CD4+ T cells upregulate expression of key transcription factors regulating subset differentiation, which in turn drive the expression of major effector cytokines associated with each particular subtype [113, 114]. Key transcription factors and cytokines involved are indicated for individual subtypes. CD4+ T cells augment the development of the CTL response [21, 24] and are required for the development of CD8+ T cell immunity (reviewed extensively here [115]) in their role as central co-ordinators of adaptive immunity. Unlike CD8+ T cells, whose primary function is to mediate cell contact-dependent cytotoxicity of infected or malignant cells, CD4+ T cells exhibit a diverse repertoire of effector functions and exhibit considerable phenotypic plasticity and heterogeneity depending on local context and microenvironment [113, 114]. CD4+ T cells activated in the periphery can also differentiate into induced Tregs (iTregs), which are able to mediate immunosuppression similar to thymic Tregs (tTregs).

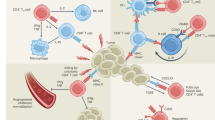
CD4+ T cells play key roles in tumour immunity through several different mechanisms. a A major role of CD4+ T cells is the provision of help for anti-tumour CTLs through both direct and indirect mechanisms (discussed in-depth here [116]). Activated CD4+ T cells secrete interleukin (IL)-2, which directly activates CD8+ CTLs expressing the high-affinity IL-2 receptor α subunit (CD25) by driving their effector function, differentiation, and proliferation. CD4+ T cells also indirectly provide help for the anti-tumour CD8+ CTL response by supporting and maintaining pro-inflammatory cross-presenting dendritic cells (DCs) [101], which in turn provide the three activating signals for CD8+ CTLs [115, 117]. This is primarily mediated by the upregulation of CD40 ligand (CD154) [22, 23, 118] on activated CD4+ T cells, which engages its cognate receptor CD40 on DCs to induce and maintain the type I profile of DCs (expression of B7 family ligands, CD70, and secretion of IL-12) [116]. These signals strongly induce anti-tumour effector functions in CD8+ CTLs such as the acquisition of cytotoxicity and the secretion of tumoricidal cytokines such as interferon-γ (IFNγ), and also stimulate the effector [117, 119, 120] and memory [121,122,123,124] phenotype differentiation of CD8+ T cells. b CD4+ T cells also produce effector cytokines such as IFNγ and tumour necrosis factor-α (TNFα), which have direct anti-tumour activity, following activation and polarisation into the TH1 phenotype [125] in response to signals from DCs, particularly IL-12. In addition, CD4+ T cells can mediate direct cytotoxicity against tumour cells in a similar manner to their CD8+ T cell counterparts under specific conditions in both preclinical mouse tumour models [45, 46, 126] and in patient-derived CD4+ T cells [127]. c CD4+ T cells are also indispensable for the induction of humoral responses against tumour antigens by providing help via CD40 ligand signalling to CD40 on B cells to drive their differentiation and maturation into affinity-matured, class-switched plasma cells. Their activity correlates with the presence of serum antibodies specific to tumour antigens [17, 128], and they likely play a role in driving local antibody responses in tertiary lymphoid structures [129] adjacent to solid tumours.
In contrast, although the development and effector functions of distinct subsets of CD4+ T cells has been recognised and described (Fig. 1), the interplay between polyfunctional CD4+ T cells and other immune cell lineages within the context of tumour immunity is less well understood. This is despite the fact that the critical role for CD4+ T cells in supporting the effector function and differentiation of CD8+ T cells had been described as early as the 1980s [21] and was already well-established by the late 1990s [22,23,24,25]. The first major turning point began in the mid-2000s when mouse models of cancer demonstrated that CD4+ T cells were necessary to maintain and sustain anti-tumour CD8+ CTL responses [26, 27] (Fig. 2). This is also validated by observations from clinical studies, most commonly in the context of cancer vaccines [28,29,30].
Parallel to this revival of interest in CD4+ T cells, there has also been significant research directed at producing a universal cancer vaccine based on promiscuous class II epitopes from self-molecules such as survivin [31,32,33] (an inhibitor of apoptosis) and telomerase reverse transcriptase (TERT) [34,35,36] (Table 1). Even at this rudimentary stage, it was already recognised that spontaneous CD4+ T cell responses towards self-antigens could be harnessed to boost anti-tumour immunity.
(Re)discovery of the importance of CD4+ T cells in driving and sustaining anti-tumour immune response
Despite these encouraging early findings, the fundamental role of CD4+ T cells in orchestrating anti-tumour responses was until recently eclipsed by the clinical success of CD8+ T cell-based immunotherapies. This greater attention to CD8+ T cells was partly due to greater availability of tools such as tetramers that could be used to monitor CD8+ responses, and also partly because CD8+ T cell numbers and function were the most proximal readouts of anti-tumour immunity. However, the last 5 years have seen many reports recognising the critical role of CD4+ T cells in driving anti-tumour immunity and in supporting anti-tumour CD8+ T cell responses.
In 2015, Linnemann et al. found that human melanomas frequently contained mutant neoepitopes recognised by CD4+ cells [37]. This was quickly followed by a report from the lab of Özlem Türeci and Ugur Sahin, which demonstrated that immunogenic tumour mutations in the ‘mutanomes’ of three separate preclinical mouse tumour models largely induced a CD4+ T cell response [38], not a CD8+ T-cell response as had been expected. Two years later, the same group and Catherine Wu’s group published back-to-back reports reporting clinical findings showing that personalised neoantigen vaccines for melanoma patients primarily induced tumour-specific responses in CD4+ rather than CD8+ T cells [39, 40]. In both cases, synthetic long peptides (SLPs) were used as the mode of vaccination. These findings serendipitously validated findings from 10 years prior that immunisation with longer peptides induced a sustained CD8+ T cell response [41], likely due to CD4+ T cell help, whereas immunisation with exact-length MHC I-restricted peptides (specifically targeting only CD8+ T cells) only gave rise to a fleeting CD8+ T cell response. Ott et al. further theorised that the unexpected preponderance of CD4+ over CD8+ T cell responses could have been due to (1) a relative paucity of the cross-presenting dendritic cell subset within tumours, which led to more efficient priming of CD4+ relative to CD8+ T cells, and (2) the relatively higher promiscuity of MHC II-restricted epitopes due to more relaxed binding requirements compared with MHC I-restricted epitopes [39].
