
Abstract
Rationale
We evaluated the effects of haloperidol and its metabolites on capsaicin-induced mechanical hypersensitivity (allodynia) and on nociceptive pain induced by punctate mechanical stimuli in mice.
Results
Subcutaneous administration of haloperidol or its metabolites I or II (reduced haloperidol) dose-dependently reversed capsaicin-induced (1 μg, intraplantar) mechanical hypersensitivity of the hind paw (stimulated with a nonpainful, 0.5-g force, punctate stimulus). The order of potency of these drugs to induce antiallodynic effects was the order of their affinity for brain sigma-1 (σ1) receptor ([3H](+)-pentazocine-labeled). Antiallodynic activity of haloperidol and its metabolites was dose-dependently prevented by the selective σ1 receptor agonist PRE-084, but not by naloxone. These results suggest the involvement of σ1 receptors, but discard any role of the endogenous opioid system, on the antiallodynic effects. Dopamine receptor antagonism also appears unlikely to be involved in these effects, since the D2/D3 receptor antagonist (−)-sulpiride, which had no affinity for σ1 receptors, showed no antiallodynic effect. None of these drugs modified hind-paw withdrawal after a painful (4 g force) punctate mechanical stimulus in noncapsaicin-sensitized animals. As expected, the control drug gabapentin showed antiallodynic but not antinociceptive activity, whereas clonidine exhibited both activities and rofecoxib, used as negative control, showed neither.
Conclusion
These results show that haloperidol and its metabolites I and II produce antiallodynic but not antinociceptive effects against punctate mechanical stimuli and suggest that their antiallodynic effect may be due to blockade of σ1 receptors but not to dopamine receptor antagonism.
Similar content being viewed by others


Introduction
Sigma (σ) receptors, initially considered a subtype of opioid receptors and later confused with phencyclidine binding sites in N-methyl-d-aspartate (NMDA) receptors, are now described as a distinct pharmacological entity (for reviews, see Guitart et al. 2004; Monnet and Maurice 2006). Two subtypes (σ1 and σ2) have been pharmacologically characterized. The σ1 receptor, cloned in several animal species and humans, has been described as a unique protein with no homology with known mammalian proteins (Guitart et al. 2004; Monnet and Maurice 2006). Drug binding to σ1 receptors is allosterically modulated by phenytoin (Quirion et al. 1992), and testing for this modulation has been proposed as a method to discriminate between σ1 receptor agonists and antagonists in vitro (Cobos et al. 2005, 2006). The pharmacology of σ1 receptors is now well-characterized, and selective agonists, such as (+)-pentazocine and PRE-084 [2-(4-morpholinethyl)1-phenylcyclohexanecarboxylate) hydrochloride], and antagonists, such as BD-1063 (1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride) and NE-100 (N, N-dipropyl-2-[4-methoxy-3-(2-phenylethoxy)phenyl]ethylamine hydrochloride), are both available (Guitart et al. 2004; Hayashi and Su 2004). Some neurosteroids, psychostimulants, and antipsychotics also bind to σ1 receptors (Maurice et al. 2001; Monnet and Maurice 2006; Cobos et al. 2008). Among the antipsychotics, haloperidol (HP) is mainly known as a D2 receptor antagonist, although it shows the same affinity for D2 and σ1 receptors (Bowen et al. 1990; Matsumoto and Pouw 2000) and exhibits σ1 receptor antagonistic activity (Maurice et al. 2001; Hayashi and Su 2004). Two major metabolic pathways for HP have been identified in experimental animals and humans (for references, see Cobos et al. 2007). One is a reversible reductive pathway that produces HP metabolite II (HP-Met-II), also called reduced HP [4-(4-chlorophenyl)-α-(4-fluorophenyl)-4-hydroxy-1-piperidinebutanol]. The other is an oxidative N-dealkylation pathway that leads to HP metabolites I (HP-Met-I, 4-(4-chlorophenyl)-4-hydroxypiperidine) and III (HP-Met-III, p-fluorobenzoylpropionic acid). Studies performed in rodent brain membranes and human neuroblastoma cells showed that metabolites I and II of HP bind to σ1 receptors with less affinity than HP, but show much lower (HP-Met-II) or no affinity (metabolite I) for D2 receptors, whereas metabolite III has no affinity for either σ1 or D2 receptors (Bowen et al. 1990; Matsumoto and Pouw 2000; Cobos et al. 2007).
Sigma-1 receptors are involved in nociception, among other processes. They are distributed in the central nervous system in areas of great importance for pain control, such as the superficial layers of the spinal cord dorsal horn, the periaqueductal gray matter, the locus coeruleus, and rostroventral medulla (Alonso et al. 2000; Kitaichi et al. 2000). Functional studies have postulated that an endogenous σ1 system tonically modulates the opioid system. The antinociception induced by agonists of opioid receptors in the tail flick test is antagonized by systemic administration of the selective σ1 agonist (+)-pentazocine, whereas it is enhanced by the σ1 antagonist HP (Chien and Pasternak 1993, 1994; Mei and Pasternak 2002, 2007). New σ1 ligands such as the σ1 antagonist (+)-MR 200 [(+)-methyl (1R,2S)-2-{[4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl]methyl}-1-phenylcyclopropanecarboxylate] and the proposed σ1 agonist (±)-PPCC [(1R,2S/1S,2R)-2-[4-hydroxy-4-phenylpiperidin-1-yl)methyl]-1-(4-methylphenyl) cyclopropanecarboxylate] also modulate opioid receptor agonist-induced antinociception (Marrazzo et al. 2006; Prezzavento et al. 2008). Sigma ligands are also able to modulate nociception per se (i.e., not associated to opioid agonists). Selective σ1 agonists induce nociception when used alone in the nociceptive flexor response test and the effects of (+)-pentazocine are reversed by selective σ1 receptor antagonists (Ueda et al. 2001). Moreover, both phases of pain behavior in the formalin test are diminished in σ1 receptor knockout mice (Cendán et al. 2005a) and after the systemic administration of the σ1 receptor antagonists HP and reduced HP (Cendán et al. 2005b). Pain behavior in the second phase of the formalin test is also reduced after intrathecal administration of the σ1 receptor antagonists BD-1047 (N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino ethylamine dihydrobromide) and BMY-14802 (α-(4-fluorophenyl)-4-(5-fluoro-2-pyrimidinyl)-1-piperazinebutanol) (Kim et al. 2006). However, the possible role of σ1 receptors in mechanical stimulus-induced pain is unknown.
The intradermal injection of capsaicin induces an immediate pain behavior response followed by longer-lasting secondary mechanical hypersensitivity (Gilchrist et al. 1996; Joshi et al. 2006). The mechanisms underlying the mechanical hypersensitivity (allodynia) produced by the intradermal injection of capsaicin and the second phase of the formalin test are comparable, involving a phenomenon of central sensitization produced and maintained mainly by NMDA receptor stimulation (South et al. 2003; Zou et al. 2000; Soliman et al. 2005). Sigma-1 receptors play an important modulatory role in NMDA receptor activity (Debonnel and de Montigny 1996; Kim et al. 2006) and even modulate acute pain induced by NMDA (Kim et al. 2008). Therefore, we hypothesized that σ1 receptor ligands might be able to modify the mechanical allodynia induced by capsaicin.
