
Abstract
Angiotensin-converting enzyme 2 (ACE2) is a new component of the renin-angiotensin system (RAS). Accumulating evidence shows that ACE2 provides protective effects in peripheral tissues and has great potential for the treatment of RAS-related diseases. The role of ACE2 in the central nervous system is not well established. However, in recent years, much more progress has been made on the studies of this carboxypeptidase in the central regulation of blood pressure and cardiovascular function in general. It has been shown that brain ACE2 interacts with the other components of the RAS (ACE, angiotensin II, and angiotensin II type 1 receptor), protects baroreflex and autonomic function, stimulates nitric oxide release, reduces oxidative stress, and prevents the development of or attenuates hypertension. These data support the critical role of ACE2 in the central regulation of cardiovascular function. This review summarizes recently published data on the central effects of ACE2 in the regulation of cardiovascular function.
Similar content being viewed by others


Introduction
The renin-angiotensin system (RAS) plays a regulatory role in cardiovascular function and a pathogenic role in the development of hypertension and associated cardiovascular disorders. In addition to the classic endocrine system, local RASs are present in various tissues throughout the body, interacting with each other and the endocrine RAS.
Angiotensinogen (AGT) is the unique known precursor of angiotensin (Ang) peptides, which is transformed by renin to generate the decapeptide Ang I. Ang I is then converted by angiotensin-converting enzyme (ACE) into the octapeptide Ang II. Ang II is one of the major players in the RAS, acting on various receptors such as Ang II type 1 (AT1R) and type 2 receptors (AT2R). By binding to AT1R, Ang II promotes vasoconstriction, cell proliferation, and fibrosis, whereas when binding to AT2R it promotes the inhibition of cell growth. Most of the effects of Ang II have been shown to be mediated by AT1R. Increased activity of the RAS is associated with various cardiovascular diseases such as hypertension and heart failure. Classical treatment of RAS-associated diseases focuses on the blockade of Ang II formation or effects through the use of ACE inhibitors and AT1R blockers [1].
ACE2 is a new component of the RAS that was discovered in 2000 [2, 3]. It is the first known human homologue of ACE but differs from ACE in substrate specificity. Although ACE2 was first shown to cleave Ang I to Ang (1-9) [3], which can be converted by ACE into the heptapeptide Ang-(1-7), another study showed that ACE2 hydrolysis of Ang II into Ang-(1-7) has a much higher efficiency (∼400-fold) than that for Ang I to Ang (1-9) [4].
It has been established that Ang-(1-7) acting on the Mas receptor mediates vasodilation, antiproliferation, and apoptosis [5], therefore opposing the effects of Ang II. Using genetic and pharmacologic approaches, such as gene knockout, knockdown, gene transfer, activators and inhibitors, ACE2 has been shown to have protective effects in various tissues and to prevent overactive RAS-associated diseases, including hypertension [6]. Thus, the new arm of the RAS, the ACE2-Ang-(1-7)-Mas axis, has been shown to be effective at counter-regulating the effects of the classic ACE-Ang II-AT1R axis.
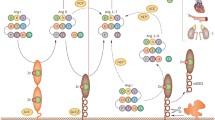
Brain RAS contains the same elements as the other tissue RAS. Besides Ang II and AT1R, all the other components of RAS such as AGT, renin, AT2R, ACE, Ang IV, Ang-(1-7), Mas, and new elements such as prorenin/renin receptor and Ang-(1-12), have been identified in the central nervous system [7, 8]. However, the distribution of the new element ACE2 in the brain was at first controversial because original reports failed to identify the carboxypeptidase in this tissue [2, 3]. A few years later, ACE2 mRNA and protein were found in the brain by our group and others [9–11]. Using a selective antibody, we reported that ACE2 is widespread throughout the mouse brain, present in nuclei involved in the central regulation of cardiovascular function such as the subfornical organ, paraventricular nucleus (PVN), nucleus of the tractus solitarius (NTS), and rostral ventrolateral medulla (RVLM), as well as in noncardiovascular areas such as the motor cortex and raphe (Fig. 1). Shortly after our observation, Lin et al. [12•] confirmed the presence of ACE2 mRNA and protein in the mouse brainstem and reported the interaction between ACE2 and AT1R in this region. These data suggest that ACE2 is actively involved in the brain RAS function.