Concurrently, findings from preclinical mouse models also highlight a larger, more fundamental role for CD4+ T cells in anti-tumour immunity than was previously thought. In 2017, Spitzer et al. reported that a unique TH1-like CD4+ subset was expanded in non-tumour peripheral tissues during an active anti-tumour response to adjuvant therapy [42], in a study conducted in the Py-MMTV mouse model of spontaneous mammary tumours. They further demonstrated that this population of CD44+ CD69+ CD62L− CD27lo T-bet+ CD4+ T cells conferred a protective benefit when transferred into treatment-naive tumour hosts. In addition, this group also found an analogous population of CD4+ T cells in the peripheral blood of melanoma patients who had received ipilimumab (α-CTLA-4 blocking antibody) combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) therapy.
In 2019, two separate preclinical studies independently highlighted the role of CD4+ T cells in enhancing the anti-tumour CD8+ T-cell response. Zander and colleagues identified a critical role for CD4+ T cell-derived interleukin (IL)-21 in driving the differentiation of a CX3CR1+ cytotoxic effector CD8+ T cell subtype with enhanced anti-viral and anti-tumour activity against murine B16F10 melanoma tumours (an immunologically-cold, aggressive tumour that is refractory to ICB therapy) [43]. Similarly, Alspach et al. found that poorly immunogenic tumours engineered to express MHC II-restricted antigens could induce TH1-polarised anti-tumour CD4+ T cell responses. These antigen-specific CD4+ T cells enhanced the efficacy of anti-tumour CD8+ T cell responses, and mediated long-lived protection against subsequent tumour re-challenge in mice that survived primary tumour challenge [44].
In addition, in a recent study, Śledzińska et al. built on previous work from a decade ago [45, 46] and found that tumour-infiltrating TH1-like CD4+ T cells acquired cytotoxicity against B16 melanoma. This development of cytotoxic capability required expression of the transcription factors T-bet and Blimp-1 [47]. Collectively, these recent preclinical studies demonstrate the critical and versatile role of polyfunctional tumour-infiltrating CD4+ T cells in the overall anti-tumour immune response.
Recent clinical evidence has also raised the importance of CD4+ T cells in generating successful anti-tumour immunity. In a meticulous deep single-cell analysis of T cell receptor (TCR)- and RNA-sequencing from colorectal cancer patient biopsies, Zemin Zhang’s group found that patients with microsatellite-instable tumours (which show a strikingly favourable response profile to ICB therapy) showed preferential enrichment for a TH1-like subset of CD4+ T cells. These unique tumour-infiltrating CD4+ T cells expressed the transcription factor BHLHE40, the effector cytokine IFNG, and the chemokine receptor CXCR5 [48]. In a second study, Galaine et al. reported the presence of anti-tumour CD4+ T cells that recognised MHC II-restricted, promiscuously-binding tumour-associated antigens in colorectal cancer patients undergoing oxaliplatin chemotherapy. In some patients, CD4+ T cell responses persisted even after 3 months of oxaliplatin treatment [49], highlighting the importance of understanding the immunomodulatory effects of oxaliplatin and other chemotherapeutic agents on CD4+ T cells.
Interestingly, the presence of specific subsets of CD4+ T cells in the peripheral circulation was also found to be predictive of good prognosis in non-small cell lung cancer (NSCLC) patients, where ICB treatment has efficacy either as a single agent or in combination therapy. A Japanese study by Kagamu et al. found that a higher level of circulating CD62Llo CD4+ T cells prior to PD-1 checkpoint blockade was significantly correlated with better response and with the presence of effector CD8+ T cells [50]. This subset of CD4+ T cells expressed T-bet and CXCR3 but not CD27 or FoxP3. Furthermore, the maintenance of high levels of these CD4+ T cells correlated significantly with patient survival, whereas a loss of this population of CD4+ T cells after ICB was correlated with resistance to ICB therapy. Separately, Laheurte et al. found that higher levels of TERT-specific TH1-type CD4+ T cells in peripheral blood was associated with better prognosis of NSCLC patients [51].
Overall, these recent clinical advances corroborate the robust findings from preclinical models that CD4+ T cells play a fundamental role in driving and sustaining meaningful anti-tumour immune responses.
Regulatory T cells in cancer immunotherapy—a plot twist
CD4+ T regulatory cells (Tregs) are a major subset of CD4+ T cells, distinct from the conventional CD4+ effector lineage (Tconvs), that mediate immunosuppressive and tolerogenic functions in both homoeostasis and inflammation [52,53,54,55,56]. CD4+ Tregs are most broadly characterised by their expression of the transcription factor FoxP3, which is a master regulator of their immunosuppressive function [53] (Fig. 1). Until recently, the prevailing paradigm was that the presence of Tregs within the tumour microenvironment (TME) was ‘bad’ for anti-tumour immunity. Tregs suppress anti-tumour immune effector responses in the TME, primarily by promoting an immunosuppressive microenvironment by their secretion of cytokines such as IL-10 and transforming growth factor-β (TGFβ) [57,58,59], and possibly by targeting anti-tumour effector immune cells and antigen-presenting cells for granzyme- and perforin-mediated killing [59,60,61]. In addition, it has also been proposed that the milieu of the tumour microenvironment converts effector CD4+ T cells into Tregs or promotes the differentiation of naïve CD4+ T cells into induced Tregs [62, 63], further exacerbating suppression of nascent anti-tumour immunity. The immunosuppressive role for tumour-infiltrating Tregs continues to be validated by observations in the clinic that increased frequencies of Tregs are associated with poorer cancer patient prognoses [64,65,66,67].
