
Abstract
More than 24 regulators have been revealed to dynamically participant in N6-methyladenosine (m6A) RNA methylation, and play critical roles in tumorigenesis and development of cancers. However, their functional roles have not been comprehensively clarified in breast cancer. Here we systematically analyzed the RNA sequencing data of 24 main m6A RNA methylation regulators in 775 breast cancer patients from The Cancer Genome Atlas dataset. Consensus clustering of the 24 m6A regulators was carried out and identified two patient subgroups, RNA methylation 1/2 (RM1/2). RM1 demonstrated generally lower RNA methylation modification than that of RM2, and had significantly shorter overall survival. The hallmarks of PI3K/AKT signaling in cancer, KRAS signaling and angiogenesis were significantly enriched in RM1. Moreover, the association between m6A regulators and antitumor immune response was also investigated in this study and revealed that RM2 was associated with significantly higher expressions of HLA-A, higher numbers of tumor-infiltrating CD8+ T cells, helper T cells and activated NK cells, but lower expressions of PD-L1, PD-L2, TIM3, and CCR4 than RM1. In conclusion, the expression pattern of m6A regulators was significantly correlated with the malignancy, prognosis and antitumor immune response in breast cancer, which might serve as potential targets and biomarkers for immunotherapy.
Similar content being viewed by others


Introduction
Breast cancers are highly heterogenous neoplasms, driven by complex signaling pathways and demonstrated distinct biological features across molecular subtypes [1]. Although incremental progress has been achieved in the prognosis of patients with early stage breast cancer over the past three decades, advanced stage breast cancer and distant disease relapse still remain incurable with the current available treatment modalities, including surgery, chemotherapy, endocrine therapy, radiotherapy, and targeted therapy [2].
Based on the improved understanding of immune invasion by cancer cells and discovery of selective immune checkpoint inhibitors, especially monoclonal antibodies against programmed death 1 (PD-1) and programmed death-ligand 1 (PD-L1), immunotherapy is emerging as a novel treatment modality in malignant tumors [3]. With regard to breast cancer, clinical trials of immunotherapy have attracted much attention in the past few years since the promising therapeutic effect of immunotherapy in combination with other strategies. As of September, 2018, the number of registered trials open to breast cancer patients that assess novel approaches harnessing the immune system has reached up to 285 [4]. However, notably, immune responses vary distinctly among different breast cancer subtypes and even individuals, which could be partially attributed to the number of tumor-infiltrating lymphocytes and heterogeneity of immune-related gene expression signatures [5]. Therefore, identifying the biological predictive markers of immune response has been highly underscored when conducting clinical trials of immunotherapy.
N6-methyladenosine (m6A), methylated at the N6 position of adenosine, has been identified to play a critical role in regulating RNA transcription, processing, translation, and metabolism [6,7,8]. These biological functions of m6A-based modification are dynamically regulated by a methyltransferases complex involving RNA methyltransferases (termed as “writers”), demethylases (termed as “erasers”), and m6A-binding proteins (termed as “readers”). Accumulating evidences demonstrated that m6A methylation participated in the pathogenesis and development of multiple cancers [9, 10], including bladder cancer [11], gastric cancer [12], acute myeloid leukemia [13, 14], glioblastoma [15, 16], and liver cancer [17, 18]. For breast cancer, methyltransferase like 3 (METTL3), a key m6A methyltransferase, has been revealed to be associated with the expression of mammalian hepatitis B X-interacting protein, and thus promote the progression of breast cancer through inhibiting tumor suppressor let-7g [19]. However, until now, it has not been comprehensively clarified the prognostic effect of m6A modification in breast cancer, and the potential roles of m6A modification in host antitumor immune response remain largely unexplored.
Based on the concerns mentioned above, we systematically analyzed the RNA sequencing data of 24 main m6A RNA methylation regulators in 775 breast cancer patients from The Cancer Genome Atlas (TCGA) dataset, exploring their expression pattern and prognostic effect in different breast cancer molecular subtypes, as well as their fundamental roles in antitumor immune response. This current study aimed to provide a better understanding of the biological function of m6A regulators in breast cancer, and shed new light on the potential predictive markers to immunotherapeutic response.
Materials and methods
Data sources
The RNA-seq transcriptome data and corresponding clinicopathological information of 775 breast cancer patients were extracted from TCGA (http://cancergenome.nih.gov/) dataset (version 2016_01_28 for BRCA), among which only 97 had information about tumor tissues and matched normal tissues. Genome, transcript sequences and annotations were obtained from the human genome (GRCh37), version 19 (Ensembl 74) (https://www.encodeproject.org/). Immune cell fraction data were downloaded through CIBERSORT (https://cibersort.stanford.edu/). And the expressions of antigen presenting genes and immunomodulator genes were obtained from TCIA (https://tcia.at/home).