The main aim of this study was to evaluate the effects of the sigma ligands HP and its metabolites on mechanical hypersensitivity induced by the intraplantar injection of capsaicin and to determine whether these effects are due to their antagonism of σ1 receptors. To this end, we correlated the effect of drugs in behavioral tests with their affinity for brain σ1 receptors labeled with [3H](+)-pentazocine and attempted to prevent the effects of HP and its metabolites by administering the prototype σ1 receptor agonist PRE-084 (Su et al. 1991; Cobos et al. 2008). To control for the influence of dopaminergic antagonism on the effects of interest, we evaluated the effect of (−)-sulpiride, a D2 and D3 receptor antagonist devoid of activity on σ1 receptors (Freedman et al. 1994; Matsumoto and Pouw 2000). Involvement of endogenous opioid system modulation in the antiallodynic effect of HP and its metabolites was tested by evaluating the possible antagonism of this effect by the opioid receptor antagonist naloxone. We also tested the effect of HP and its metabolites on pain induced by mechanical punctate stimuli in animals not sensitized with capsaicin. The activity of these drugs was compared with that of control drugs (gabapentin, clonidine, and rofecoxib) with known effects on capsaicin-induced mechanical hypersensitivity or mechanical pain. Finally, we used the rotarod test to explore the possible role of motor incoordination in the effects of the drugs tested.
Materials and methods
Experimental animals
Female CD-1 mice (Charles River, Barcelona, Spain) weighing 25–30 g were used for all experiments. The animals were housed in a temperature-controlled room (21 ± 1°C) with air exchange every 20 min and an automatic 12-h light/dark cycle (0800 to 2000 hours). They were fed a standard laboratory diet and tap water ad libitum until the beginning of the experiments. The experiments were performed during the light phase (0900–1500 hours). Naive animals were used throughout the study. Mice were always handled in accordance with the European Communities Council Directive of 24 November 1986 (86/609/ECC). The experimental protocol was approved by the Research Ethics Committee of the University of Granada, Spain.
Drugs and drug administration
The σ ligands used (and their suppliers) were the nonselective σ1 antagonists haloperidol (HP), haloperidol metabolite I [HP-Met-I, 4-(4-chlorophenyl)-4-hydroxypiperidine], haloperidol metabolite II [HP-Met-II, 4-(4-chlorophenyl)-α-(4-fluorophenyl)-4-hydroxy-1-piperidinebutanol], and haloperidol metabolite III (HP-Met-III, p-fluorobenzoylpropionic acid) (all from Sigma-Aldrich Química S.A., Madrid, Spain), as well as the selective σ1 antagonist BD-1063 (1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride) and the selective σ1 agonist PRE-084 [2-(4-morpholinethyl)1-phenylcyclohexanecarboxylate) hydrochloride] (both from Tocris Cookson, Bristol, UK). As a control of dopaminergic antagonism, we used the D2 and D3 antagonist (−)-sulpiride (Sigma-Aldrich Química S.A., Madrid, Spain). We also used naloxone hydrochloride (Sigma-Aldrich Química S.A.) and morphine hydrochloride (General Directorate of Pharmacy and Drugs, Spanish Ministry of Health) to evaluate the involvement of opioidergic system modulation in the antiallodynic effect of HP and its metabolites. Three additional drugs (all from Sigma-Aldrich Química S.A.) were used as controls in the behavioral assays: (a) clonidine, which has antinociceptive and antiallodynic effects (Paqueron et al. 2003); (b) gabapentin, which has antiallodynic but not antinociceptive effects (Joshi et al. 2006, Tanabe et al. 2005); and (c) rofecoxib, an anti-inflammatory drug (Moore et al. 2005), which is devoid of antinociceptive and antiallodynic activity in animals without inflammation (Bingham et al. 2005; Padi and Kulkarni 2004).
The radioligand used for binding assays was [3H](+)-pentazocine with a specific activity of 33.6 Ci/mmol (PerkinElmer Life Sciences, Boston, MA, USA). Dilutions from the stock [3H](+)-pentazocine solution were prepared with ice-cold incubation buffer (50 mM HCl–Tris buffer pH 8 at 30°C). Haloperidol, HP metabolites I, II, III, and (−)-sulpiride were dissolved in absolute ethanol to make up a stock solution of 1 mM from which further dilutions were prepared with incubation buffer to yield a final maximal concentration of ethanol in the incubation medium of 1% (v/v). We previously verified that this final concentration of ethanol did not affect the binding of [3H](+)-pentazocine. The other cold drugs (PRE-084, gabapentin, clonidine, and rofecoxib) used in competition binding assays were dissolved in deionized ultrapure water.
The drugs were suspended in 5% gum arabic (Sigma-Aldrich Química S.A.) in water for in vivo assays, and all were injected subcutaneously (s.c.) into the interscapular region. An equal volume of vehicle was used in control animals. When PRE-084 was used to reverse the effects of HP or its metabolites, it was s.c. injected immediately before the other drug solution. Each injection was performed in different areas to avoid mixture of the drug solutions and any interference with results due to physicochemical interaction. The chemical algogen used was capsaicin (Sigma-Aldrich Química S.A.), which was dissolved in 1% dimethyl sulfoxide (DMSO; Merck KGaA, Darnstadt, Germany) in physiological sterile saline to a concentration of 0.05 µg/µL. Capsaicin solution was injected intraplantarly (i.pl.) into the right hind paw in a volume of 20 µL, using a 1710 TLL Hamilton microsyringe (Teknokroma, Barcelona, Spain) with a 301/2-gauge needle. Control animals were injected with the same volume of capsaicin solvent (DMSO 1% in saline).
Mice brain membrane preparations
Crude synaptosomal membranes (P2 fraction) were prepared for [3H](+)-pentazocine binding as previously described (Cobos et al. 2006) with slight modifications. Mice were killed by cervical dislocation and the brain was rapidly removed and homogenized in 15 volumes (w/v) of 0.32 M sucrose–10 mM Tris–HCl, pH 7.4, with a Polytron homogenizer (model PT10-35, Kinematica AG, Basel, Switzerland). The homogenates were centrifuged (Avanti 30, Beckman Coulter España S.A., Madrid, Spain) at 1,000×g for 13 min, the resulting pellets were discarded, and the supernatants were centrifuged at 21,000×g for 15 min to obtain the P2 pellets; each pellet, obtained from two whole brains, was resuspended in 15 mL of 10 mM Tris–HCl, pH 7.4, and centrifuged again at 21,000×g for 15 min. The entire process was performed at 4°C. Finally, each pellet was resuspended in 1 mL of 10 mM Tris–HCl, pH 7.4, and frozen in aliquots (protein concentration 12–14 mg/mL) at −80°C. The binding characteristics of the tissue were stable for at least 1 month when stored at −80°C. Protein concentrations were measured by the method of Lowry et al. (1951) with some modifications, using bovine serum albumin as the standard.
[3H](+)-Pentazocine binding assays
To test the affinities of drugs for mice brain σ1 receptors, we performed [3H](+)-pentazocine competition binding assays. Aliquots of mice brain membranes were slowly thawed and resuspended in fresh incubation buffer and [3H](+)-pentazocine binding assays were performed as previously described (Cobos et al. 2007) with slight modifications. Resuspended membrane preparations (460 µL) were incubated at a final protein concentration of 0.8 mg/mL with 20 µL of several concentrations of the cold drug or its solvent and with 20 µL [3H](+)-pentazocine (final concentration of 5 nM) for 240 min at 30°C, pH 8. Nonspecific binding was defined as the binding retained in the presence of HP 10 µM and was always less than 20% of the total binding.
To stop [3H](+)-pentazocine binding to the mouse brain membranes, 5 mL ice-cold filtration buffer (Tris 10 mM, pH 7.4) was added to the tubes. The bound and free radioligand were separated by rapid filtration under a vacuum using a Brandel cell harvester (Model M-12 T, Brandel Instruments, SEMAT Technical, St. Albans, Hertfordshire, UK) over Whatman GF/B glass fiber filters (SEMAT Technical, St. Albans, Hertfordshire, UK) presoaked with 0.5% polyethylenimine in Tris 10 mM, pH 7.4, for at least 1 h prior to use, to reduce nonspecific binding. The filters were washed under a vacuum twice with 5-mL volumes of the ice-cold filtration buffer and transferred to scintillation counting vials. Then, 4 mL liquid scintillation cocktail (CytoScint scintillation counting solution, MP Biomedicals, Irvine, CA, USA) was added to each vial and the mixture was equilibrated for at least 20 h. The radioactivity retained in the filter was measured with a liquid scintillation spectrometer (Beckman Coulter España S.A.) with an efficiency of 52%. Each assay was conducted in triplicate.
