Compensatory growth renders Tcf7l1a dispensable for eye formation despite its requirement in eye field specification
- University College London, United Kingdom
- Wellcome Genome Campus, United Kingdom
- University of Cambridge, United Kingdom
- Facultad de Ciencias, Universidad de Chile, Chile
Abstract
The vertebrate eye originates from the eye field, a domain of cells specified by a small number of transcription factors. In this study, we show that Tcf7l1a is one such transcription factor that acts cell-autonomously to specify the eye field in zebrafish. Despite the much-reduced eye field in tcf7l1a mutants, these fish develop normal eyes revealing a striking ability of the eye to recover from a severe early phenotype. This robustness is not mediated through genetic compensation at neural plate stage; instead, the smaller optic vesicle of tcf7l1a mutants shows delayed neurogenesis and continues to grow until it achieves approximately normal size. Although the developing eye is robust to the lack of Tcf7l1a function, it is sensitised to the effects of additional mutations. In support of this, a forward genetic screen identified mutations in hesx1, cct5 and gdf6a, which give synthetically enhanced eye specification or growth phenotypes when in combination with the tcf7l1a mutation.
https://doi.org/10.7554/eLife.40093.001eLife digest
Left and right eyes develop independently, yet they consistently grow to roughly the same size in humans and other creatures. How they do this remains a mystery, though scientists have learned that both eyes originate from a single group of cells in the developing nervous system called the eye field. As development progresses, the eye field splits in two, and buds into the two separate compartments from which each eye forms. As the eyes grow, the cells in each compartment specialize, or ‘differentiate’, to make working left and right eyes. Scientists often study eye development in zebrafish embryos because it is easy to see each step in the process.
Now, Young at al. show that zebrafish with a mutation that causes the eye field to be half its normal size go on to form normal-sized eyes. Somehow these developing embryos overcome this deleterious mutation. It turns out that the eyes of zebrafish with this mutation grow for a longer period of time than typical zebrafish eyes. This change allows the mutant fish’s eyes to catch up and reach normal size. When Young et al. removed some cells from one of the forming eyes of normal zebrafish embryos they found that same thing happened. The smaller eye developed for a longer time and delayed its differentiation until both eyes were the same size. Conversely, when eyes developed from a larger than normal eye field, growth stopped prematurely and differentiation began early preventing the eyes from ending up oversized.
Though the fish were able to overcome the effects of one mutation to develop normal-sized eyes, adding a second mutation that affected eye development led to unusual sized eyes or absence of eyes. Together the experiments identify genes and mechanisms essential for the formation and size of the eyes. Given that the processes underlying eye formation are very similar in many animals, this new information should help scientists to better understand eye abnormalities in humans.
https://doi.org/10.7554/eLife.40093.002Introduction
The paired optic vesicles originate from the eye field, a single, coherent group of cells located in the anterior neural plate (Cavodeassi, 2018). During early neural development, the specification and relative sizes of prospective forebrain territories, including the eye field, depend on the activity of the Wnt/β-Catenin and other signalling pathways (Beccari et al., 2013; Cavodeassi, 2014; Wilson and Houart, 2004). Enhanced Wnt/β-Catenin activity leads to embryos with small or no eyes (Cavodeassi et al., 2005; Kim et al., 2000; Heisenberg et al., 2001; Houart et al., 2002). In contrast, decreasing activity of Wnt/β-Catenin signalling generates embryos with bigger forebrain and eyes (Cavodeassi et al., 2005; Glinka et al., 1998; Lekven et al., 2001; Houart et al., 2002). Although much research has focused on the molecular mechanisms involved in the specification of the eye field, little is known about what happens to the eyes if eye field size is disrupted.
A number of genes have been identified as encoding a transcription factor network that specifies the eye field (Beccari et al., 2013; Zuber et al., 2003). These genes have been defined based on conserved cross species expression patterns in the anterior neuroectoderm and on phenotypes observed when overexpressed or when function is compromised (Beccari et al., 2013). Perhaps surprisingly, to date, there are relatively few mutations that lead to complete loss of eyes suggesting that early stages of eye development are robust to compromised function of genes involved in eye development. Indeed, in humans, eye phenotypes are often highly variable in terms of penetrance and expressivity even between left and right eyes (Reis and Semina, 2015; Williamson and FitzPatrick, 2014). This again raises the possibility that the developing eye is robust and can sometimes cope with mutations in genes involved in eye formation.