Selection and expression of m6A RNA methylation regulators
We first collated a list of m6A RNA methylation regulators from previous published literature, and then restricted the list to the genes with available RNA expression data in TCGA dataset, which ultimately yielded a total of 24 m6A regulators in this study [20,21,22]. These regulators were known as three categories based on their functions (Supplementary table S1). Then, we systematically compared the expression levels of m6A regulators between tumor tissues and matched normal tissues, as well as among different breast cancer subtypes. Each m6A regulator gene was dichotomized into low or high expression level, with cutoff value defining as the median expression value. The criterion for breast cancer subtype classification was according to the PAM50 [23].
Cluster and principal component analysis
Considering the interaction across m6A regulators, we clustered the patients into different subgroups using the R package “ConsensusClusterPlus” (version 1.46.0, 50 iterations, resample rate of 80%, and Pearson correlation) based on the expression levels of these 24 m6A regulators. And principal component analysis, was used to identify the most important contributing factor for each subgroup.
GO pathway and GSEA analysis
To better understating the association between m6A regulators and malignancy of breast cancer, Gene Ontology (GO) pathway analysis and gene set enrichment analysis (GSEA) were performed to functionally annotate genes that were differentially expressed in different subgroups. Differentially expressed genes were regarded as genes with a fold-change > 2.0 and adjusted p-value < 0.05. A p-value (the significance threshold of the hypergeometric test) <0.05 was used as the cutoff criteria for GO analysis. |Normalized enrichment score | > 1, p-value < 0.05 and false discovery rate q-value < 0.25 were considered as statistically significant in GSEA.
CIBERSORT analysis
CIBERSORT, a bioinformatic algorithm for characterizing cell composition of complex tissues from their gene expression profiles, was performed to estimate tumor-infiltrating cell composition in tumor tissues by running with the validated LM22 gene signature matrix of leukocytes [24].
Statistical analysis
One-way ANOVA and t-test were carried out to compare the expression levels of m6A regulators in different breast cancer subtypes, and between normal and matched tumor tissues, respectively. Chi-square test was used to evaluate the differences of clinicopathological characteristics between subgroups, and Kaplan–Meier plots and Log-rank test were performed to estimate and compare their survival rates. Comparison of immune cell fractions, antigen presenting genes and immunomodulator genes expressions between subgroups was analyzed by Wilcoxon signed-rank test. All statistical analyses were performed using R software (version 3.5.2, http://www.r-project.org/) and Bioconductor (version 3.0, http://bioconductor.org/). A two-sided p-value < 0.05 was considered statistically significant in all analyses.
Results
Inconsistent expression pattern of m6A regulators across different clinicopathological features
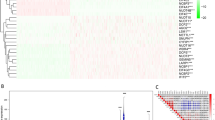
The expression pattern of the 24 m6A regulators between 97 tumor tissues and matched normal tissues was inconsistent, which was the same among different clinicopathological features in 775 breast cancer patients (Fig. 1, Supplementary Figs. S1 and S2). For example, compared to normal tissues, the expressions of METTL3, METTL14, WTAP, ZCCHC4, and FTO increased significantly in matched tumor tissues, while the expressions of YTHDF1, YTHDF2, and YTHDF3 decreased (Fig. 1a). Besides, basal-like subtype had significantly higher expressions of WTAP, YTHDF2, HNRNPD, and RBM15, but lower expressions of METTL3, METTL14, YTHDC2, and MSI2 than other subtypes (Fig. 1b). In addition, patients with lymph node metastasis had significantly higher expression of YTHDF1 and lower expressions of WTAP, HNRNPD, and RBM15 than those without lymph node metastasis (Fig. 1c). Compared to patients without distant metastasis, HNRNPC, HNRNPD, TRA2A, and GNL3 expressed significantly lower in those with distant metastasis (Fig. 1d). Altogether, the expression pattern of m6A regulators was inconsistent among different clinicopathological features, which indicated the crucial but complicated roles of m6A regulators in tumorigenesis and development of breast cancer.

a Box plots showed the expression distribution of METTL3, METTL14, WTAP, ZCCHC4, FTO, YTHDF1, YTHDF2, and YTHDF3 between tumor tissues and matched normal tissues. b Box plots showed the pairwise comparison of expression levels of WTAP, YTHDF2, HNRNPD, RBM15, METTL3, METTL14, YTHDC2, and MSI2 between base-like subtype and other breast cancer subtypes. *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. c Expression distribution of WTAP, YTHDF1, HNRNPD, and RBM15 in patients with or without lymph node metastasis. d Expression distribution of HNRNPC, HNRNPD, TRA2A, and GNL3 in patients with or without distant metastasis.