Evaluation of mechanical punctate nociceptive pain and capsaicin-induced mechanical hypersensitivity
Animals were placed in the experimental room (under low illumination) to allow them to acclimatize to the study room for 1 h before the experiments were begun. After that time, the animals were placed into individual test compartments for 2 h before the test to habituate them to the test conditions. The test compartments had black walls and were situated on an elevated mesh-bottomed platform with a 0.5-cm2 grid to provide access to the ventral surface of the hind paws. After this period, the animals were carefully removed from the compartment, injected i.pl. with 1 µg capsaicin (or its solvent) in the right hind paw proximate to the heel, and immediately returned to the compartment. In all experiments, punctate mechanical stimulation was applied with a Dynamic Plantar Aesthesiometer (Ugo Basile, Varese, Italy) at 15 min after the administration of capsaicin (time to maximum effect, data not shown) or its solvent. Briefly, a nonflexible filament (0.5 mm diameter) was electronically driven into the ventral side of the paw previously injected with capsaicin or solvent (i.e., the right hind paw), at least 5 mm away from the site of the injection towards the fingers. When a paw withdrawal response occurred, the stimulus was automatically terminated and the response latency time was automatically recorded. A cut-off time of 50 s was used. In all experiments, the filament was applied to the right hind paw of each mouse three times, separated by intervals of 0.5 min, and the mean value of the three trials was considered the withdrawal latency time of the animal.
Responses to mechanical stimuli were compared between control (DMSO-treated) and capsaicin-sensitized mice by applying the filament at a wide range of electronically controlled intensities from 0.05 to 8 g force, recording the paw withdrawal latency time for each force applied as described above. Each animal was tested using only one intensity of mechanical stimulation in order to maintain a strictly constant time (15 min) between administration of capsaicin or its solvent and the behavioral evaluation. As expected, shorter withdrawal latency times were obtained as higher forces were applied (see Fig. 2 and the “Results” section). This approach allowed us to construct a force–response curve (i.e., intensity of the stimulus versus paw withdrawal latency time) and to quantify the degree of mechanical punctate nociceptive pain in DMSO-treated animals and the mechanical sensitization in capsaicin-treated animals.
When the effects of drugs were tested, the drug under study (or its solvent) was administered s.c. 30 min before the i.pl. administration of capsaicin or DMSO 1% (i.e., 45 min before we evaluated the response to the mechanical punctate stimulus). The antinociceptive effects of drugs were assessed in DMSO-treated animals using a mechanical stimulation of 4 g force, which induced a marked reduction in paw withdrawal latency time in these nonsensitized mice (for details, see Fig. 2 and the “Results” section). The antiallodynic effects of the drugs were evaluated in capsaicin-sensitized mice using a mechanical stimulation of 0.5 g force. This intensity of the mechanical stimulus did not induce paw withdrawal in DMSO-treated mice, but markedly reduced paw withdrawal latency time in capsaicin-sensitized mice. We chose these forces because the latency time in capsaicin-sensitized animals stimulated with 0.5 g was similar to that in nonsensitized animals (DMSO-treated) stimulated at 4 g force (see Figs. 2 and 6).
Rotarod test
Changes in motor coordination were tested with an accelerating rotarod (Cibertec, Madrid, Spain) as previously described (Nieto et al. 2008) with slight modifications. Briefly, mice were required to walk against the motion of an elevated rotating drum at speeds increasing from 4 to 40 rpm over a 300-s period. The latency to fall down was recorded automatically with a cut-off time of 300 s. Twenty-four hours prior to each experiment with drugs, mice were acclimatized to the apparatus in three training sessions on the rotarod separated by 30-min intervals. On the day of the test, rotarod latencies were measured immediately before (time 0) and 45 min after the drug or vehicle was given. This time was chosen because it was the time used to test the effects of drugs on mechanical hypersensitivity.
Data analysis
We estimated the concentration of unlabeled drug that inhibited 50% of [3H](+)-pentazocine specific binding (IC50) values and their standard errors from the inhibition curves with nonlinear regression analysis of the equation for a sigmoid plot, assuming one-site competition; the SigmaPlot v. 8.0 (2002) program was used for all estimates. The force of mechanical stimulus applied that produced half the maximal reduction in paw withdrawal latency time (EF50) values were calculated from the force–response curves using nonlinear regression analysis of the equation for a sigmoid plot. The degree of effect on capsaicin-induced mechanical hypersensitivity was calculated as: \( \% \,{\text{reduction}}\,{\text{mechanical}}\,{\text{hypersensitivity}} = \left[ {{{\left( {{\text{LTD}} - {\text{LTS}}} \right)} \mathord{\left/ {\vphantom {{\left( {{\text{LTD}} - {\text{LTS}}} \right)} {\left( {{\text{CT}}--{\text{LTS}}} \right)}}} \right. } {\left( {{\text{CT}}--{\text{LTS}}} \right)}}} \right] \times 100 \) where LTD is the latency time for paw withdrawal in drug-treated animals, LTS is the latency time in solvent-treated animals, and CT is the cut-off time (50 s). The dose of drug that produced half the maximal inhibition of mechanical allodynia (ED50) and maximum antiallodynic effect (E max) values were calculated from the dose–response curves using nonlinear regression analysis of the equation for a sigmoid plot. The EF50, ED50, and E max values obtained from sigmoid plots and their standard errors were calculated as the best-fit values±standard errors of regression with the SigmaPlot v. 8.0 (2002) program.
The values obtained in several experimental groups were compared with one-way or two-way analysis of variance (ANOVA) followed by the Bonferroni test. Differences between two means were assessed by the Student’s t test. The differences were considered significant when the value of P was below 0.05.
Results
Affinity of drugs for [3H](+)-pentazocine binding sites in the mouse brain
We used competition binding assays to test the affinities of the drugs under study for the σ1 receptor, labeled with [3H](+)-pentazocine, in mouse brain membranes (P2 fraction). Specific binding of [3H](+)-pentazocine (which always represented more than 80% of the total binding) was concentration-dependently inhibited by the unlabeled ligands with the following order of potency (IC50 values): HP (5.45 ± 0.48 nM) > HP-Met-II (12.80 ± 0.69 nM) > PRE-084 (171.40 ± 18.36 nM) > HP-Met-I (254.18 ± 18.40 nM) >>> HP-Met-III or (−)-sulpiride (the last two compounds had negligible affinity for [3H](+)-pentazocine binding sites, with IC50 > 10,000 nM) (Fig. 1 and Table 1). The drugs used as controls in the behavioral tests (gabapentin, clonidine, and rofecoxib) also did not substantially decrease [3H](+)-pentazocine binding, showing IC50 values higher than 10,000 nM (data not shown).

Inhibition by unlabeled drugs of [3H](+)-pentazocine specific binding to membranes (P2 fraction) obtained from whole mouse brain. [3H](+)-pentazocine (5 nM) was incubated with 0.8 mg/mL brain membrane protein at 30°C, pH 8, for 240 min and increasing concentrations of haloperidol (HP, filled circles), haloperidol metabolite I (HP-Met-I, filled squares), haloperidol metabolite II (HP-Met-II, filled triangles), haloperidol metabolite III (HP-Met-III, filled inverted triangles), PRE-084 (open squares), or (−)-sulpiride (filled diamonds). Data shown are the average of two experiments carried out in triplicate
Evaluation of mechanical punctate nociceptive pain and capsaicin-induced mechanical hypersensitivity
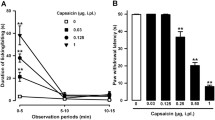
We applied the filament at different intensities ranging from innocuous to noxious stimuli (0.05–8 g force) in mice treated with capsaicin (1 µg, i.pl.) or its solvent. As expected, paw withdrawal latency time decreased as the force applied increased in both experimental situations (Fig. 2). Forces higher than 1 g were needed to induce paw withdrawal in nonsensitized (DMSO-treated) mice, and the shortest withdrawal latency time was obtained at 8 g force (Fig. 2). In contrast, a weak mechanical stimulus (0.2 g) was sufficient to induce paw withdrawal in capsaicin-sensitized mice, and withdrawal latency time was minimal at forces as low as 0.5–1 g (Fig. 2). Treatment with capsaicin produced a 9.4-fold decrease in the EF50 value for inducing paw withdrawal in comparison to control mice (EF50 values of 2.59 ± 0.08 and 0.30 ± 0.02 g for DMSO-treated and capsaicin-treated animals, respectively), which clearly indicates that capsaicin induced mechanical hypersensitivity to punctate stimuli.

