



Abstract
Adipocytes (AdCs) and osteoblasts (OBs) are derived from mesenchymal stem cells (MSCs) and differentiation toward either lineage is both mutually exclusive and transcriptionally controlled. Recent studies implicate the mammalian target of rapamycin (mTOR) pathway as important in determining MSC fate, with inhibition of mTOR promoting OB differentiation and suppressing AdC differentiation. mTOR functions within two distinct multiprotein complexes, mTORC1 and mTORC2, each of which contains the unique adaptor protein, raptor or rictor, respectively. While compounds used to study mTOR signaling, such as rapamycin and related analogs, primarily inhibit mTORC1, prolonged exposure can also disrupt mTORC2 function, confounding interpretation of inhibitor studies. As a result, the relative contribution of mTORC1 and mTORC2 to MSC fate determination remains unclear. In this study, we generated primary mouse MSCs deficient in either Rptor (RapKO) or Rictor (RicKO) using the Cre/loxP system. Cre-mediated deletion of Rptor or Rictor resulted in impaired mTORC1 and mTORC2 signaling, respectively. Under lineage-inductive culture conditions, RapKO MSCs displayed a reduced capacity to form lipid-laden AdCs and an increased capacity to form a mineralized matrix. In contrast, RicKO MSCs displayed reduced osteogenic differentiation capacity and enhanced adipogenic differentiation potential. Taken together, our findings reveal distinct roles for mTORC1 and mTORC2 in MSC lineage commitment. Stem Cells 2015;33:1359–1365
Introduction
Mesenchymal stem cells (MSCs) are multipotent progenitor cells that, in response to extracellular factors and environmental cues, differentiate into several specialized cell types including adipocytes (AdCs) and osteoblasts (OBs). In vitro, lineage specification can be guided by inductive culture conditions. Murine MSC cultures stimulated with glucocorticoid, insulin, and indomethacin differentiate into mature lipid-laden AdCs, whereas cultures supplemented with a source of inorganic phosphate and glucocorticoid differentiate into mineralized matrix-producing OBs.
Several signaling pathways have been shown to regulate MSC differentiation in vitro [1-5]. The mammalian target of rapamycin (mTOR) pathway is the primary nutrient-sensing pathway that controls cell growth and metabolism. mTOR plays an important role in the adipogenic program, as rapamycin, an allosteric mTOR inhibitor, inhibits adipogenesis [6-10] whilst deletion of TSC2, a negative regulator of mTOR, promotes adipogenesis [10].
The role of mTOR in MSC commitment to other lineages is less clear. Most notably, rapamycin has been shown to have both inhibitory [11-16] and stimulatory [17-20] effects on osteogenesis, a disparity which may reflect the nature of the target cells used. Furthermore, rapamycin sequesters FKBP12 [21], a negative regulator of TGFβ1 signaling, and thus the pro-osteogenic effects of rapamycin may be the indirect result of TGFβ1 activation.
mTOR exists in two distinct complexes, mTORC1 and mTORC2, each of which is defined by the unique adaptor proteins raptor and rictor, respectively. mTORC1 controls protein synthesis, ribosome biogenesis, and nutrient transport [22, 23] whereas mTORC2 regulates cell survival, metabolism, and cytoskeletal organization [24]. While rapamycin primarily inhibits mTORC1, prolonged exposure can also disrupt mTORC2 function [25, 26], confounding data interpretation with respect to MSC differentiation and the role played by mTORC1 and mTORC2 in osteogenesis. In this study, we used a genetic approach to specifically delete Rptor or Rictor in primary MSCs and examine the roles of mTORC1 and mTORC2 in osteogenic and adipogenic MSC differentiation.
Results
Generation of Rptor and Rictor Knockout MSCs
Primary compact bone-derived MSCs were isolated from Rptorfl/fl or Rictorfl/fl mice, and infected with a tamoxifen-inducible self-deleting Cre recombinase (CreERT2) or empty vector as a control. Tamoxifen (4-OHT) treatment induced the nuclear translocation of CreERT2, resulting in Rptor or Rictor deficient MSCs, termed RapKO and RicKO, respectively (Fig. 1A).