Angiotensin-converting enzyme 2 (ACE2) expression in the mouse brain. ACE2 is widely expressed throughout the mouse brain, including in key brain regions involved in the regulation of blood pressure and body fluid homeostasis. This shows examples of ACE2 expression (red) in the subfornical organ (SFO), the neurons of the paraventricular nucleus (PVN), the nucleus of tractus solitarii (NTS), the dorsal motor nucleus of the vagus (DMNX), and the rostral ventrolateral medulla (RVLM). Green: MAP2 (neuronal marker); blue: DAPI (nuclei marker). CVLM—caudal ventrolateral medulla; IML—intermediolateral cell column; NA—nucleus ambiguus
In the central nervous system, Ang II acting on AT1R results in cardiac baroreflex desensitization and resetting, increased sympathetic outflow, vasopressin release, and stimulation of the water intake, leading to the increase in blood pressure [13]. It has been shown that upregulated AGT, ACE, Ang II, and AT1R in the brain are involved in the development and maintenance of hypertension and heart failure [7, 14]. Similar to the observations in the periphery, Ang-(1-7) shows opposite properties to Ang II in the central nervous system, serving as an important neuromodulator in the central control of cardiovascular function. It has been well demonstrated that central Ang-(1-7) increases cardiac baroreflex sensitivity [15, 16], reduces norepinephrine release [17] and decreases blood pressure [18, 19] in hypertensive rats. The depressor effects of Ang-(1-7) in the brain have also been shown to be mediated by increase in bradykinin levels [20], potentiating the hypotensive effects of bradykinin [21] and stimulating nitric oxide (NO) release [22]. ACE2 provides an important route for the metabolism of Ang II and, as the principal forming enzyme for Ang-(1-7), shifts the balance between the two biological active peptides.
However, compared with the well-established central effects of Ang II and Ang-(1-7), there are very few studies supporting the central nervous system effects of ACE2. Most of the studies have focused on the role of ACE2 in the kidney, heart, lung, liver, and other peripheral tissues, as evidenced by the numerous reviews on ACE2 [23–27]. However, with the discovery of ACE2 in the brain, the role of ACE2 in the central nervous system gained increasing attention from the scientific community. In recent years, growing evidence has demonstrated the important role of ACE2 in the central nervous system, such as in blood pressure control [28, 29•, 30, 31••].We previously reviewed the central effects of ACE2 and discussed the potential for this carboxypeptidase as a new target in the treatment of cardiovascular diseases [6]. In this report, we updated this information by reviewing the recent discoveries of the past 1 year to 2 years, specifically focusing on the role of ACE2 in the brain RAS and its effects on central regulation of cardiovascular function.
ACE2 Interacts with Other Components of the Brain RAS
Over the past 1 year to 2 years, more studies from various groups have demonstrated that ACE2 can affect or be affected by other components of the RAS in the central nervous system. An in vitro study from Xiao et al. [32] showed that overexpression of ACE2 in neurons attenuates the Ang II–induced upregulation of AT1R, which could result from inhibition of the well-known feed-forward effect of Ang II on its receptor. In vivo, our group showed that overexpression of ACE2 in the mouse brain not only reduces AT1R expression as previously suggested [30] but also increases Mas and AT2R [33••], whereas blockade of AT1R increases brain ACE2 activity in hypertensive mice [31••]. Using a gene-silencing approach, Lin et al. [12•] reported that downregulation of AT1R mRNA is associated with downregulation of ACE2 mRNA in the mouse brainstem. In addition, downregulated ACE2 is accompanied by upregulation of ACE levels in the brain of a heart failure animal model [34] and in multiple sclerosis patients [35], indicating a special link between ACE2 and ACE in the brain. These data confirmed and extend previous findings that ACE2 interacts with other RAS components in the brain and strengthens the concept that ACE2 is a significant player in the brain RAS.
ACE2 and Cardiovascular Diseases
Alteration of ACE2 expression or activity in the brain has been found in pathologic conditions related to cardiovascular diseases. Kawajiri et al. [35] reported decreased ACE2 and increased ACE concentration in cerebrospinal fluid from patients with multiple sclerosis compared to those with non-neurologic diseases. Similarly, downregulated ACE2 and upregulated ACE protein and mRNA expression were observed in various brain nuclei (including PVN, NTS, and RVLM ) from pacing-induced chronic heart failure (CHF) rabbits with increased sympathetic nerve activity [34]. These data suggest that the alteration of ACE/ACE2 in the brain is involved in the development of cardiovascular diseases. Moreover, we recently showed decreased ACE2 activity in the brain of a chronically hypertensive mouse model with increased Ang II level [31••]. These observations support the critical role of ACE2 in the central nervous system in the maintenance of normal cardiovascular function.