Withdrawal latency time of the hind paw stimulated with a filament at different forces, 15 min after the intraplantar injection of capsaicin (1 μg, filled circles) or its solvent (DMSO 1%, open circles). Each point and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 6–8 per group). Each group was tested with only one stimulation force. Statistically significant differences between the latency time values obtained with each applied force under the two experimental conditions: **P < 0.01 (two-way ANOVA followed by Bonferroni test)
Effect of haloperidol and its metabolites on capsaicin-induced mechanical hypersensitivity
The effects of s.c. administration of drugs was investigated as the change in mechanical hypersensitivity induced by intraplantar capsaicin. Injection of the σ1 ligands HP (0.004–0.25 mg/kg), HP-Met-II (0.04–2 mg/kg), and HP-Met-I (8–128 mg/kg) induced a dose-dependent increase in withdrawal latency in the capsaicin-injected hind paw when stimulated with a filament at 0.5 g force, i.e., they produced antiallodynic effects (Fig. 3). The order of potency in these effects (ED50 values) was: HP (0.026 ± 0.006 mg/kg) > HP-Met-II (0.135 ± 0.03 mg/kg) >> HP-Met-I (31.05 ± 7.83 mg/kg), the same as the order of potency of these compounds for displacing [3H](+)-pentazocine binding in mouse brain membranes (Table 1).

Effects of different doses of subcutaneously administered haloperidol (HP, filled circles), haloperidol metabolite II (HP-Met-II, filled triangles), haloperidol metabolite I (HP-Met-I, filled squares), haloperidol metabolite III (HP-Met-III, filled inverted triangles), (−)-sulpiride (filled diamonds), or their vehicle (open diamonds) on mechanical hypersensitivity (allodynia) induced by intraplantar injection of capsaicin (1 µg) to mouse hind paw. The results represent the percentage reduction in capsaicin-induced mechanical hypersensitivity (calculated as explained in the “Materials and methods” section). Each point and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 8–10 per group). Each group was treated with only one dose of drug or solvent. Statistically significant differences between the values obtained in solvent-injected and drug-injected groups: **P < 0.01 (one-way ANOVA followed by Bonferroni test)
The maximum effect (E max) for each drug was calculated by regression analysis. Haloperidol and its metabolite II produced the maximum possible effect in this model (E max = 100%); however, the E max for HP-Met-I was lower (62.27 ± 6.99%).
Interestingly, HP-Met-III and (−)-sulpiride, which had no affinity for [3H](+)-pentazocine-labeled σ1 receptor (Fig. 1; Table 1), showed no antiallodynic effects even at very high doses (128 and 100 mg/kg, s.c., respectively) (Fig. 3 and Table 1).
Reversion of the antiallodynic effects of haloperidol and its metabolites by the selective σ1 receptor agonist PRE-084
The known σ1 receptor agonist PRE-084 did not modify paw withdrawal latency in capsaicin-treated animals at either dose (32 or 64 mg/kg s.c.) (Fig. 4 and Table 1). However, PRE-084 (16–64 mg/kg, s.c.) produced a dose-dependent reversion of the antiallodynic effect of HP (0.06 mg/kg, s.c.) (Fig. 4). The highest dose of PRE-084 abolished the effect of HP, and paw withdrawal latency was similar to that in animals treated with the drug solvent (Fig. 4). PRE-084 (32–64 mg/kg, s.c.) also completely and dose-dependently prevented the antiallodynic effects of HP-Met-II (0.25 mg/kg, s.c.) and HP-Met-I (64 mg/kg, s.c.) (Fig. 4). As a control, we evaluated the ability of PRE-084 to reverse the effect of BD-1063, a selective antagonist of σ1 receptors (Matsumoto et al. 1995; Hayashi and Su 2004). We found that BD-1063 (20 mg/kg, s.c.) produced an increase in paw withdrawal latency similar to that induced by HP and its metabolites I and II and that the antiallodynic effect of BD-1063 was completely antagonized by PRE-084 (32 mg/kg, s.c.) (Fig. 4).

Effects of the association of subcutaneous administration of PRE-084 (16-64 mg/kg) or its solvent with haloperidol (HP, 0.06 mg/kg), haloperidol metabolite II (HP-Met-II, 0.25 mg/kg), haloperidol metabolite I (HP-Met-I, 64 mg/kg), BD-1063 (20 mg/kg), or their solvent (all injected s.c.) on mechanical hypersensitivity induced by the intraplantar injection of capsaicin (1 µg) to the mouse hind paw. The results represent the latency to hind-paw withdrawal of the capsaicin-sensitized paw after ipsilateral stimulation with a filament at 0.5 g force (see the “Materials and methods” section for details). Each bar and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 8–10 per group). Each group was treated with only one of the drugs (or their solvent) associated to one dose of PRE-084 or its solvent. Statistically significant differences between the values obtained in each drug-treated group with respect to the solvent-treated group: *P < 0.05; **P < 0.01. Statistically significant differences between the values obtained in the groups treated with each drug (HP, HP-Met-II, HP-Met-I, or BD-1063) associated with PRE-084 with respect to those in the groups treated with each drug associated with PRE-084 solvent: ##P < 0.01 (two-way ANOVA followed by Bonferroni test)
Effect of naloxone on the antiallodynic activity of morphine, haloperidol, and its metabolites
As expected, morphine (0.25 mg/kg, s.c.) inhibited mechanical hypersensitivity induced by capsaicin, and its effect was reversed by the opioid receptor antagonist naloxone (1 mg/kg, s.c.) (Fig. 5). In contrast, the same dose of naloxone did not significantly modify the antiallodynic effect of HP (0.06 mg/kg, s.c.), HP-Met-II (0.25 mg/kg, s.c.), or HP-Met-I (64 mg/kg, s.c.) (Fig. 5).

Effects of the association of subcutaneous administration of naloxone (1 mg/kg) or its solvent with morphine (0.25 mg/kg), haloperidol (HP, 0.06 mg/kg), haloperidol metabolite II (HP-Met-II, 0.25 mg/kg), and haloperidol metabolite I (HP-Met-I, 64 mg/kg) on mechanical hypersensitivity induced by the intraplantar injection of capsaicin (1 µg) to the mouse hind paw. The results represent the latency to hind-paw withdrawal of the capsaicin-sensitized paw after ipsilateral stimulation with a filament at 0.5 g force (see the “Materials and methods” section for details). Each bar and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 8–10 per group). Each group was treated with only one of the drugs under study associated to naloxone or its solvent. Statistically significant differences in values were only found between the morphine-treated and morphine+naloxone-treated groups: **P < 0.01 (nonpaired Student’s t test)
Comparison of the antiallodynic and antinociceptive effects of haloperidol, its metabolites, and control drugs
To compare the effects of drugs on mechanical punctate stimuli-induced nociceptive pain (in nonsensitized animals) and mechanical allodynia (in capsaicin-treated animals), we chose mechanical intensity stimuli that produced similar withdrawal latency times in both experimental situations. Thus, when animals were pretreated with drug solvent, the withdrawal latency time obtained after stimulation with a 4-g force in nonsensitized (1% DMSO-treated) mice (6.58 ± 1.17 s) was very similar to that obtained in animals treated with capsaicin (1 µg, i.pl.) and stimulated at 0.5 g force (8.03 ± 0.84 s; Fig. 6).