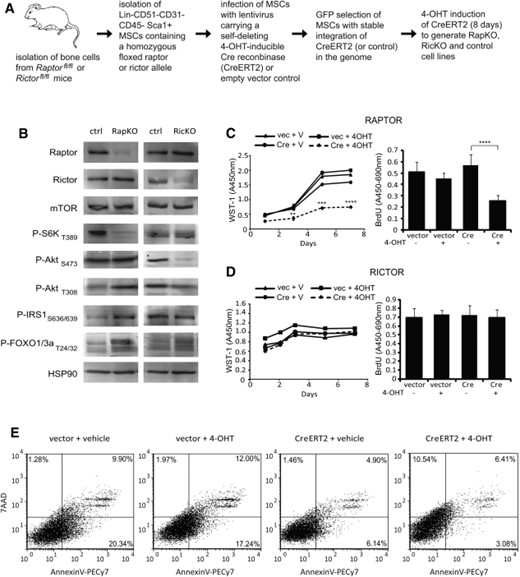
Cre-mediated loss of Rptor and Rictor expression in RapKO and RicKO MSCs. (A): Workflow illustrating the generation of RapKO, RicKO, and control MSC lines. (B): Rptorfl/fl and Rictorfl/fl MSCs infected with the CreERT2 lentivirus were treated with 4-OHT, to induce Rptor or Rictor knockout, or vehicle as a control. Lysates were prepared from control, RapKO, and RicKO MSCs and Western blot analysis performed using antibodies directed against the proteins indicated. (C, D): RapKO, RicKO, and control MSCs were cultured for 7 days and proliferation assessed using WST-1 and BrdU assays. Both cumulative WST-1 data (left) and BrdU data from the Day 7 time point (right) are shown, expressed as the mean ± SD of quintuplicate wells from a representative experiment of three. **, p < .005; *** p < 0.0005; ****, p < .0001, t test. (E): RapKO cells were stained for the apoptotic markers Annexin V and 7-AAD, and analyzed immediately by flow cytometry. Abbreviations: MSC, mesenchymal stem cell; mTOR, mammalian target of rapamycin, 4-OHT, tamoxifen.
Protein levels of RPTOR and RICTOR and the activation status of mTORC1 and mTORC2 downstream effectors were assessed by Western immunoblotting. As previously reported [27-30], low levels of residual RPTOR or RICTOR protein expression in RapKO and RicKO cells were observed (Fig. 1B). Consistent with disruption of mTORC1 function, Rptor deletion resulted in decreased S6K phosphorylation, and hyperphosphorylation of AktThr308, IRS-1 and FOXO1/3a (Fig. 1B). Rictor deletion resulted in ablation of AktSer473 phosphorylation, in keeping with mTORC2 substrate specificity (Fig. 1B).
Loss of Rptor, But Not Rictor, Impairs MSC Proliferation
The effect of Rptor or Rictor knockout on MSC growth was examined using WST-1 and BrdU assays. RapKO cells displayed a significant decrease in growth after 3, 5, and 7 days compared to controls (Fig. 1C). In contrast, no difference in proliferation was observed in response to Rictor deletion (Fig. 1D).
To determine whether the inhibition of RapKO cell proliferation was associated with an induction of apoptosis, RapKO and control MSCs were stained with Annexin V and 7-AAD. Whilst no significant difference in Annexin V labelling was observed, a twofold increase in 7-AAD positive cells was evident in RapKO MSCs compared to controls (Fig. 1E).
The Roles of Rptor and Rictor in Osteogenic and Adipogenic MSC Differentiation
We next examined the ability of RapKO and RicKO cells to differentiate into mature OBs and form a mineralized extracellular matrix [30, 31]. Under osteoinductive conditions, RapKO cells displayed a fivefold increase in mineral formation relative to controls (Fig. 2A), whereas RicKO cells displayed a twofold decrease in mineral formation (Fig. 2B).
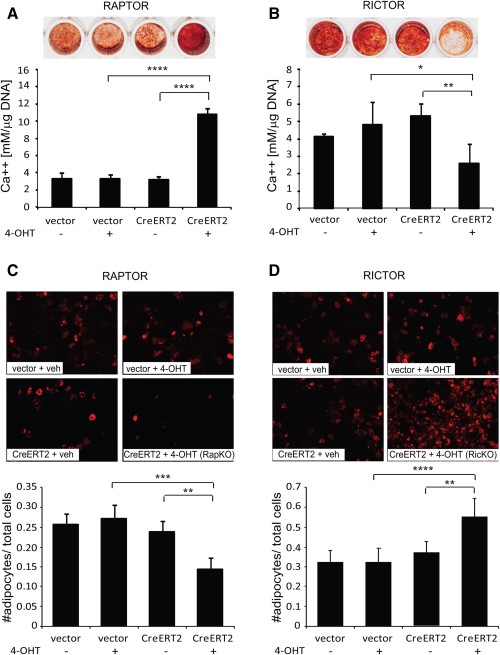
Differential roles for Rptor and Rictor in osteogenic and adipogenic mesenchymal stem cell (MSC) differentiation. Rptorfl/fl and Rictorfl/fl MSCs infected with the CreERT2 lentivirus or empty vector were treated for 8 days with 4-OHT (+) or vehicle (−). (A, B): Treated cells were cultured under osteoinductive conditions for 21 days and the amount of acid-solubilized calcium was quantitated and normalized to cell number. Data are expressed as the mean ± SD of quadruplicate wells from a representative experiment of three. Representative images of Alizarin Red-stained wells are shown. (C, D): Treated cells were cultured under adipogenic conditions for 8 days. Adipocytes were visualized using Nile Red, and the number of adipocytes quantitated and normalized to total cell number. Data are expressed as the mean ± SD of quadruplicate wells from a representative experiment of three. *, p < .05; **, p < .005; ***, p < .0005; ****, p < .0001, t test.