Because decreased ACE2 in the central nervous system is associated with multiple cardiovascular diseases, compensation by ACE2, as a result of increasing or activating this enzyme, in the brain could provide beneficial effects on these diseases. Several studies showed the effects of ACE2 on prevention or attenuation of hypertension. Using adenovirus coding for ACE2, Sriramula et al. [36] showed that PVN-targeted ACE2 overexpression attenuates the Ang II–induced pressor response in rats. Using a new transgenic mouse model (syn-hACE2) with neuron-targeted ACE2 overexpression, we found that the development of Ang II–induced neurogenic hypertension was blunted [33••]. In additional experiments, the syn-hACE2 mice were bred with chronically hypertensive transgenic mice expressing both human renin and AGT genes [37], to generate a triple-transgenic model (SARA) with elevated Ang II throughout the body and overexpression of human ACE2 selectively in the brain. Following chronic measurement of blood pressure in SARA mice, we observed that central overexpression of ACE2 significantly decreases baseline blood pressure in these chronically hypertensive mice [31••]. However, the blood pressure was not normalized due to the chronic and widespread expression of Ang II in the periphery. Nevertheless, our observations are consistent with previous reports from Yamazato et al. [28] and our group [30] showing the antihypertensive effects of ACE2 in specific brain nuclei, such as RVLM and subfornical organ. In addition to these gene transfer approaches, Cangussu et al. [38] showed that increases in ACE2 and Mas mRNA expression in the hypothalamus following exercise training in 2K1C rats are associated with a reduction of the increased systolic arterial pressure in these animals, although ACE2 levels were not altered in these hypertensive rats before exercise training compared with sham controls. Besides the role of ACE2 in the regulation of blood pressure, Lima et al. [39] showed that central activation of ACE2 by intracerebroventricular injection of XNT, an activator of endogenous ACE2, attenuates air jet stress-induced tachycardia in rats, suggesting the beneficial effects of ACE2 on cardiac function.
ACE2 Modulates Baroreflex and Autonomic Functions
Several studies have focused on the mechanisms involved in ACE2 regulation of blood pressure and other cardiovascular functions. Baroreflex sensitivity and autonomic function are important central mechanisms modulating blood pressure and other cardiovascular functions. It has been shown that inhibition of ACE2 activity in the NTS, by injecting a specific inhibitor in this region, reduces the sensitivity of the baroreceptor reflex control of heart rate to increases in arterial pressure [29•]. Similarly, we previously showed that ACE2 gene deletion on a C57bl/6 background mouse resulted in impairment of the spontaneous baroreflex sensitivity (SBRS), increased sympathetic tone, and decreased parasympathetic tone [40, 41]. Moreover, exercise training–induced increase in endogenous ACE2 in the brain of CHF rabbits was associated with an attenuation of sympathetic nerve activity in these animals [34]. Also, overexpressing ACE2 in the PVN, by virus-mediated gene transfer, improves renal sympathetic nerve activity in coronary ligation-induced CHF rats [42]. Global overexpression of ACE2 in the brain also restored the impaired SBRS, normalized the increased sympathetic tone, and decreased parasympathetic tone in transgenic hypertensive mice, contributing to the attenuation of hypertension in these animals [31••]. In low-dose Ang II infusion, a model of neurogenic hypertension, although ACE2 overexpression in the brain did not alter the increased sympathetic tone, it significantly prevented the decrease in both SBRS and parasympathetic tone and attenuated the development of hypertension [33••]. Similar observations were made by Gao et al. [43] showing that global overexpression of exogenous ACE2 in the brain prevents impairment of baroreflex sensitivity and sympathoexcitation in the CHF state.