Effects of s.c. administration of haloperidol (HP, 0.125 mg/kg), haloperidol metabolite II (HP-Met-II, 0.5 mg/kg), haloperidol metabolite I (HP-Met-I, 64 mg/kg), clonidine (0.5 mg/kg), gabapentin (64 mg/kg), rofecoxib (32 mg/kg), or their solvent on latency time to paw withdrawal under two experimental conditions: capsaicin-treated paw stimulated at 0.5 g force, i.e., mechanical allodynia (left panel), and 1% DMSO-treated paw stimulated at 4 g force, i.e., mechanical nociceptive pain (right panel) (see the “Materials and methods” section for details). Each bar and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 8–10 per group). Each group was treated with only one of the drugs (or their solvent) and tested under only one of the two experimental conditions. Statistically significant differences between the values obtained in the drug-treated and solvent-treated group under the same experimental conditions: **P < 0.01; statistically significant differences between the effects of each drug tested under the two experimental conditions (0.5 g force in capsaicin-treated mice versus 4 g force in DMSO-treated mice): ## P < 0.01 (two-way ANOVA followed by Bonferroni test)
The subcutaneous administration of HP, HP-Met-II, or HP-Met-I (0.125, 0.5, or 64 mg/kg, respectively) markedly increased paw withdrawal latency time in capsaicin-sensitized animals stimulated with the filament at 0.5 g force. Therefore, they exerted antiallodynic effects, which were maximal for HP and HP-Met-II (Fig. 6, left panel). The control drugs gabapentin (64 mg/kg, s.c.) and clonidine (0.5 mg/kg, s.c.) also induced a marked antiallodynic effect in capsaicin-treated animals (Fig. 6, left panel). However, as expected, rofecoxib was inactive in this behavioral test (Fig. 6, left panel).
Nonsensitized (DMSO-treated) mice treated with the σ1 receptor antagonists HP, HP-Met-I, and HP-Met-II (at the same doses that showed maximal antiallodynic effects) did not increase paw withdrawal latencies after stimulation with a filament at 4 g force, i.e., these drugs did not exert any antinociceptive effect on punctate mechanical nociceptive pain (Fig. 6, right panel). Gabapentin and rofecoxib were also devoid of effect against this noxious punctate mechanical stimulus (Fig. 6, right panel). However, clonidine (0.5 mg/kg, s.c.) increased the paw withdrawal latency time up to the cut-off time in nonsensitized mice stimulated with the filament at 4 g force (Fig. 6, right panel), inducing a clear antinociceptive effect.
Effect of drugs on the rotarod test
The latency to fall down from the rotarod before the drug or solvent was injected (time 0) was similar in all experimental groups (data not shown). The s.c. administration of HP (0.06 mg/kg), HP-Met-I (64 mg/kg), and HP-Met-II (0.25 mg/kg) had no effect on rotarod latency in comparison to control (solvent-treated) animals 45 min after administration (Fig. 7a), i.e., they did not interfere with motor coordination at times and doses that produced around 70% of the maximum antiallodynic effect (compare Figs. 4 and 7a). The s.c. administration of (−)-sulpiride (100 mg/kg), HP-Met-III (128 mg/kg), or PRE-084 (64 mg/kg) was also devoid of any effect on rotarod latency (Fig. 7a, b).

Rotarod latency times obtained 45 min after s.c. administration of drugs or their solvent. a Effects of the solvent, haloperidol (HP, 0.06 mg/kg), haloperidol metabolite I (HP-Met-I, 64 mg/kg), haloperidol metabolite II (HP-Met-II, 0.25 mg/kg), haloperidol metabolite III (HP-Met-III, 128 mg/kg), or (−)-sulpiride (100 mg/kg). Each bar and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 8 per group). Each group was treated with only one of the drugs (or their solvent) under study. There were no statistically significant differences between the values obtained in solvent-treated and drug-treated groups (one-way ANOVA). b Effects of solvent, PRE-084 (64 mg/kg), and haloperidol (HP, 0.125 mg/kg), or haloperidol metabolite II (HP-Met-II, 1 mg/kg), both alone or associated with PRE-084 (64 mg/kg). Each bar and vertical line represent the mean±SEM of the values obtained in an independent group of animals (n = 8 per group). Each group was treated with only one of the associations of drugs (or their solvent) with PRE-084 (or its solvent). Statistically significant differences between the values obtained in the drug-treated and solvent-treated groups: *P < 0.05; **P < 0.01. Statistically significant differences with respect to the group treated with PRE-084 alone: ## P < 0.01. NS nonsignificant differences (two-way ANOVA followed by Bonferroni test)
On the other hand, animals treated with higher doses of HP (0.125 mg/kg, s.c.) or HP-Met-II (1 mg/kg, s.c.), which produced 100% of the antiallodynic effect (see Fig. 3), showed a significant decrease in rotarod latencies in comparison to both their own values at time 0 (data not shown) and the values in solvent-treated animals at 45 min after administration (Fig. 7b). Interestingly, the effects of HP and HP-Met-II in the rotarod test were not prevented by PRE-084 at a dose (64 mg/kg, s.c.) that completely prevented the antiallodynic effect of both drugs (compare Figs. 4 and 7b).
Discussion
The main findings of this study were that the subcutaneous administration of HP or HP metabolites I or II dose-dependently antagonized capsaicin-induced mechanical allodynia (probably by blocking σ1 receptors) but did not modify nociceptive pain induced by a painful punctate stimulus in nonsensitized animals. These effects have not been previously reported.
We found that the intraplantar injection of capsaicin produced mechanical allodynia (sensitization to an innocuous mechanical punctate stimulus), which was manifested by both a decrease in the force necessary to induce hind-paw withdrawal and a decrease in the latency time for paw withdrawal when a determined force was applied. These results agree with those of previous studies that also reported capsaicin-induced mechanical hypersensitivity to punctate stimulus in rodents (Gilchrist et al. 1996; Joshi et al. 2006; Brenchat et al. 2009). Under our experimental conditions, HP, HP-Met-I, and HP-Met-II exerted dose-dependent antiallodynic effects. It is noteworthy that the antiallodynic effect of HP is produced at a range of doses similar to those producing positive effects in animal models predictive of antipsychotic activity (Natesan et al. 2008; Millan et al. 2008). The control drugs gabapentin and clonidine were able to inhibit capsaicin-induced mechanical allodynia, whereas rofecoxib was inactive. These results agree with those previously described for gabapentin and clonidine, both of which exerted antiallodynic effects (Joshi et al. 2006; Paqueron et al. 2003), and with the lack of effect of rofecoxib against capsaicin-induced mechanical hypersensitivity (Bingham et al. 2005).
We also evaluated the effects of the drugs on the response to mechanical stimuli in nonsensitized (DMSO-treated) animals. The punctate stimulus at 4 g force is painful in nonsensitized animals, since it exceeds the threshold for activation of Aδ- and C-nociceptors in mouse glabrous skin (Cain et al. 2001) and induces paw withdrawal. We chose this force (4 g) in nonsensitized animals because it produced a paw withdrawal latency similar to that found in capsaicin-sensitized animals stimulated with an innocuous force (0.5 g) and allowed us to evaluate the effect of drugs on nociceptive pain induced by a type of stimulus qualitatively similar to that used in capsaicin-sensitized animals. Haloperidol and its metabolites did not affect the response to punctate stimuli in nonsensitized animals, which suggests that the drugs tested did not alter mechanical stimulus perception or the motor mechanisms necessary to produce paw withdrawal. Previous studies have also found that HP and its metabolites did not modify heat-induced nociceptive pain (tail flick test) (Chien and Pasternak 1994; Cendán et al. 2005b), but data on their effect on mechanical nociceptive pain have not been previously reported. Regarding the drugs used as controls, our results fully agree with those previously reported, since neither gabapentin nor rofecoxib affected mechanical nociception in nonsensitized animals (Tanabe et al. 2005; Padi and Kulkarni 2004), whereas clonidine reduced mechanical pain in these animals (Honda et al. 2002; Paqueron et al. 2003).