Consequently, most therapeutic modalities targeting Tregs involve depletion by specific chemotherapeutic agents such as cyclophosphamide, or by antibody-dependent cellular cytotoxicity (ADCC) mechanisms initiated by the targeted labelling of Tregs with antibodies specific for surface markers strongly expressed on Tregs such as CD25 and CTLA-4 (comprehensively reviewed here [68]). Other approaches include blocking of Treg recruitment into the TME by blocking the binding of chemokine receptors such as CCR4 involved in their trafficking to tumour sites [68, 69], or inhibiting Treg immunosuppressive function [59, 68]. Of note, CD4+ Tregs constitutively express high levels of surface receptors that are only upregulated by conventional T cells in response to activation, including PD-1 and CTLA-4, as well as a host of TNF receptor superfamily members such as OX-40 (CD134) and GITR [57, 58, 70]. These receptors are potential targets for antibody-mediated depletion. Of note, the therapeutic efficacy of the anti-CTLA-4 antibody ipilimumab is likely due in part to its depleting effects on intratumoral Tregs [71].
However, despite advances in technology, the function and stability of Tregs within the tumour microenvironment have been poorly characterised beyond the minimum knowledge necessary to remove Tregs or inhibit their function.
Early reports as far back as 2009 indicated potential involvement of FoxP3+ CD4+ Tregs in the anti-tumour response in patients that had been treated with a MHC II-restricted MAGE-A3 peptide vaccine [18]. A subsequent study in murine B16F10 melanoma found that administration of glucocorticoid-induced TNF receptor (GITR) agonist antibodies resulted in the selective expansion of tumour-specific Tregs and was accompanied by a broadening of the TCR repertoire of the Teg population, but not of the Tconv TCR repertoire [72]. Furthermore, Tregs were substantially increased in HER2+ breast cancer patients that showed tumour rejection following treatment with the drug-antibody conjugate trastuzumab emtansine (Kadcyla®) [73]. Although tumour-infiltrating Tregs were known to be immunosuppressive, distinct from anti-tumour CD4+ Tconvs [67, 74, 75], these early studies raised the question of whether tumour antigen-specific Tregs could potentially acquire an effector phenotype under certain conditions and contribute towards anti-tumour immunity.
Recent literature has raised the interesting possibility that Tregs can indeed be converted into anti-tumour effector cells. Several molecular mechanisms controlling the stability of the suppressive phenotype of Tregs have been identified, including OX-40 signalling [76, 77], GITR signalling [78], NF-κB signalling [79], the histone methyltransferase Ezh2 [80], and the Ikaros family transcription factor Helios (IKZF2) [81, 82]. These molecules may well function as gatekeepers for the conversion of Tregs into anti-tumour effector CD4+ T cells. The application of such findings to develop Treg conversion as a potential cancer immunotherapy could thus potentially deliver a potent one-two combo by minimising Treg immunosuppression while simultaneously generating more anti-tumour effector T cells.
In one notable study, Shimon Sakaguchi’s group showed that CD4+ Tregs isolated from colorectal cancer patients could be subdivided into two functionally distinct populations based on levels of FoxP3 expression. These tumour-infiltrating Tregs consisted of a FoxP3hi population suppression-competent population, and a second poorly suppressive FoxP3lo population that was induced by the TH1-polarising cytokine IL-12 and that also secreted the pro-inflammatory cytokines IFNγ and IL-17 [65]. Interestingly, patients with higher infiltrates of FoxP3lo cells showed better clinical prognoses than did patients with lower infiltrates of the same cells, suggesting that these cells could be anti-tumour effectors. More recently, Steven Rosenberg’s group found that tumour-infiltrating CD4+ Tregs expressed a distinct TCR repertoire that was enriched in tumours across several cancer types [83] by using deep TCR-sequencing to compare the TCR repertoires of tumour-infiltrating CD4+ Tregs and Tconvs with those of CD4+ T cell populations in autologous peripheral blood. Intriguingly, the authors also found two patient-derived TCRs that were reactive to tumour neoantigens and stimulated production of the effector cytokine IFNγ. These findings, while not definitive statements about tumour-reactive Tregs, provide support for Treg conversion as a paradigm relook at cancer immunotherapy.
Recent advances in CD4+ CAR T cells
The observation that CD4+ T cells synergise with CD8+ T cells in the immune response against tumours also extends to CAR T cell adoptive immunotherapy. Carl June’s research group recently reported preliminary observations that a higher CD4/CD8 ratio in the leukapheresis products used to generate CAR T cells directed against multiple myeloma correlated with better clinical response [84]. These observations, while not necessarily indicative of any particular mechanism, are nonetheless consistent with the known function of CD4+ T cells in co-ordinating and sustaining the immune response against tumours. Furthermore, in a recent glioblastoma (GBM) study, Wang et al. found that the maintenance of CD4+ CAR T cells correlated positively with the recursive killing ability of CAR T cell products derived from GBM patients (so-called “serial killers [8]”). These CD4+ CAR T cells also showed anti-tumour effector activity independent of CD8+ CAR T cells [85].
Reflecting the increasing appreciation for the role of CD4+ T cells in sustaining the efficacy of CAR T cell therapy, a number of recent studies have characterised molecular regulators of CD4+ CAR T function. In an elegant study, Yang et al. showed that CD4+ and CD8+ T cells transduced with a second-generation CD19-targeting CAR (with a CD28 co-stimulatory signalling domain) showed similar in vitro and in vivo efficacy against a murine model of pre-B cell acute lymphoblastic leukaemia. Strikingly, unlike CD8+ CAR T cells that become exhausted or apoptotic when exposed to activating signals through both their CARs and native TCRs, CD4+ CAR T cells retained their in vivo efficacy in controlling leukaemia [86]. In another study using murine CAR T cells specific for B7H6 (a tumour-specific activating ligand for the NKp30 receptor [87]), the TH1 phenotype-associated transcription factor T-bet was found to increase the efficacy of their CAR T cells in controlling NKp30+ RMA tumours in mice [88].