Consensus clustering of m6A regulators identified two clusters with distinct survival outcomes
Considering the interaction across different functional groups of m6A regulators, consensus clustering of the 24 m6A regulators was performed to investigate their association with malignancy of breast cancer. According to the expression similarity of m6A regulators, k = 2 seemed to be the optimal selection when clustering stability increased from k = 2 to 10 in the TCGA dataset (Supplementary Fig. S3). Therefore, we divided the 775 breast cancer patients into two subgroups by making 2 as the k value, namely, RNA methylation 1/2 (RM1/2). RM2 was significantly associated with younger age at diagnosis (p = 0.033), more basal-like molecular subtype (p = 0.010), lower lymph node stage (p = 0.022), and less distant metastasis events (p = 0.035) than RM1 (Table 1). Furthermore, patients in RM2 had significantly longer overall survival than those in RM1 (median overall survival 17.69 vs 10.65 years, p = 0.007, Fig. 2a). Specifically, we found that lower expressions of KIAA1429, YTHDF1, YTHDF3, or MSI2 were significantly correlated with better clinical outcomes (Fig. 2b–e). Therefore, consensus clustering of m6A regulators might serve as a potential prognostic factor for breast cancer.

a Overall survival (OS) curves for patients in RM1 and RM2 subgroups. b–e Overall survival (OS) curves for patients with high and low expressions of KIAA1429, YTHDF1, YTHDF3, or MSI2. RM RNA methylation.
RM1/2 subgroups were closely associated with malignancy of breast cancer
Figure 3a displayed the general RNA methylation modification of RM1/2 subgroups. Results demonstrated that the expressions of METTL3, a key m6A methyltransferase, and FTO, an important m6A demethylases, were significantly lower and higher, respectively, in RM1 than RM2, implying that the RNA methylation modification of RM1 might be generally lower than that of RM2. Then, principal component analysis was further performed to compare the transcriptional profile between RM1 and RM2 subgroups, and a prominent distinction was demonstrated between them (Supplementary Fig. S4). In particular, YTHDC1 and RBM27 were identified as the variables with the largest contribution to the first and second principal components of RM1 and RM2 subgroups, respectively. Furthermore, GO pathway analysis indicated that upregulated genes in RM2 were enriched in toll-like receptor signaling pathway, RAGE receptor binding, innate immune response-activating signal transduction and leukocyte migration involved in inflammatory response (Fig. 3b). GSEA revealed that malignant hallmarks of tumors, including KRAS signaling (NES = 1.840, normalized P = 0.008), ANGIOGENESIS (NES = 1.760, normalized P = 0.021), and PI3K/AKT signaling in cancer (NES = 1.670, normalized P = 0.011) were significantly enriched in RM1 (Fig. 3c). All these results suggested that RM1/2 subgroups identified by consensus clustering of m6A regulators were closely linked to the malignancy of breast cancer, and thus probably contributed to different survival outcomes.

a General RNA methylation modification in RM1/2 subgroups. Box plots showed the expression distribution of METTL3, YTHDC1, YTHDF2, HNRNPC, FTO, YTHDC2, YTHDF3, and RBM27 between RM1 and RM2 subgroups. b Representative functional annotation of upregulated genes in RM2 compared with RM1 by using GO in terms of biological process (BP), cellular component (CC), and molecular function (MF). Upregulated genes were defined as genes in RM2 with log2Fold-change > 1 and adjusted p-value < 0.05 compared with those in RM1. c GSEA revealed that upregulated genes in RM1 were enriched for hallmarks of malignant tumors. RM RNA methylation.
RM1/2 subgroups were significantly correlated with host antitumor immune response
To elucidate the correlation between expression pattern of m6A regulators and host antitumor immune response in breast cancer, we assessed the immune cell infiltration, expressions of antigen presenting genes and immunomodulator genes in tumor tissues between RM1/2 subgroups.
As demonstrated by Fig. 4a, RM2 had significantly higher numbers of tumor-infiltrating CD8+ T cells (p = 0.014), helper T cells (p < 0.001), and activated NK cells (p = 0.006) than RM1, suggesting an enhanced immunosurveillance in RM2. In parallel, the fraction of macrophage M2, which contributed to tumor promotion inflammation, was significantly lower in RM2 than in RM1 (p = 0.004). Of note, the regulatory T cell displayed a significantly higher fraction in RM2 (p = 0.017).