Haloperidol and HP-Met-II showed affinity for σ1 receptors at nanomolar concentrations (Cobos et al. 2007; this study) and for D2 and D3 receptors at nanomolar (HP) and low micromolar (HP-Met-II) concentrations (Bowen et al. 1990; Jaen et al. 1993). Therefore, their antiallodynic activity might be due to their effects on σ1 or dopamine receptors or on both. Interestingly, (−)-sulpiride, which shows nanomolar affinity for D2 and D3 receptors but no affinity for σ1 receptors (Freedman et al. 1994; Matsumoto and Pouw 2000; this study), is devoid of antiallodynic effects, whereas HP-Met-I, which has affinity for σ1 receptors but not for D2 receptors (Bowen et al. 1990; Cobos et al. 2007), shows antiallodynic activity. Therefore, dopamine receptor antagonism seems unlikely to be involved in the antiallodynic effects of HP and its metabolites. Because HP also has nanomolar affinity for α1-adrenoceptors (Millan et al. 2000), these receptors could play a role in its antiallodynic effect. However, the highly selective agonist of σ1 receptors PRE-084 has no affinity for α1-adrenoceptors and many other receptors (Su et al. 1991) but totally inhibits the antiallodynic effects of HP and its metabolites. This strongly suggests that α1-adrenoceptors are not involved in their antiallodynic effects.
In contrast, there are several arguments indicating that the antiallodynic effects of HP and its metabolites are due to their activity on σ1 receptors. Firstly, their order of potency for inducing antiallodynic effects correlates with their order of affinity for σ1 binding sites in mouse brain (this study), rat brain (Bowen et al. 1990; Matsumoto and Pouw 2000), guinea pig brain, and human neuroblastoma cells (Cobos et al. 2007). Secondly, HP-Met-III and (−)-sulpiride, which had negligible affinity for σ1 receptors in this and other studies (Bowen et al. 1990; Cobos et al. 2007; Matsumoto and Pouw 2000), exerted no antiallodynic activity in this model. Thirdly, the selective σ1 receptor agonist PRE-084 (Su et al. 1991; Hayashi and Su 2004), at doses that do not produce any effect per se on capsaicin-induced mechanical sensitization, reversed the antiallodynic effects of HP, HP-Met I, and HP-Met II. Fourthly, the selective σ1 receptor antagonist BD-1063 (Matsumoto et al. 1995; Hayashi and Su 2004) produced a response pattern very similar to that of HP and its metabolites, i.e., an antiallodynic effect that was fully reversed by the σ1 receptor agonist PRE-084. Therefore, the antiallodynic effects of the drugs studied seem to be due to their antagonistic activity on σ1 receptors.
Haloperidol and other σ1 receptor antagonists increase opioid receptor agonist-induced antinociception (see the “Introduction” for references). Hence, their ability to inhibit capsaicin-induced mechanical hypersensitivity might be due to an increase in the effects of endogenous opioids. We found that a dose of naloxone that reversed the antiallodynic effect of morphine was unable to modify the antiallodynic effects of HP and its metabolites. These experiments rule out any influence on our results of modulation of the endogenous opioid system by HP or its metabolites.
Previous studies have reported that HP and its metabolites I and II, as well as other antagonists of σ1 receptors such as BD-1047 and BMY-14802, inhibited the second phase of formalin-induced pain (Cendán et al. 2005b; Kim et al. 2006). The mechanisms underlying the second phase of the formalin test and the mechanical hypersensitivity induced by capsaicin involve a process of central sensitization in which the activity of glutamatergic NMDA receptors plays a pivotal role (see Baron 2000 and the “Introduction” for references). Therefore, either a direct effect on NMDA receptors or an indirect modulation of NMDA-mediated effects could explain our results. Haloperidol and reduced HP show affinity for NMDA receptors, but their affinity for NMDA receptors is very low (K i in the micromolar range) (Whittemore et al. 1997; Coughenour and Cordon 1997) in comparison to their affinity for σ1 receptors, which is 1,000 times higher (in the nanomolar range) (Matsumoto and Pouw 2000; Cobos et al. 2007; present study). Therefore, a direct effect of HP or its metabolites on NMDA receptors might be ruled out as an explanation for their antiallodynic effect in the present study. On the other hand, it has been reported that σ1 receptor agonists facilitated several NMDA receptor-mediated electrophysiological effects and that HP and other σ1 receptor antagonists inhibited these effects of σ1 receptor agonists (Debonnel and de Montigny 1996; Chen et al. 2006; Martina et al. 2007). Interestingly, low doses of HP (0.01–0.1 mg/kg), within the range of doses used in the present study, antagonized the potentiation induced by σ1 receptor agonists of NMDA-mediated response in vivo (Monnet et al. 1992; Maurice and Privat 1997; Bermack and Debonnel 2005). Therefore, the antiallodynic effects of HP and its metabolites might be explained by their ability to indirectly modulate NMDA-mediated effects through an action on σ1 receptors.
Unspecific interference on motor coordination does not appear to explain the antiallodynic effects because doses of HP, HP-Met-I, and HP-Met-II that lacked activity in the rotarod test exerted around 70% of the maximum antiallodynic activity. Higher doses of HP and HP-Met-II (that produced 100% of the antiallodynic effect) affected the response in the rotarod test, but these motor effects were not reversed by PRE-084 (i.e., were not due to antagonism of σ1 receptors), whereas PRE-084 reversed the antiallodynic effects. Moreover, neither HP nor HP-Met-II affected paw withdrawal latency in nonsensitized animals (stimulated at a force of 4 g) at doses that interfered with rotarod test responses. Taken together, these results indicate dissociation between the motor and neurochemical mechanisms involved in the effects of HP and its metabolites in paw withdrawal induced by mechanical stimulation and the rotarod tests.
It is known that σ1 receptor agonists and antagonists modulate pain processing, acting at both spinal (Kim et al. 2006, 2008) and supraspinal sites (Chien and Pasternak 1994; Mei and Pasternak 2002, 2007; Marrazzo et al. 2006). Therefore, HP and its metabolites might act at any of these sites to produce their antiallodynic effects. Since we administered the drugs subcutaneously, our data do not shed light on the level of central pain processing (spinal or supraspinal) at which HP and its metabolites produce their antiallodynic effects, and new experiments will be needed to identify their level of action.
Regarding the therapeutic potential of our findings, it has been reported that HP (2–5 mg) is useful in humans to treat migraine (Honkaniemi et al. 2006), chronic pain (Raft et al. 1979), and reduce nalbuphine-induced hyperalgesia (Gear et al. 2006). Moreover, a case report described complete acute relief of neuropathic pain with a 2-mg dose of HP (Shir et al. 1990). The affinity of HP for σ1 receptors is high enough to bind to them in humans at therapeutic doses. In fact, a dose of 3 mg HP occupied around 80% of σ1 receptors in different areas of the human brain (Ishiwata et al. 2006). Interestingly, this dose of HP occupies more σ1 than D2 receptors in the human brain (Ishiwata et al. 2006). This raises the possibility that the analgesic effects observed with HP in humans might be related to its ability to bind to σ1 receptors and that the antiallodynic activity of this compound may have clinical applications. New studies are necessary to test these possibilities.
In summary, our results show that haloperidol and its metabolites I and II inhibit capsaicin-induced mechanical hypersensitivity and suggest that the antagonism of σ1 receptors produced by these drugs is responsible for their antiallodynic effects in this model. These findings raise the possibility of new therapeutic applications for this antipsychotic drug, which is routinely used for other indications in humans.