Finally, in a comprehensive study combining multiple single-cell analysis techniques, Xhangolli and co-authors profiled the transcriptional responses of human T cells expressing a third-generation CAR specific for CD19 (containing both 4-1BB and CD28 signalling domains) in response to CAR stimulation. In agreement with the work of Yang et al, 2 years prior, they found that CD4+ and CD8+ CAR T cells were indeed equally effective at killing human CD19+ Raji cells. Furthermore, while both CD4+ and CD8+ CAR T cells were highly polyfunctional, with more than 50% secreting >5 cytokines (including the TH1-characteristic cytokine IFNγ), CD4+ CAR T cells were slightly more polyfunctional than CD8+ CAR T cells, and exhibited a mixed TH1/TH2 transcriptional and cytokine secretion profile [89].
Altogether, these recent studies highlight a role for CD4+ T cell-derived CAR T cells distinct from their CD8+ T cell-derived counterparts. Although molecular evidence is currently scant and preliminary, these pioneer studies also suggest that a polyfunctional TH1-like phenotype (characterised by secretion of IFNγ and possibly driven by the activity of the transcription factor T-bet) may be beneficial for the overall efficacy of the final CAR T cell product. However, given the diversity of CD4+ T cell functional subtypes (Fig. 1) and the current lack of information regarding potential cross-talk between CAR signalling and CD4+ T cell-intrinsic gene programmes, it is very likely that CD4+ T cell-derived CAR T cells polarised towards non-TH1 phenotypes may also mediate effective anti-tumour immunity in a context-dependent manner. In addition, there is also the possibility that CD4+ CAR T cells could be influenced by the TME to acquire Treg-like immunosuppressive phenotypes. Further investigation into all these open questions would be essential to further refine CAR T cell engineering to increase its treatment efficacy and safety profile.
Self-reactive anti-tumour CD4+ T cells—the next frontier in cancer immunotherapy?
The evolving themes we have highlighted illustrate a renewed appreciation of the central role of CD4+ T cells directed against cancer. While these recent studies have clearly established that CD4+ T cells provide critical help for anti-tumour immune responses, the repertoire of antigens that CD4+ T cells recognise within the tumour microenvironment remains relatively unexplored.
In principle, tumour-derived MHC II-restricted epitopes could be derived either from tumour-specific mutations or from self-antigens. The empirical evidence thus far suggests that the majority of the anti-tumour CD4+ response is directed against self-derived epitopes, regardless of whether Tconv or Treg cells respond. This is likely due to the fact that self-reactive CD4+ T cells are less likely to be deleted during formation of central tolerance in the thymus (as are self-reactive developing CD8+ T cells), but rather are tolerised in the periphery [55] or develop into regulatory T cells [53, 54, 56].
This raises three interesting questions that could potentially open up new approaches towards more effectively incorporating CD4+ T cells into the cancer immunotherapy arsenal (Fig. 3).

a The majority of tumour-reactive CD4+ T cells have been found to recognise self-derived antigens, but have thus far only been shown to become activated in the tumour microenvironment (TME) and not in surrounding tissues, suggesting that there may be mechanisms specific to within the TME that permit the breaking of self-tolerance. Another possible avenue for loss of CD4+ T cell self-tolerance is the conversion of self-specific Tregs into conventional effector T cells within the TME. b Because CD4+ T cells are MHC II-restricted, their activation within the TME requires antigen presentation in the context of MHC II molecules. In principle, MHC II+ tumours could directly present antigen and activate CD4+ T cells, or antigen presentation could occur indirectly via antigen-presenting cells (APCs) resident within tumours or tumour-draining lymphoid sites. c The mutually reinforcing interaction between myeloid-derived suppressor cells (MDSCs) and regulatory CD4+ T cells (Tregs) (above) is a negative mirror image of the APC-effector CD4+ T cell synergy that drives the generation of effective immunity (below). Understanding the molecular circuitry is crucial to developing targeted strategies to disrupt and convert these negative interactions into cycles that drive anti-tumour immunity.
First, what breaks peripheral tolerance in self-reactive CD4+ T cells within the tumour microenvironment (Fig. 3a)? Many of the anti-tumour CD4+ T cell responses described in the early days of cancer immunotherapy were specific for highly immunogenic self-derived antigens [14, 31, 32, 90,91,92,93]. This in turn implies that, in the case of self-reactive anti-tumour CD4+ T cells, the self-tolerance mechanisms that would normally check such aberrant self-directed autoimmunity are either disrupted or negated within the TME. Recent evidence in mouse models has shown that Tregs (likely to be self-reactive) can become anti-tumour effector cells in response to epigenetic modulator drugs [80] or agonist antibodies specific for TNF receptor superfamily members [82]. Investigating the upstream signalling cues that trigger and maintain a pro-inflammatory CD4+ T cell phenotype could lead to the identification of therapeutic regimens that favour the generation and/or maintenance of self-antigen-biased, anti-tumour CD4+ T cell immunity.
Furthermore, we posit that elucidating the origin of self-antigen-directed CD4+ T cell immunity would also be particularly relevant to the engineering of more sophisticated CAR T cell products for cellular therapy. Certainly, it would open up the possibility of using self-antigen-specific CARs to redirect autologous CD4+ T cells (possibly even CD4+ Tregs) with less risk of the immune-related adverse events (irAEs) that currently limit therapy with CAR T cells. Another possible translational application of such knowledge would be in pre-selecting autologous self-antigen-specific CD4+ T cells (including Tregs) as the starting material for CAR T cell generation, in order to favour the development of more therapeutically efficacious final CAR T cell product for patient infusion. The use of Tregs as a source material for CAR T cell development could also potentially lead to the exciting prospect of generating “dual programme” CAR T cells, with the option of selecting between effector T cell (anti-tumour) and regulatory T cell (immunosuppressive, to prevent irAEs) functional programmes as appropriate to the clinical status of the patient.