a Comparisons of cell composition fractions of tumor-infiltrating CD8+ T cells, CD4+ T cells, helper T cells, regulatory T cells, activated NK cells, monocytes, macrophage M1, macrophage M2, activated dendritic cells (activated DCs), and neutrophils between RM1 and RM2 subgroups. b Expressions of antigen presenting genes of HLA-A, HLA-B, HLA-C, TAP1, and B2M between RM1 and RM2 subgroups. c Expressions of immunomodulator genes of PD-1, PD-L1, PD-L2, LAG3, TIM3, CTLA-4, CCR4, TIGIT, CD27, and IDO1 between RM1 and RM2 subgroups. RM RNA methylation.
With regard to antigen presenting genes, we found that RM2 had a significantly higher expression of HLA-A than RM1 (P = 0.009, Fig. 4b), which is an important gene that regulates human MHC class I cell surface receptor and thus activates cytotoxic T cells. This finding suggested that tumors in RM2 had stronger immunogenicity, which was consistent with the higher numbers of active infiltrated immune cells presenting in RM2. Otherwise, there were no significant differences in expressions of HLA-B, HLA-C, TAP1, and B2M between RM1/2 subgroups.
In terms of immunomodulator genes, results implied that compared with RM1, RM2 was significantly associated with lower expressions of PD-L1, PD-L2, TIM3, and CCR4, and higher expressions of LAG3 (Fig. 4c), all of which are key genes of T-cell exhaustion markers. Taken together, the above results indicated the critical roles of m6A regulators in host antitumor immune response in breast cancer.
Discussion
RNA methylation has been revealed to be a widespread phenomenon and play a pivotal role in transcript expression, since the identification of the first RNA demethylase in a few years ago [25,26,27,28]. This new layer of regulation is termed as “epitranscriptomics”, differentiated from the traditional epigenetics, which is limited to DNA or protein modification [29, 30]. Increasing evidences have suggested that m6A modification participants in tumor proliferation, differentiation, invasion, and metastasis [31]. However, until now, the role of m6A regulators in the malignancy and prognosis of breast cancer has not been comprehensively clarified, especially in the field of host antitumor immune response.
To this end, the current study aimed at investigating the association between m6A regulators and the malignancy, prognosis and antitumor immune response of breast cancer, via performing consensus clustering of 24 commonly reported m6A regulators based on the RNA expression data from TCGA. Results revealed that RM1/2 subgroups identified by consensus clustering were significantly correlated with clinicopathological features, hallmarks of malignant tumor, and overall survival of breast cancer, as well as the immune cell infiltration, expressions of antigen presenting genes, and immunomodulator genes. To the best of our knowledge, this is the first study to systemically clarify the association of m6A regulators and host antitumor immune response in breast cancer, especially in terms of T-cell exhaustion markers.
m6A RNA modification is a dynamic and reversible process coordinated by a large methyltransferases complex made up of 3 different functional categories [25], whose roles are elucidated as “writers” (m6A methyltransferases), “erasers” (m6A demethylases), and “readers” (effectors recognizing m6A). However, these functional categories are closely interacted with each other [32], and even regulators in the same category can have distinct impacts in different cells [33], which makes it complicated and difficult to investigate the biological function of m6A regulators in tumorigenesis and prognosis of malignant tumors. Therefore, in this current study, we performed consensus clustering of the m6A regulators based on their expression similarity. Clustering is a widely used exploratory tool to identify similar groups with a consistent property, which allows more detailed biological insights into global regulation of gene expression and cellular function [34, 35]. Our results revealed that RM1/2 subgroups identified by consensus clustering demonstrated different extent of RNA methylation, and were significantly correlated with distinct clinicopathological features, survival outcomes and expressions of immune response genes in breast cancer, which was in line with the conclusions of previous studies [36, 37]. What’s more, a recently published study indicated that FTO, a key m6A demethylase, was negatively correlated with BNIP3 expression, a proapoptosis gene, and thus significantly associated with survival outcomes in breast cancer patients [38]. To sum up, our findings suggested that consensus clustering of m6A regulators was closely linked to the malignancy of breast cancer, which might serve as a potential prognostic factor in clinical therapeutic guidance.