In parallel experiments, the potential of RapKO, RicKO, and control MSCs to differentiate into functional AdCs was also assessed. Quantitation of lipid-laden, Nile Red-stained adipocytes, relative to total cell number, showed that deletion of Rptor induced a 1.8-fold decrease in AdC formation, whereas deletion of Rictor resulted in a 1.8-fold increase in AdC formation (Fig. 2C, 2D).
Loss of Rptor Promotes Osteogenesis and Inhibits Adipogenesis
To gain insight into the molecular mechanisms by which Rptor deletion promotes osteogenic MSC differentiation, temporal gene expression analyses were performed. Loss of Rptor induced a strong upregulation of genes involved in OB differentiation including the osteogenic transcription factors Cbfa-1 (Runx2) and osterix (Sp7), and the marker of OB function, alkaline phosphatase (Alpl) (Fig. 3A). In inductive differentiation media, Rptor deletion was also associated with an increase in the expression of BMP-2 and other osteogenic TGFβ superfamily members such as inhibinβ A chain (Inhba) and inhibinβ E chain (Inhbe). In contrast, expression of integrin binding sialoprotein (Ibsp) was strongly downregulated in RapKO cultures, as was the expression of noggin (Nog), a negative regulator of BMP signaling (Fig. 3A).
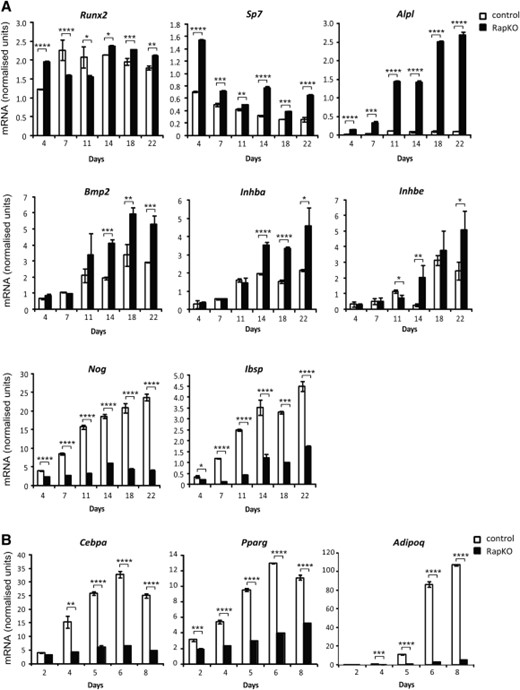
Loss of Rptor promotes osteogenesis and inhibits adipogenesis. Raptorfl/fl mesenchymal stem cells infected with the CreERT2 lentivirus were treated with vehicle (control) or 4-OHT (RapKO) for 8 days. (A): Treated cells were cultured under osteogenic conditions for 22 days and RNA harvested at the indicated time points. The mRNA level of osteogenic genes was examined by real-time PCR and data were normalized to β-actin. (B): Treated cells were cultured under adipogenic conditions for 8 days and RNA harvested at the indicated time points. The mRNA level of adipogenic genes was examined by real-time PCR and data were normalized to β-actin. *, p < .05; **, p < .005; ***, p < .0005; ****, p < .0001, t test.
In parallel experiments, the effect of Rptor deletion on molecular markers of AdC differentiation and function was also examined. Consistent with the marked inhibition of AdC formation in RapKO MSCs (Fig. 2C), the expression of Cebpa and Pparg, transcription factors essential for adipogenesis, and adiponectin (Adipoq), a marker of mature adipocytes, was downregulated in RapKO MSCs at all time points examined (Fig. 3B).
Loss of Rictor Promotes Adipogenesis and Inhibits Osteogenesis
We also examined the temporal regulation of adipogenic genes in RicKO and control MSCs during adipogenic differentiation. Consistent with the induction of AdC formation in RicKO MSCs (Fig. 2D), the expression of Cebpa, Pparγ, and Adipoq was strongly upregulated in RicKO MSCs at all time points examined (Fig. 4A). Under osteoinductive culture conditions, loss of Rictor was associated with a downregulation of Runx2, Sp7, Alpl, Bmp2, and Inhba expression (Fig. 4B), consistent with the inhibition of osteogenesis observed in RicKO cells (Fig. 2B).