Second, which antigen-presenting cells are responsible for activating tumour-specific CD4+ T cells in the tumour microenvironment (Fig. 3b)? Although higher MHC II expression in tumours correlates with better clinical outcome in MHC II+ cancers [94,95,96], MHC II expression across cancer types is highly variable and context-dependent [97,98,99]. It may be that tumour-infiltrating CD4+ T cells recognise antigen on MHC II molecules present on “professional” antigen-presenting cells (myeloid cells of the macrophage or DC lineage and B cells) within the tumour or tumour-draining lymphatics. This could possibly occur in a similar manner to what naturally occurs in secondary lymphoid organs during an infection [100,101,102]. Furthermore, under certain conditions, non-haemotopoietic cells (e.g. epithelial cells) may also acquire the ability to present antigen on MHC II complexes [103,104,105]. Understanding these mechanisms of antigen priming and/or re-stimulation within the specific tumour microenvironments would be critical to harness the full potential of CD4+ T cells in cancer immunotherapy, and may inform rational combinations with therapies targeting antigen-presenting cells.
Finally, as the fundamental role of anti-tumour CD4+ T cell responses becomes increasingly apparent, one under-explored area that should be examined would be the dynamics of the interaction between CD4+ T cells and myeloid-derived suppressor cells (MDSCs) within the tumour microenvironment (Fig. 3c). MDSCs are myeloid-lineage cells within the tumour microenvironment that exhibit an immature, tolerogenic phenotype. MDSCs are capable of promoting the differentiation and expansion of regulatory T cell populations within the tumour microenvironment [106,107,108], possibly by presenting self-antigen on MHC II molecules. Conceptually, this situation is the mirror image of the synergistic positive feedback interactions between effector CD4+ T cells and activated DCs presenting non-self-antigens that form the nexus driving immunity. Identifying molecular drivers that disrupt the immunosuppressive interactions between MDSCs [109, 110] and Tregs, possibly by simultaneously converting these cells into activated DCs and effector CD4+ T cells, respectively, would be highly informative and beneficial to designing more effective immunotherapeutic strategies.
Conclusions and perspectives
In this review, we present recent literature showing that CD4+ T cells are a critical cornerstone of optimal anti-tumour immunity. Engagement of the CD4+ T cell compartment is associated with the generation of an effective anti-tumour response, even when CD4+ T cells themselves are not the primary immune cell subtype targeted by therapy. We highlight the emerging consensus that the majority of these tumour-specific CD4+ T cells are self-reactive, and juxtapose this against observations from preclinical studies that self-antigen-biased regulatory T cells can themselves mount anti-tumour responses when appropriately conditioned. These recent ongoing developments also present fascinating prospects for the engineering of CD4+ T cells for ACT. In conclusion, harnessing the full potential of the immune system for cancer immunotherapy will require a deeper understanding of, and rational targeting of self-reactive CD4+ T cells to sustain a durable, robust anti-tumour response towards clinical benefit while minimising autoimmunity.
References
Wilson RAM, Evans TRJ, Fraser AR, Nibbs RJB. Immune checkpoint inhibitors: new strategies to checkmate cancer. Clin Exp Immunol. 2018;191:133–48. https://doi.org/10.1111/cei.13081.
Linhares DeSousa, Leitner A, Grabmeier-Pfistershammer J, Steinberger K, Not All P. Immune checkpoints are created equal. Front Immunol. 2018;9:1909. https://doi.org/10.3389/fimmu.2018.01909.
Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33:581–98. https://doi.org/10.1016/j.ccell.2018.03.005.
Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8. https://doi.org/10.1186/s40425-018-0316-z.
Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–47. https://doi.org/10.1146/annurev-med-060512-150254.
Frigault MJ, Maus MV. Chimeric antigen receptor-modified T cells strike back. Int Immunol. 2016;28:355–63. https://doi.org/10.1093/intimm/dxw018.
Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5:16. https://doi.org/10.1186/s40425-017-0218-5.
Watanabe K, Kuramitsu S, Posey AD Jr., June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. 2018;9:2486. https://doi.org/10.3389/fimmu.2018.02486.
RIbas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5.
Curran MA, Glisson BS. New hope for therapeutic cancer vaccines in the era of immune checkpoint modulation. Annu Rev Med. 2019;70:409–24. https://doi.org/10.1146/annurev-med-050217-121900.
Martin NT, Bell JC. Oncolytic virus combination therapy: killing one bird with two stones. Mol Ther. 2018;26:1414–22. https://doi.org/10.1016/j.ymthe.2018.04.001.
Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18:313–22. https://doi.org/10.1038/nrc.2018.6.
Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, et al. Advances in cancer immunotherapy 2019—latest trends. J Exp Clin Cancer Res. 2019;38:268. https://doi.org/10.1186/s13046-019-1266-0.
Hiltbold EM, Ciborowski P, Finn OJ. Naturally processed class II epitope from the tumor antigen MUC1 primes human CD4+ T cells. Cancer Res. 1998;58:5066–70.
Campi G, Crosti M, Consogno G, Facchinetti V, Conti-Fine BM, Longhi R, et al. CD4+ T cells from healthy subjects and colon cancer patients recognize a carcinoembryonic antigen-specific immunodominant epitope. Cancer Res. 2003;63:8481–6.
Jager E, Jager D, Karbach J, Chen YT, Ritter G, Nagata Y, et al. Identification of NY-ESO-1 epitopes presented by human histocompatibility antigen (HLA)-DRB4*0101–3 and recognized by CD4(+) T lymphocytes of patients with NY-ESO-1-expressing melanoma. J Exp Med. 2000;191:625–30. https://doi.org/10.1084/jem.191.4.625.
Gnjatic S, Atanackovic D, Jäger E, Matsuo M, Selvakumar A, Altorki NK, et al. Survey of naturally occurring CD4+ T cell responses against NY-ESO-1 in cancer patients: correlation with antibody responses. Proc Natl Acad Sci USA. 2003;200:8862–7.