Nowadays, the importance of epigenetic modification in antitumor immune response is an emerging focus of investigation. A recently published study found that loss of YTHDF1 in classical dendritic cells could enhance the cross-presentation of tumor antigen, cross-priming of CD8+ T cells and furthermore enhance the therapeutic efficacy of PD-L1 checkpoint blockade in melanoma [39]. Another study also demonstrated that METTL3 promoted dendritic cells maturation and function in T-cell activation in a m6A catalytic activity-dependent manner [40]. Consistent with the previous findings, our study revealed that RM1/2 subgroups were significantly correlated with the numbers of tumor infiltrated CD8+ T cells, helper T cells, activated NK cells, macrophage M2, and regulatory T cells, suggesting the critical roles of m6A regulators in host antitumor immune response. Furthermore, the expression pattern of m6A regulators was also significantly correlated with the expressions of PD-L1, TIM3, LAG3, and CCR4, which are well-known T-cell exhaustion markers and important targets in immunotherapy. In this aspect, our findings might provide potential targets for improving the response to immunotherapy in breast cancer.
One limitation of this study needs to be taken into consideration. All the analyses in the current study were based on the bioinformatics tools, therefore, further experimental validation is warranted to confirm the results of our study.
In conclusion, we demonstrated that the expression pattern of m6A regulators was significantly correlated with the clinicopathological features, survival outcomes and expressions of immunomodulator genes in breast cancer. This current study provided comprehensive evidence for further study of m6A regulators in breast cancer, and shed new light on the epigenetic regulation of antitumor immune response.
References
Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389(10074):1134–50.
Ponde NF, Zardavas D, Piccart M. Progress in adjuvant systemic therapy for breast cancer. Nat Rev Clin Oncol. 2019;16(1):27–44.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–86.
Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):e175–86.
Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32(27):2959–66.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. 2016;61(4):507–19.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 2015;161(6):1388–99.
Vu LP, Cheng Y, Kharas MG. The biology of m(6)A RNA methylation in normal and malignant hematopoiesis. Cancer Discov. 2019;9(1):25–33.
Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, et al. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol Cancer. 2019;18(1):137.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18(1):110.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193–205.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18(1):127.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–19.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol cell. 2019;74(4):640–50.
Yang Y, Hsu PJ, Chen Y-S, Yang Y-G. Dynamic transcriptomic mA decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616–24.
Meyer KD, Jaffrey SR. Rethinking mA readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506–19.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Nachtergaele S, He C. Chemical modifications in the life of an mRNA transcript. Annu Rev Genet. 2018;52:349–72.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–200.
Hsu PJ, Shi H, He C. Epitranscriptomic influences on development and disease. Genome Biol. 2017;18(1):197.
Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79(7):1285–92.
Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S, et al. Cross-talk among writers, readers, and erasers of m(6)A regulates cancer growth and progression. Sci Adv. 2018;4:eaar8263. https://doi.org/10.1126/sciadv.aar8263.
Chai RC, Wu F, Wang QX, Zhang S, Zhang KN, Liu YQ, et al. m(6)A RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging (Albany NY). 2019;11(4):1204–25.
Swift S, Tucker A, Vinciotti V, Martin N, Orengo C, Liu X, et al. Consensus clustering and functional interpretation of gene-expression data. Genome Biol. 2004;5(11):R94.
Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering—a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52(1-2):91–118.
Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2019;722:144076.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19(1):326.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270–4.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898.
Acknowledgements
We gratefully acknowledge Jean J. Zhao (Dana Farber Cancer Institute, Boston, Massachusetts, USA) for her suggestion in revising the paper, and the contributions from TCGA, TCIA, and CIBERSORT networks.
Funding
This work was supported by National Natural Science Foundation of China (81201747), Natural Science Foundation of Guangdong Province, China (S2012040006323, 2014A030313023), and the open Foundation of State Key Laboratory of Oncology in South China (HN2013-07).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
He, X., Tan, L., Ni, J. et al. Expression pattern of m6A regulators is significantly correlated with malignancy and antitumor immune response of breast cancer. Cancer Gene Ther 28, 188–196 (2021). https://doi.org/10.1038/s41417-020-00208-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-00208-1
This article is cited by
-
Guanylate binding protein 4 shapes an inflamed tumor microenvironment and identifies immuno-hot tumors
Journal of Cancer Research and Clinical Oncology (2024)
-
Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy
Molecular Cancer (2023)
-
m6A regulator-mediated methylation modification patterns and tumor microenvironment immune infiltration with prognostic analysis in esophageal cancer
Scientific Reports (2023)
-
Molecular phenotypic linkage between N6-methyladenosine methylation and tumor immune microenvironment in hepatocellular carcinoma
Journal of Cancer Research and Clinical Oncology (2023)
-
Targeting RNA N6-methyladenosine modification: a precise weapon in overcoming tumor immune escape
Molecular Cancer (2022)