References
Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, Maurice T (2000) Immunocytochemical localization of the sigma1 receptor in the adult rat central nervous system. Neuroscience 97:155–170
Baron R (2000) Capsaicin and nociception: from basic mechanisms to novel drugs. Lancet 356:785–787
Bermack JE, Debonnel G (2005) Distinct modulatory roles of sigma receptor subtypes on glutamatergic responses in the dorsal hippocampus. Synapse 55:37–44
Bingham S, Beswick PJ, Bountra C, Brown T, Campbell IB, Chessell IP, Clayton N, Collins SD, Davey PT, Goodland H, Gray N, Haslam C, Hatcher JP, Hunter AJ, Lucas F, Murkitt G, Naylor A, Pickup E, Sargent B, Summerfield SG, Stevens A, Stratton SC, Wiseman J (2005) The cyclooxygenase-2 inhibitor GW406381X[2-(4-ethoxyphenyl)-3-[4-(methylsulfonyl) phenyl]-pyrazolo[1, 5-b]pyridazine ] is effective in animal models of neuropathic pain and central sensitization. J Pharmacol Exp Ther 312:1161–1169
Bowen WD, Moses EL, Tolentino PJ, Walker JM (1990) Metabolites of haloperidol display preferential activity at σ receptors compared to dopamine D-2 receptors. Eur J Pharmacol 177:111–118
Brenchat A, Romero L, García M, Pujol M, Burgueño J, Torrens A, Hamon M, Baeyens JM, Buschmann H, Zamanillo D, Vela JM (2009) 5-HT7 receptor activation inhibits mechanical hypersensitivity secondary to capsaicin sensitization in mice. Pain 141:239–247
Cain DM, Khasabov SG, Simone DA (2001) Response properties of mechanoreceptors and nociceptors in mouse glabrous skin: an in vivo study. J Neurophysiol 85:1561–1574
Cendán CM, Pujalte JM, Portillo-Salido E, Montoliu L, Baeyens JM (2005a) Formalin-induced pain is reduced in σ1 receptor knockout mice. Eur J Pharmacol 511:73–74
Cendán CM, Pujalte JM, Portillo-Salido E, Baeyens JM (2005b) Antinociceptive effects of haloperidol and its metabolites in the formalin test in mice. Psychopharmacology (Berl) 182:485–493
Chen L, Dai XN, Sokabe M (2006) Chronic administration of dehydroepiandrosterone sulfate (DHEAS) primes for facilitated induction of long-term potentiation via sigma 1 (σ1) receptor: optical imaging study in rat hippocampal slices. Neuropharmacology 50:380–392
Chien CC, Pasternak GW (1993) Functional antagonism of morphine analgesia by (+)-pentazocine: evidence for an anti-opioid σ 1 system. Eur J Pharmacol 250:R7–R8
Chien CC, Pasternak GW (1994) Selective antagonism of opioid analgesia by a sigma system. J Pharmacol Exp Ther 271:1583–1590
Cobos EJ, Baeyens JM, Del Pozo E (2005) Phenytoin differentially modulates the affinity of agonist and antagonist ligands for σ1 receptors of guinea pig brain. Synapse 55:192–195
Cobos EJ, Lucena G, Baeyens JM, Del Pozo E (2006) Differences in the allosteric modulation by phenytoin of the binding properties of the σ1 ligands [3H](+)-pentazocine and [3H]NE-100. Synapse 59:152–161
Cobos EJ, Del Pozo E, Baeyens JM (2007) Irreversible blockade of sigma-1 receptors by haloperidol and its metabolites in guinea pig brain and SH-SY5Y human neuroblastoma cells. J Neurochem 102:812–825
Cobos EJ, Entrena JM, Nieto FR, Cendán CM, Del Pozo E (2008) Pharmacology and therapeutic potential of sigma1 receptor ligands. Curr Neuropharmacol 6:344–366
Coughenour LL, Cordon JJ (1997) Characterization of haloperidol and trifluperidol as subtype-selective N-methyl-D-aspartate (NMDA) receptor antagonists using [3H]TCP and [3H]ifenprodil binding in rat brain membranes. J Pharmacol Exp Ther 280:584–592
Debonnel G, de Montigny C (1996) Modulation of NMDA and dopaminergic neurotransmissions by sigma ligands: possible implications for the treatment of psychiatric disorders. Life Sci 58:721–734
Freedman SB, Patel S, Marwood R, Emms F, Seabrook GR, Knowles MR, McAllister G (1994) Expression and pharmacological characterization of the human D3 dopamine receptor. J Pharmacol Exp Ther 268:417–426
Gear RW, Lee JS, Miaskowski C, Gordon NC, Paul SM, Levine JD (2006) Neuroleptics antagonize nalbuphine antianalgesia. J Pain 7:187–191
Gilchrist HD, Allard BL, Simone DA (1996) Enhanced withdrawal responses to heat and mechanical stimuli following intraplantar injection of capsaicin in rats. Pain 67:179–188
Guitart X, Codony X, Monroy X (2004) Sigma receptors: biology and therapeutic potential. Psychopharmacology (Berl) 174:301–319
Hayashi T, Su TP (2004) Sigma-1 receptor ligands: potential in the treatment of neuropsychiatric disorders. CNS Drugs 18:269–284
Honda K, Koga K, Moriyama T, Koguchi M, Takano Y, Kamiya HO (2002) Intrathecal α2 adrenoceptor agonist clonidine inhibits mechanical transmission in mouse spinal cord via activation of muscarinic M1 receptors. Neurosci Lett 322:161–164
Honkaniemi J, Liimatainen S, Rainesalo S, Sulavuori S (2006) Haloperidol in the acute treatment of migraine: a randomized, double-blind, placebo-controlled study. Headache 46:781–787
Ishiwata K, Oda K, Sakata M, Kimura Y, Kawamura K, Oda K, Sasaki T, Naganawa M, Chihara K, Okubo Y, Ishii K (2006) A feasibility study of [11C]SA4503-PET for evaluating sigmal receptor occupancy by neuroleptics: the binding of haloperidol to sigma1 and dopamine D2-like receptors. Ann Nucl Med 20:569–573
Jaen JC, Caprathe BW, Pugsley TA, Wise LD, Akunne H (1993) Evaluation of the effects of the enantiomers of reduced haloperidol, azaperol, and related 4-amino-1-arylbutanols on dopamine and σ receptors. J Med Chem 36:3929–3936
Joshi SK, Hernandez G, Mikusa JP, Zhu CZ, Zhong C, Salyers A, Wismer CT, Chandran P, Decker MW, Honore P (2006) Comparison of antinociceptive actions of standard analgesics in attenuating capsaicin and nerve-injury-induced mechanical hypersensitivity. Neuroscience 143:587–596
Kim HW, Kwon YB, Roh DH, Yoon SY, Han HJ, Kim KW, Beitz AJ, Lee JH (2006) Intrathecal treatment with σ 1 receptor antagonists reduces formalin-induced phosphorylation of NMDA receptor subunit 1 and the second phase of formalin test in mice. Br J Pharmacol 148:490–498
Kim HW, Roh DH, Yoon SY, Seo HS, Kwon YB, Han HJ, Kim KW, Beitz AJ, Lee JH (2008) Activation of the spinal sigma-1 receptor enhances NMDA-induced pain via PKC- and PKA-dependent phosphorylation of the NR1 subunit in mice. Br J Pharmacol 154:1125–1134
Kitaichi K, Chabot JG, Moebius FF, Flandorfer A, Glossmann H, Quirion R (2000) Expression of the purported sigma1 (σ1) receptor in the mammalian brain and its possible relevance in deficits induced by antagonism of the NMDA receptor complex as revealed using an antisense strategy. J Chem Neuroanat 20:375–387
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurements with the folin phenol reagent. J Biol Chem 193:265–271
Marrazzo A, Parenti C, Scavo V, Ronsisvalle S, Scoto GM, Ronsisvalle G (2006) In vivo evaluation of (+)-MR200 as a new selective sigma ligand modulating MOP, DOP and KOP supraspinal analgesia. Life Sci 78:2449–2453