Francois V, Ottaviani S, Renkvist N, Stockis J, Schuler G, Thielemans K, et al. The CD4(+) T-cell response of melanoma patients to a MAGE-A3 peptide vaccine involves potential regulatory T cells. Cancer Res. 2009;69:4335–45. https://doi.org/10.1158/0008-5472.CAN-08-3726.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. https://doi.org/10.1158/1078-0432.CCR-11-0116.
Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–5.
Keene JA, Forman J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med. 1982;155:768–82. https://doi.org/10.1084/jem.155.3.768.
Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–3.
Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–80.
Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997;186:65–70. https://doi.org/10.1084/jem.186.1.65.
Ossendorp F, Mengede E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med. 1998;187:693–702. https://doi.org/10.1084/jem.187.5.693.
Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–601. https://doi.org/10.4049/jimmunol.174.5.2591.
Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368–77. https://doi.org/10.1158/0008-5472.CAN-10-1322.
Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol Ther. 2011;19:990–9. https://doi.org/10.1038/mt.2010.289.
Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, et al. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res. 2013;73:19–29. https://doi.org/10.1158/0008-5472.CAN-12-1127.
Disis ML, Wallace DR, Gooley TA, Dang Y, Slota M, Lu H, et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J Clin Oncol. 2009;27:4685–92. https://doi.org/10.1200/JCO.2008.20.6789.
Piesche M, Hildebrandt Y, Zettl F, Chapuy B, Schmitz M, Wulf G, et al. Identification of a promiscuous HLA DR-restricted T-cell epitope derived from the inhibitor of apoptosis protein survivin. Hum Immunol. 2007;68:572–6. https://doi.org/10.1016/j.humimm.2007.03.007.
Wang XF, Kerzerho J, Adotevi O, Nuyttens H, Badoual C, Munier G, et al. Comprehensive analysis of HLA-DR- and HLA-DP4-restricted CD4+ T cell response specific for the tumor-shared antigen survivin in healthy donors and cancer patients. J Immunol. 2008;181:431–9. https://doi.org/10.4049/jimmunol.181.1.431.
Berinstein NL, Karkada M, Oza AM, Odunsi K, Villella JA, Nemunaitis JJ, et al. Survivin-targeted immunotherapy drives robust polyfunctional T cell generation and differentiation in advanced ovarian cancer patients. Oncoimmunology. 2015;4:e1026529. https://doi.org/10.1080/2162402X.2015.1026529.
Brunsvig PF, Kyte JA, Kersten C, Sundstrom S, Moller M, Nyakas M, et al. Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res. 2011;17:6847–57. https://doi.org/10.1158/1078-0432.CCR-11-1385.
Godet Y, Fabre E, Dosset M, Lamuraglia M, Levionnois E, Ravel P, et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: potential synergistic effect with chemotherapy response. Clin Cancer Res. 2012;18:2943–53. https://doi.org/10.1158/1078-0432.CCR-11-3185.
Adotevi O, Dosset M, Galaine J, Beziaud L, Godet Y, Borg C. Targeting antitumor CD4 helper T cells with universal tumor-reactive helper peptides derived from telomerase for cancer vaccine. Hum Vaccin Immunother. 2013;9:1073–7. https://doi.org/10.4161/hv.23587.
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81–85. https://doi.org/10.1038/nm.3773.
Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–6. https://doi.org/10.1038/nature14426.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–21. https://doi.org/10.1038/nature22991.
Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–6. https://doi.org/10.1038/nature23003.
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–40. https://doi.org/10.4049/jimmunol.179.8.5033.
Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487–502. https://doi.org/10.1016/j.cell.2016.12.022.
Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4(+) T cell help is required for the formation of a cytolytic CD8(+) T cell subset that protects against chronic infection and cancer. Immunity. 2019;51:1028–42. https://doi.org/10.1016/j.immuni.2019.10.009.
Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. https://doi.org/10.1038/s41586-019-1671-8.
Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50. https://doi.org/10.1084/jem.20091918.
Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–67. https://doi.org/10.1084/jem.20091921.
Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T cells restrain interleukin-2- and Blimp-1-dependent acquisition of cytotoxic function by CD4(+) T cells. Immunity. 2020;52:e156. https://doi.org/10.1016/j.immuni.2019.12.007.
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–72. https://doi.org/10.1038/s41586-018-0694-x.
Galaine J, Turco C, Vauchy C, Royer B, Mercier-Letondal P, Queiroz L, et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int J Cancer. 2019;145:3112–25. https://doi.org/10.1002/ijc.32620.
Kagamu H, Kitano S, Yamaguchi O, Yoshimura K, Horimoto K, Kitazawa M, et al. CD4(+) T-cell immunity in the peripheral blood correlates with response to anti-PD-1 therapy. Cancer Immunol Res. 2019. https://doi.org/10.1158/2326-6066.CIR-19-0574.
Laheurte C, Dosset M, Vernerey D, Boullerot L, Gaugler B, Gravelin E, et al. Distinct prognostic value of circulating anti-telomerase CD4(+) Th1 immunity and exhausted PD-1(+)/TIM-3(+) T cells in lung cancer. Br J Cancer. 2019;121:405–16. https://doi.org/10.1038/s41416-019-0531-5.
Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC. Regulatory T cells in the treatment of disease. Nat Rev Drug Disco. 2018;17:823–44. https://doi.org/10.1038/nrd.2018.148.
Owen DL, Sjaastad LE, Farrar MA. Regulatory T cell development in the thymus. J Immunol. 2019;203:2031–41. https://doi.org/10.4049/jimmunol.1900662.
Savage PA, Leventhal DS, Malchow S. Shaping the repertoire of tumor infiltrating effector and regulatory T cells. Immunol Rev. 2014;259:245–58.
Legoux FP, Lim JB, Cauley AW, Dikiy S, Ertelt J, Mariani TJ, et al. CD4+ T cell tolerance to tissue-restricted self antigens is mediated by antigen-specific regulatory T cells rather than deletion. Immunity. 2015;43:896–908. https://doi.org/10.1016/j.immuni.2015.10.011.