These data suggest that ACE2 is involved in baroreflex control mechanisms and is able to modulate sympathetic and parasympathetic tones, thus participating in long-term and short-term regulation of blood pressure as well as cardiac function. The modulation of baroreflex and autonomic function by ACE2 could be independent of Ang II level, since measurement of Ang II in the brain and plasma showed no change in adult ACE2-deficient mice in which baroreflex and autonomic function were impaired [41]. However, the ACE2-mediated changes in baroreflex and autonomic function could also be resulting from, at least partially, metabolism of Ang II in the brain in some conditions, as decreased water intake following ACE2 overexpression in the brain was observed in both of the two hypertensive models mentioned above [31••, 33••]. Moreover, similar to the effects of Ang-(1-7) [15, 16], intracerebroventricular infusion of a novel ACE2 product peptide, Ala1-Ang-(1-7), increases baroreflex sensitivity for reflex bradycardia [44], suggesting the modulation of ACE2 on baroreflex function could also be triggered by its products.
ACE2 Mechanisms Involve NO Synthase and Reactive Oxygen Species Signaling
In the central nervous system, it has been shown that NO reduces, whereas oxidative stress increases, sympathetic activity [45–47]. Overexpressing ACE2 in the PVN normalized the decreased neuronal nitric oxide synthase (NOS) protein levels in this region in CHF rats and was accompanied with improved sympathetic nerve activity, suggesting the inhibitory effects of ACE2 on sympathoexcitation involve an NO mechanism [42]. Similarly, Gao et al. [43] showed that overexpression of ACE2 in the brain prevented the downregulation of NOS expression in the CHF mice, contributing to the improvement of baroreflex sensitivity and the reduction of sympathoexcitation. This is confirmed by our observation showing that overexpression of ACE2 in the brain upregulated NOS expression and increased NO levels in the cerebrospinal fluid [33••]. In addition, ACE2 overexpression in the brain prevented the Ang II–mediated decrease in NOS expression in regions modulating blood pressure regulation [33••]. Furthermore, we observed that ACE2 gene deletion leads to increased superoxide levels in various brain nuclei of aged mice [48], supporting the antioxidant capacity of ACE2 in the brain. These data indicate that the protective effects of ACE2 could be linked to an increased NO availability and decreased oxidative stress in the brain.
Conclusions and Perspectives
Clear progress has been made in our understanding of the role of ACE2 in the brain. The many beneficial effects of this carboxypeptidase on the central regulation of cardiovascular function could be independent or at least partially result from the modification of angiotensin peptides. The potential therapeutic implications of ACE2 suggest that it could be used to treat hypertension, heart failure, and other cardiovascular diseases. Increasing brain ACE2 by stimulating endogenous ACE2 activity and/or expression, or administration of exogenous ACE2 may provide beneficial effects in some pathologic conditions and this could be a new strategy for future development of novel therapeutic agents. Further understanding of its role in the central nervous system is needed because it would lead to the exploration of novel therapies for neurogenic hypertension and other cardiovascular as well as neurologic diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Paul M, Poyan Mehr A, Kreutz R: Physiology of local renin-angiotensin systems. Physiol Rev 2006, 86:747–803.
Tipnis SR, Hooper NM, Hyde R, et al.: A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000, 275:33238–33243.
Donoghue M, Hsieh F, Baronas E, et al.: A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000, 87:E1–E9.
Vickers C, Hales P, Kaushik V, et al.: Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 2002, 277:14838–14843.
Ferrario CM, Trask AJ, Jessup JA: Advances in the biochemical and functional roles of angiotensin converting enzyme 2 and angiotensin-(1-7) in the regulation of cardiovascular function. Am J Physiol Heart Circ Physiol 2005, 289:H2281–H2290.
Xia H, Lazartigues E: Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 2008, 107:1482–1494.
Phillips MI, de Oliveira EM: Brain renin angiotensin in disease. J Mol Med 2008, 86:715–722.
Nagata S, Kato J, Sasaki K, et al.: Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun 2006, 350:1026–1031.
Harmer D, Gilbert M, Borman R, et al.: Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 2002, 532:107–110.
Hamming I, Timens W, Bulthuis MLC, et al.: Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004, 203:631–637.
Doobay MF, Talman LS, Obr TD, et al.: Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 2007, 292:R373–R381.
• Lin Z, Chen Y, Zhang W, et al.: RNA interference shows interactions between mouse brainstem angiotensin AT1 receptors and angiotensin-converting enzyme 2. Exp Physiol 2008, 93:676–684. Using adenovirus vectors carrying small, hairpin RNA for AT1a or AT1b, the authors demonstrated that downregulated AT1a mRNA in the mouse brainstem resulted in decreased ACE2 mRNA expression in this region, whereas reduction in AT1b mRNA had no effect on ACE2.