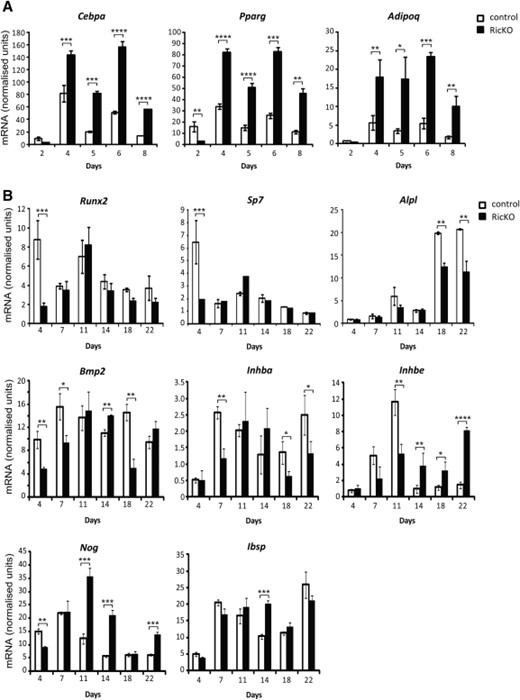
Loss of Rictor promotes adipogenesis and inhibits osteogenesis. Rictorfl/fl mesenchymal stem cells infected with the CreERT2 lentivirus were treated with vehicle (control) or 4-OHT (RicKO) for 8 days. (A): Treated cells were cultured under adipogenic conditions for 8 days and RNA harvested at the indicated time points. The mRNA level of adipogenic genes was examined by real-time PCR and data were normalized to β-actin. (B): Treated cells were cultured under osteogenic conditions for 22 days and RNA harvested at the indicated time points. The mRNA level of osteogenic genes was examined by real-time PCR and data were normalized to β-actin. *, p < .05; **, p < .005; ***, p < .0005; ****, p < .0001, t test.
Discussion
Using Cre-mediated gene deletion and well-established in vitro differentiation assays, we have shown that mTORC1 and mTORC2 have distinct roles in MSC fate determination. Consistent with previous studies (reviewed in [32]), deletion of Rptor in MSCs reduced their adipogenic potential. Under osteoinductive conditions however, Rptor deletion promoted osteogenic differentiation. This switching between differentiation programs could reflect the mutual exclusivity of these programs (i.e., enhanced osteogenesis in the absence of mTORC1-mediated PPARγ activation). Alternatively, loss of mTORC1 may maintain the osteogenic potential of MSCs, in line with studies showing that rapamycin attenuates stem cell aging, maintaining their self-renewal and osteogenic potential [33-36], and with in vivo studies showing that deletion of p70S6K (mTORC1 effector) protects against age-related bone loss [37].
While further studies are required to characterize the precise mechanism(s) by which mTORC1 inhibition promotes osteogenesis, our PCR data suggest that BMP signaling is involved. Many BMPs are strongly pro-osteogenic (reviewed in [38]); however, their role in osteogenesis is complex [39, 40], influenced by variables such as dose [41] and BMP receptor subtype [39, 42].
In contrast to RapKO cells, Rictor deletion promoted adipogenesis and inhibited osteogenesis, which is consistent with a recent siRNA study which showed that mTORC2 regulation of cytoskeletal organization plays a role in MSC fate determination in response to mechanical stress [43]. These findings suggest that the decreased osteogenic potential of RicKO cells may be associated with impaired cytoskeletal organization. Overall, these findings provide new insight into the role of mTOR signaling in MSC lineage commitment.
Acknowledgments
We thank Dr. Kristoffer Riecken (University Medical Centre Hamburg) for the LeGO-iG2 vector and Dr. Pierre Chambon (IGBMC, France) for the Cre-ERT2 expression vector. This work was supported by grants awarded by the National Health and Medical Research Council of Australia (APP1030528), a Royal Adelaide Hospital Mary Overton Early Career Fellowship (SKM), and an Australian Postgraduate Award (MPM). We gratefully acknowledge the assistance of Kate Pilkington and Rebecca Salmon (Detmold Family Imaging Facility, SA Pathology) with cell sorting.
Author Contributions
S.K.M.: conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, and final approval of this manuscript; S.F.: conceived and designed the experiments, performed the experiments, analyzed the data, and final approval of this manuscript; A.C.W.Z.: conceived and designed the experiments and final approval of this manuscript; A.K.D.: performed the experiments, analyzed the data, and final approval of this manuscript; M.P.M.: performed the experiments and final approval of this manuscript; M.E.M.: analyzed the data and final approval of this manuscript; C.R.W., M.A.R., M.N.H., and S.G.: contributed reagents, expertise, and materials, and final approval of this manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
{kind=link}
{kind=link}
{kind=link}
{kind=link}