Savage PA, Klawon DEJ, Miller CH. Regulatory T cell development. Annu Rev. Immunol. 2020. https://doi.org/10.1146/annurev-immunol-100219-020937.
Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. https://doi.org/10.1016/j.coi.2013.12.005.
Jorgensen N, Persson G, Hviid TVF. The Tolerogenic Function of Regulatory T Cells in Pregnancy and Cancer. Front Immunol. 2019;10:911. https://doi.org/10.3389/fimmu.2019.00911.
Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N. Y Acad Sci. 2018;1417:104–15. https://doi.org/10.1111/nyas.13625.
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–46. https://doi.org/10.1016/j.immuni.2007.08.014.
Guipouy D, Gertner-Dardenne J, Pfajfer L, German Y, Belmonte N, Dupre L. Granulysin- and granzyme-dependent elimination of myeloid cells by therapeutic ova-specific type 1 regulatory T cells. Int Immunol. 2019;31:239–50. https://doi.org/10.1093/intimm/dxy083.
Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol. 2007;178:2883–92. https://doi.org/10.4049/jimmunol.178.5.2883.
Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res. 2006;66:4488–95. https://doi.org/10.1158/0008-5472.CAN-05-4217.
Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, et al. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. 2015;64:419–27. https://doi.org/10.1007/s00262-014-1651-7.
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679–84. https://doi.org/10.1038/nm.4086.
Su S, Liao J, Liu J, Huang D, He C, Chen F, et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017;27:461–82. https://doi.org/10.1038/cr.2017.34.
Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest. 2009;119:3311–21. https://doi.org/10.1172/JCI39608.
Han S, Toker A, Liu ZQ, Ohashi PS. Turning the tide against regulatory T cells. Front Oncol. 2019;9:279. https://doi.org/10.3389/fonc.2019.00279.
Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. 2019;25:6614–22. https://doi.org/10.1158/1078-0432.CCR-19-1090.
Montler R, Bell RB, Thalhofer C, Leidner R, Feng Z, Fox BA, et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin Transl Immunol. 2016;5:e70. https://doi.org/10.1038/cti.2016.16.
Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018;28:416–32. https://doi.org/10.1038/s41422-018-0011-0.
Scirka B, Szurek E, Pietrzak M, Rempala G, Kisielow P, Ignatowicz L, et al. Anti-GITR antibody treatment increases TCR repertoire diversity of regulatory but not effector T cells engaged in the immune response against B16 melanoma. Arch Immunol Ther Exp (Warsz). 2017;65:553–64. https://doi.org/10.1007/s00005-017-0479-1.
Müller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Trans Med. 2015;7:ra188.
Ayyoub M, Pignon P, Classe JM, Odunsi K, Valmori D. CD4+ T effectors specific for the tumor antigen NY-ESO-1 are highly enriched at ovarian cancer sites and coexist with, but are distinct from, tumor-associated Treg. Cancer Immunol Res. 2013;1:303–8. https://doi.org/10.1158/2326-6066.CIR-13-0062-T.
Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, et al. Analysis of the T-cell receptor repertoires of tumor-infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen-induced tumors. Cancer Res. 2011;71:736–46. https://doi.org/10.1158/0008-5472.CAN-10-1797.
Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Trans Med. 2018;10:eaan4488.
Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. OX40 costimulation inhibits Foxp3 expression and treg induction via BATF3-dependent and independent mechanisms. Cell Rep. 2018;24:607–18. https://doi.org/10.1016/j.celrep.2018.06.052.
Xiao X, Shi X, Fan Y, Zhang X, Wu M, Lan P, et al. GITR subverts Foxp3(+) Tregs to boost Th9 immunity through regulation of histone acetylation. Nat Commun. 2015;6:8266. https://doi.org/10.1038/ncomms9266.
Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell. 2017;170:1096–108. https://doi.org/10.1016/j.cell.2017.08.004.
Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 2018;23:3262–74. https://doi.org/10.1016/j.celrep.2018.05.050.
Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci USA. 2016;113:6248–53. https://doi.org/10.1073/pnas.1604765113.
Yates K, Bi K, Haining WN, Cantor H, Kim HJ. Comparative transcriptome analysis reveals distinct genetic modules associated with Helios expression in intratumoral regulatory T cells. Proc Natl Acad Sci USA. 2018;115:2162–7. https://doi.org/10.1073/pnas.1720447115.
Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanovic S, Robbins PF et al. Tumor-infiltrating human CD4(+) regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. 2019;4. https://doi.org/10.1126/sciimmunol.aao4310.
Garfall AL, Dancy EK, Cohen AD, Hwang WT, Fraietta JA, Davis MM, et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019;3:2812–5. https://doi.org/10.1182/bloodadvances.2019000600.
Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. 2018;3. https://doi.org/10.1172/jci.insight.99048.
Yang Y, Kohler ME, Chien CD, Sauter CT, Jacoby E, Yan C, et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Trans Med. 2017;9:eaag1209.
Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med. 2009;206:1495–503. https://doi.org/10.1084/jem.20090681.
Gacerez AT, Sentman CL. T-bet promotes potent antitumor activity of CD4(+) CAR T cells. Cancer Gene Ther. 2018;25:117–28. https://doi.org/10.1038/s41417-018-0012-7.
Xhangolli I, Dura B, Lee G, Kim D, Xiao Y, Fan R. Single-cell analysis of CAR-T cell activation reveals a mixed TH1/TH2 response independent of differentiation. Genomics Proteom Bioinforma. 2019;17:129–39. https://doi.org/10.1016/j.gpb.2019.03.002.
Tsuji T, Matsuzaki J, Ritter E, Miliotto A, Ritter G, Odunsi K, et al. Split T cell tolerance against a self/tumor antigen: spontaneous CD4+ but not CD8+ T cell responses against p53 in cancer patients and healthy donors. PLoS ONE. 2011;6:e23651. https://doi.org/10.1371/journal.pone.0023651.