Phillips MI, Sumners C: Angiotensin II in central nervous system physiology. Regul Pept 1998, 78:1–11.
Tan J, Wang H, Leenen FH: Increases in brain and cardiac AT1 receptor and ACE densities after myocardial infarct in rats. Am J Physiol Heart Circ Physiol 2004, 286:H1665–H1671.
Campagnole-Santos MJ, Heringer SB, Batista EN, et al.: Differential baroreceptor reflex modulation by centrally infused angiotensin peptides. Am J Physiol 1992, 263:R89–R94.
Heringer-Walther S, Batista EN, Walther T, et al.: Baroreflex improvement in SHR after ace inhibition involves angiotensin-(1-7). Hypertension 2001, 37:1309–1314.
Gironacci MM, Valera MS, Yujnovsky I, et al.: Angiotensin-(1-7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension 2004, 44:783–787.
Dobruch J, Paczwa P, Lon S, et al.: Hypotensive function of the brain angiotensin-(1-7) in Sprague Dawley and renin transgenic rats. J Physiol Pharmacol 2003, 54:371–381.
Höcht C, Gironacci MM, Mayer MA, et al.: Involvement of angiotensin-(1-7) in the hypothalamic hypotensive effect of captopril in sinoaortic denervated rats. Regul Pept 2008, 146:58–66.
Lu J, Zhang Y, Shi J: Effects of intracerebroventricular infusion of angiotensin-(1-7) on bradykinin formation and the kinin receptor expression after focal cerebral ischemia-reperfusion in rats. Brain Res 2008, 1219:127–135.
Bomtempo CA, Santos GF, Santos RA, et al.: Interaction of bradykinin and angiotensin-(1-7) in the central modulation of the baroreflex control of the heart rate. J Hypertens 1998, 16:1797–1804.
Gironacci MM, Vatta M, Rodriguez-Fermepin M, et al.: Angiotensin-(1-7) reduces norepinephrine release through a nitric oxide mechanism in rat hypothalamus. Hypertension 2000, 35:1248–1252.
Dean RG, Burrell LM: ACE2 and diabetic complications. Curr Pharm Des 2007, 13:2730–2735.
Raizada MK, Ferreira AJ: ACE2: A new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol 2007, 50:112–119.
Ferreira AJ, Raizada MK: Genomic and proteomic approaches for targeting of angiotensin-converting enzyme 2 for cardiovascular diseases. Curr Opin Cardiol 2008, 23:364–369.
Lambert DW, Hooper NM, Turner AJ: Angiotensin-converting enzyme 2 and new insights into the renin-angiotensin system. Biochem Pharmacol 2008, 75:781–786.
Ingelfinger JR: Angiotensin-converting enzyme 2: implications for blood pressure and kidney disease. Curr Opin Nephrol Hypertens 2009, 18:79–84.
Yamazato M, Yamazato Y, Sun C, et al.: Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension 2007, 49:926–931.
• Diz DI, Garcia-Espinosa MA, Gegick S, et al.: Injections of angiotensin-converting enzyme 2 inhibitor MLN4760 into nucleus tractus solitarii reduce baroreceptor reflex sensitivity for heart rate control in rats. Exp Physiol 2008, 93:694–700. Using an ACE2 inhibitor, MLN4760, in Sprague Dawley rats, the authors showed that inhibition of ACE2 activity in the NTS impaired the baroreflex sensitivity for control of heart rate in response to increases in arterial pressure. Moreover, there was no further reduction of these effects following combined Ang-(1-7) receptor blockade and ACE2 inhibition, suggesting that ACE2 is the main enzyme forming Ang-(1-7).
Feng Y, Yue X, Xia H, et al.: Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res 2008, 102:729–736.
•• Xia H, Feng Y, Obr TD, et al.: Angiotensin II type 1 receptor mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension 2009, 53:210–216. Using a chronically hypertensive model and a triple-transgenic model with chronic overexpression of Ang II and brain ACE2, the authors found that AT1R exerts an inhibitory effect on ACE2 activity in the brain of hypertensive mice, and that brain selective overexpression of ACE2 attenuated the hypertension by improving arterial baroreflex and autonomic function.