Martina M, Turcotte ME, Halman S, Bergeron R (2007) The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J Physiol 578:143–157
Matsumoto RR, Bowen WD, Tom MA, Vo VN, Truong DD, De Costa BR (1995) Characterization of two novel σ receptor ligands: antidystonic effects in rats suggest σ receptor antagonism. Eur J Pharmacol 280:301–310
Matsumoto RR, Pouw B (2000) Correlation between neuroleptic binding to σ1 and σ2 receptors and acute dystonic reactions. Eur J Pharmacol 401:155–160
Maurice T, Privat A (1997) SA4503, a novel cognitive enhancer with σ1 receptor agonist properties, facilitates NMDA receptor-dependent learning in mice. Eur J Pharmacol 328:9–18
Maurice T, Urani A, Phan VL, Romieu P (2001) The interaction between neuroactive steroids and the σ1 receptor function: behavioral consequences and therapeutic opportunities. Brain Res Brain Res Rev 37:116–132
Mei J, Pasternak GW (2002) σ1 Receptor modulation of opioid analgesia in the mouse. J Pharmacol Exp Ther 300:1070–1074
Mei J, Pasternak GW (2007) Modulation of brainstem opiate analgesia in the rat by sigma1 receptors: a microinjection study. J Pharmacol Exp Ther 322:1278–1285
Millan MJ, Brocco M, Rivet JM, Audinot V, Newman-Tancredi A, Maiofiss L, Queriaux S, Despaux N, Peglion JL, Dekeyne A (2000) S18327 (1-[2-[4-(6-fluoro-1, 2-benzisoxazol-3-yl) piperid-1-yl]ethyl]3-phenyl imidazolin-2-one), a novel, potential antipsychotic displaying marked antagonist properties at α1- and α2-adrenergic receptors: II. Functional profile and a multiparametric comparison with haloperidol, clozapine, and 11 other antipsychotic agents. J Pharmacol Exp Ther 292:54–66
Millan MJ, Loiseau F, Dekeyne A, Gobert A, Flik G, Cremers TI, Rivet JM, Sicard D, Billiras R, Brocco M (2008) S33138 (N-[4-[2-[(3aS, 9bR)-8-cyano-1, 3a, 4, 9b-tetrahydro[1] benzopyrano[3, 4-c]pyrrol-2(3H)-yl)-ethyl]phenyl-acetamide), a preferential dopamine D3 versus D2 receptor antagonist and potential antipsychotic agent: III. Actions in models of therapeutic activity and induction of side effects. J Pharmacol Exp Ther 324:1212–1226
Monnet FP, Debonnel G, de Montigny C (1992) In vivo electrophysiological evidence for a selective modulation of N-methyl-D-aspartate-induced neuronal activation in rat CA3 dorsal hippocampus by sigma ligands. J Pharmacol Exp Ther 261:123–130
Monnet FP, Maurice T (2006) The sigma1 protein as a target for the non-genomic effects of neuro(active) steroids: molecular, physiological, and behavioral aspects. J Pharmacol Sci 100:93–118
Moore RA, Derry S, Makinson GT, McQuay HJ (2005) Tolerability and adverse events in clinical trials of celecoxib in osteoarthritis and rheumatoid arthritis: systematic review and meta-analysis of information from company clinical trial reports. Arthritis Res Ther 7:R644–R665
Natesan S, Reckless GE, Barlow KB, Odontiadis J, Nobrega JN, Baker GB, George SR, Mamo D, Kapur S (2008) The antipsychotic potential of l-stepholidine—a naturally occurring dopamine receptor D1 agonist and D2 antagonist. Psychopharmacology (Berl) 199:275–289
Nieto FR, Entrena JM, Cendan CM, Pozo ED, Vela JM, Baeyens JM (2008) Tetrodotoxin inhibits the development and expression of neuropathic pain induced by paclitaxel in mice. Pain 137:520–531
Padi SS, Kulkarni SK (2004) Differential effects of naproxen and rofecoxib on the development of hypersensitivity following nerve injury in rats. Pharmacol Biochem Behav 79:349–358
Paqueron X, Conklin D, Eisenach JC (2003) Plasticity in action of intrathecal clonidine to mechanical but not thermal nociception after peripheral nerve injury. Anesthesiology 99:199–204
Prezzavento O, Parenti C, Marrazzo A, Ronsisvalle S, Vittorio F, Arico G, Scoto GM, Ronsisvalle G (2008) A new sigma ligand, (±)-PPCC, antagonizes kappa opioid receptor-mediated antinociceptive effect. Life Sci 82:549–553
Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP (1992) A proposal for the classification of sigma binding sites. Trends Pharmacol Sci 13:85–86
Raft D, Toomey T, Gregg JM (1979) Behavior modification and haloperidol in chronic facial pain. South Med J 72:155–159
Shir Y, Shenkman Z, Kaplan L (1990) Neuropathic pain unrelieved by morphine, alleviated by haloperidol. Harefuah 118:452–454
Soliman AC, Yu JS, Coderre TJ (2005) mGlu and NMDA receptor contributions to capsaicin-induced thermal and mechanical hypersensitivity. Neuropharmacology 48:325–332
South SM, Kohno T, Kaspar BK, Hegarty D, Vissel B, Drake CT, Ohata M, Jenab S, Sailer AW, Malkmus S, Masuyama T, Horner P, Bogulavsky J, Gage FH, Yaksh TL, Woolf CJ, Heinemann SF, Inturrisi CE (2003) A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury-induced pain. J Neurosci 23:5031–5040
Su TP, Wu XZ, Cone EJ, Shukla K, Gund TM, Dodge AL, Parish DW (1991) Sigma compounds derived from phencyclidine: identification of PRE-084, a new, selective sigma ligand. J Pharmacol Exp Ther 259:543–550
Tanabe M, Takasu K, Kasuya N, Shimizu S, Honda M, Ono H (2005) Role of descending noradrenergic system and spinal α2-adrenergic receptors in the effects of gabapentin on thermal and mechanical nociception after partial nerve injury in the mouse. Br J Pharmacol 144:703–714
Ueda H, Inoue M, Yoshida A, Mizuno K, Yamamoto H, Maruo J, Matsuno K, Mita S (2001) Metabotropic neurosteroid/σ-receptor involved in stimulation of nociceptor endings of mice. J Pharmacol Exp Ther 298:703–710
Whittemore ER, Ilyin VI, Woodward RM (1997) Antagonism of N-methyl-d-aspartate receptors by σ site ligands: potency, subtype-selectivity and mechanisms of inhibition. J Pharmacol Exp Ther 282:326–338
Zou X, Lin Q, Willis WD (2000) Enhanced phosphorylation of NMDA receptor 1 subunits in spinal cord dorsal horn and spinothalamic tract neurons after intradermal injection of capsaicin in rats. J Neurosci 20:6989–6997
Acknowledgments
J.M. Entrena and F.R. Nieto were supported by Formación de Profesorado Universitario grants (M. E. C.). C.M. Cendán was supported by the Research Program of the University of Granada. This research was done in partial fulfillment of the requirements for the doctoral thesis of J.M. Entrena. We thank K. Shashok for revising the English style of the manuscript. This study was partially supported by Grant CTS 109 from the Junta de Andalucía, Grant SAF 2006-06122 from the Spanish Ministerio de Educación y Ciencia (MEC), FEDER funds, a grant from Laboratorios Dr. Esteve, S.A., and a grant from the Centro para el Desarrollo Tecnológico Industrial (Genius Pharma Project).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Entrena, J.M., Cobos, E.J., Nieto, F.R. et al. Antagonism by haloperidol and its metabolites of mechanical hypersensitivity induced by intraplantar capsaicin in mice: role of sigma-1 receptors. Psychopharmacology 205, 21–33 (2009). https://doi.org/10.1007/s00213-009-1513-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1513-8