Ohue Y, Eikawa S, Okazaki N, Mizote Y, Isobe M, Uenaka A, et al. Spontaneous antibody, and CD4 and CD8 T-cell responses against XAGE-1b (GAGED2a) in non-small cell lung cancer patients. Int J Cancer. 2012;131:E649–658. https://doi.org/10.1002/ijc.27359.
Quandt J, Schlude C, Bartoschek M, Will R, Cid-Arregui A, Scholch S, et al. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. Oncoimmunology. 2018;7:e1500671. https://doi.org/10.1080/2162402X.2018.1500671.
Vauchy C, Gamonet C, Ferrand C, Daguindau E, Galaine J, Beziaud L, et al. CD20 alternative splicing isoform generates immunogenic CD4 helper T epitopes. Int J Cancer. 2015;137:116–26. https://doi.org/10.1002/ijc.29366.
Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. https://doi.org/10.1038/ncomms10582.
Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Trans Med. 2018;10:eaar3442.
Johnson DB, Nixon MJ, Wang Y, Wang DY, Castellanos E, Estrada MV, et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight. 2018;3. https://doi.org/10.1172/jci.insight.120360.
Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC-II expression by tumor cells in cancer. Clin Cancer Res. 2019;25:2392–402. https://doi.org/10.1158/1078-0432.Ccr-18-3200.
de Charette M, Marabelle A, Houot R. Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? Eur J Cancer. 2016;68:134–47. https://doi.org/10.1016/j.ejca.2016.09.010.
Thibodeau J, Bourgeois-Daigneault MC, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology. 2012;1:908–16. https://doi.org/10.4161/onci.21205.
Calabro S, Liu D, Gallman A, Nascimento MS, Yu Z, Zhang TT, et al. Differential intrasplenic migration of dendritic cell subsets tailors adaptive immunity. Cell Rep. 2016;16:2472–85. https://doi.org/10.1016/j.celrep.2016.07.076.
Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity. 2015;43:554–65. https://doi.org/10.1016/j.immuni.2015.07.020.
Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell. 2015;162:1322–37. https://doi.org/10.1016/j.cell.2015.08.004.
Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol. 2018;9:2144. https://doi.org/10.3389/fimmu.2018.02144.
Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hulsdunker J, Varelias A, et al. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity. 2019;51:885–98 e887. https://doi.org/10.1016/j.immuni.2019.08.011.
Arebro J, Tengroth L, Razavi R, Kumlien Georen S, Winqvist O, Cardell LO. Antigen-presenting epithelial cells can play a pivotal role in airway allergy. J Allergy Clin Immunol. 2016;137:957–60. https://doi.org/10.1016/j.jaci.2015.08.053.
Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185–98. https://doi.org/10.1016/j.semcancer.2015.03.004.
Muenst S, Laubli H, Soysal SD, Zippelius A, Tzankov A, Hoeller S. The immune system and cancer evasion strategies: therapeutic concepts. J Intern Med. 2016;279:541–62. https://doi.org/10.1111/joim.12470.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–77. https://doi.org/10.1038/nrc3258.
Sun NY, Chen YL, Wu WY, Lin HW, Chiang YC, Chang CF, et al. Blockade of PD-L1 enhances cancer immunotherapy by regulating dendritic cell maturation and macrophage polarization. Cancers (Basel). 2019;11. https://doi.org/10.3390/cancers11091400.
Benencia F, Muccioli M, Alnaeeli M. Perspectives on reprograming cancer-associated dendritic cells for anti-tumor therapies. Front Oncol. 2014;4:72. https://doi.org/10.3389/fonc.2014.00072.
König R, Huang L-Y, Germain RN. MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature. 1992;356:796–8. https://doi.org/10.1038/356796a0.
Spits H. Development of alphabeta T cells in the human thymus. Nat Rev Immunol. 2002;2:760–72. https://doi.org/10.1038/nri913.
Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–55. https://doi.org/10.1016/j.immuni.2009.05.001.
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2020;28:445–89. https://doi.org/10.1146/annurev-immunol-030409-10121.
Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol. 2016;16:102–11. https://doi.org/10.1038/nri.2015.10.
Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635–47. https://doi.org/10.1038/s41577-018-0044-0.
Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, et al. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5:1143–8. https://doi.org/10.1038/ni1129.
Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–79. https://doi.org/10.1084/jem.194.6.769.
Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. CD4(+) T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. 2017;47:848. https://doi.org/10.1016/j.immuni.2017.10.009.
Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–71. https://doi.org/10.4049/jimmunol.171.10.5165.
Janssen EM, Lemmens E, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6.
Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–9.
Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–42.
Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol. 2014;44:69–79. https://doi.org/10.1002/eji.201343718.
Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–44.
Takeuchi A, Badr Mel S, Miyauchi K, Ishihara C, Onishi R, Guo Z, et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J Exp Med. 2016;213:123–38. https://doi.org/10.1084/jem.20150519.
Matsuzaki J, Tsuji T, Luescher IF, Shiku H, Mineno J, Okamoto S. et al. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. 2015;5:14896. https://doi.org/10.1038/srep14896.
Reed CM, Cresce ND, Mauldin IS, Slingluff CL Jr., Olson WC. Vaccination with melanoma helper peptides induces antibody responses associated with improved overall survival. Clin Cancer Res. 2015;21:3879–87. https://doi.org/10.1158/1078-0432.CCR-15-0233.
Dieu-Nosjean M-C, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. 2016;271:260–75.
Author information
Authors and Affiliations
Contributions
R.E.T., E.K.R. and H.C.T. did literature research and wrote the paper. H.C.T. provided editorial supervision.
Corresponding author
Ethics declarations
Conflict of interest
H.C.T. is the Chief Medical Officer of Tessa Therapeutics Limited.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tay, R.E., Richardson, E.K. & Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther 28, 5–17 (2021). https://doi.org/10.1038/s41417-020-0183-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-0183-x