Xiao L, Gao L, Zucker IH: Angiotensin-converting enzyme 2 attenuates the angiotensin II-induced upregulation of angiotensin II type 1 receptor in CATH.a neurons. Hypertension 2009, 54:e70.
•• Feng Y, Xia H, Cai Y, et al.: Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res 2010, 106:373–382. Using a new transgenic mouse model (syn-hACE2) with neuron-targeted ACE2 overexpression, the investigators demonstrated that ACE2 overexpression attenuates the development of Ang II–induced hypertension by preventing the decrease in baroreflex sensitivity and parasympathetic tone. In addition, these protective effects could be mediated by increased NO availability in the brain, likely resulting from Mas and AT2R upregulation.
Kar S, Gao L, Zucker IH: Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing induced chronic heart failure. J Appl Physiol. 2010 Jan 21 [Epub ahead of print].
Kawajiri M, Mogi M, Higaki N, et al.: Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult Scler 2009, 15:262–265.
Sriramula S, Cardinate JP, Lazartigues E, et al.: Bilateral ACE2 overexpression in the PVN attenuates angiotensin II-induced blood pressure response. Hypertension 2009, 54:e97.
Merrill DC, Thompson MW, Carney CL, et al.: Chronic hypertension and altered baroreflex responses in transgenic mice containing the human renin and human angiotensinogen genes. J Clin Invest 1996, 97:1047–1055.
Cangussu LM, Silva JR, Alzamora AC, et al.: Increased hypothalamus expression of ACE2 and Mas receptor mRNA in renovascular hypertensive rats submitted to exercise training. [abstract No. 930]. Presented at the International American Society of Hypertension 17th Scientific Sessions. Belo Horizonte, Brazil; August 5–8, 2009.
Lima AM, Xavier CH, Santos RAS, et al.: Central activation of the ACE2-Ang-(1-7)-Mas axis markedly reduces the tachycardia evoked by acute stress exposure. Hypertension 2009, 54:e98.
Feng Y, Xia H, Lazartigues E: Alteration of baroreflex and autonomic function precedes the development of high blood pressure in angiotensin-converting enzyme 2 deficient mice. Hypertension 2009, 54:e97.
Xia H, Feng Y, Seth D, et al.: Impaired baroreflex and autonomic function in ACE2 knockout mice. Hypertension 2008, 52:e80.
Zheng H, Liu X, Moser JS, et al.: Gene transfer of angiotensin converting enzyme 2 to the paraventricular nucleus improves attenuated nitric oxide mechanism in rats with chronic heart failure. FASEB J 2009, 23:956.2.
Gao L, Farrar R, Wang W, et al.: Selective over expression of central ACE2 prevents baroreflex dysfunction in the chronic heart failure. FASEB J 2009, 23:610.2.
Villela DC, Verano TB, Campagnole-Santos MJ, et al.: Changes in the baroreflex control of heart rate produced by central infusion of a novel ACE2 product peptide, Ala1-Ang-(1-7). [abstract No. 1072]. Presented at the International American Society of Hypertension 17th Scientific Sessions. Belo Horizonte, Brazil; August 5–8, 2009.
Zanzinger J: Role of nitric oxide in the neural control of cardiovascular function. Cardiovasc Res 1999, 43:639–649.
Zanzinger J, Czachurski J: Chronic oxidative stress in the RVLM modulates sympathetic control of circulation in pigs. Pflugers Arch 2000, 439:489–494.
Gao L, Wang W, Li YL, et al.: Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res 2004, 95:937–944.
Xia H, Cai Y, Bindom S, et al.: ACE2 gene deletion mediates an age-dependent increase in oxidative stress in the central nervous system. Hypertension 2009, 54:e98.
Acknowledgments
This work was supported, in part, by an American Physiological Society Postdoctoral Fellowship to Dr. Huijing Xia and National Institutes of Health grants NS052479, RR018766, and HL093178 to Dr. Eric Lazartigues.
Disclosure
No potential conflict of interest relevant to this article was reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xia, H., Lazartigues, E. Angiotensin-Converting Enzyme 2: Central Regulator for Cardiovascular Function. Curr Hypertens Rep 12, 170–175 (2010). https://doi.org/10.1007/s11906-010-0105-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-010-0105-7