Genetic robustness is the capacity of organisms to withstand mutations, such that they show little or no phenotype, or compromised viability (Félix and Barkoulas, 2015; Wagner, 2005). This inherent property of biological systems is wired in the genetic and proteomic interactomes and enhances the chance of viability of individuals in the face of mutations. High-throughput reverse mutagenesis projects and the emergence of CRISPR/Cas9 gene editing techniques have highlighted the fact that homozygous loss of function mutations in many genes generate viable mutants with no overt phenotype (Varshney et al., 2015; Dickinson et al., 2016; Meehan et al., 2017). Across phyla, mutations in single genes are more likely to give rise to viable organisms than to show overt or lethal phenotypes. For instance, it is estimated that zygotic homozygous null mutations in just ~7% of zebrafish genes compromise viability before 5 days post-fertilisation (Kettleborough et al., 2013) and 8–10% between day 5 and 3 months (Shawn Burgess, personal communication); and compromised viability is predicted following loss of function for about 35% of mice genes (Dickinson et al., 2016; Meehan et al., 2017). Furthermore, apparently healthy viable homozygous or compound heterozygous ‘gene knockouts’ have been found for 1171 genes in the Icelandic human population (Sulem et al., 2015) and for 1317 genes in the Pakistani population (Saleheen et al., 2017).
In some cases, the lack of overt phenotype may be due to redundancy in gene function based on functional compensation by paralogous or related genes (Barshir et al., 2018; Hurles, 2004; Wagner, 1996). We can assume that genes that do not express a phenotype when mutated are not lost to genetic drift because in some way they enhance the fitness of the species. For instance, even though two paralogous Lefty genes encoding Nodal signalling feedback effectors have been shown to be individually dispensable for survival, they do make embryonic development robust to signalling noise and perturbation (Rogers et al., 2017).
Genetic compensation for deleterious mutations is a cross-species feature (El-Brolosy and Stainier, 2017), and mRNAs that undergo nonsense-mediated decay due to mutations that lead to premature termination codons can upregulate the expression of paralogous and other related genes (El-Brolosy et al., 2018). However, only a fraction of genes have paralogues and other compensatory mechanisms must contribute to the ability of the embryo to cope with potentially deleterious mutations. One such mechanism is distributed robustness, which can emerge in gene regulatory networks (Wagner, 2005). This kind of robustness relies on the ability of the network to regulate the expression of genes and/or the activity of proteins within the network, such that homeostasis is preserved when one of its components is compromised (Davidson, 2010; Peter and Davidson, 2016).
Maternal-zygotic tcf7l1a mutant zebrafish have been previously described as lacking eyes (Kim et al., 2000). In this study, we show that expression of this phenotype is dependent on the genetic background. We find that tcf7l1a mutants can develop functional eyes and are viable, and that this is not due to compensatory upregulation of other lef/tcf genes. Despite the presence of functional eyes, the eye field in tcf7l1a mutants is only half the size of the eye field of wildtype embryos, indicating an early requirement for tcf7l1a during eye field specification. We further show that this requirement is cell autonomous, revealing a striking dissociation between the early role and requirement for Tcf7l1a in eye field specification and the later absence of an overt eye phenotype. Subsequent to compromised eye field specification, tcf7l1a mutant eyes recover their size by delaying neurogenesis and prolonging growth in comparison to wildtype eyes. This compensatory ability of the developing eye was also observed when cells were removed from wild-type optic vesicles. Altogether, our study suggests that the loss of Tcf7l1a does not trigger any genetic compensation or signalling pathway changes that restore eye field specification; instead, the developing optic vesicle shows a remarkable ability to subsequently modulate its development to compensate for the early, severe loss of eye field progenitors.
The penetrance and expressivity of eye phenotypes appears to be dependent on complex genetic and environmental interactions (Gestri et al., 2009; Kaukonen et al., 2018; Prokudin et al., 2014). Thus, we speculated that tcf7l1a mutant eyes may be sensitised to the effects of additional mutations. Here, we show this is indeed the case and describe the isolation of three mutations from a recessive synthetic modifier screen in tcf7l1a homozygous mutant zebrafish that lead to enhanced/novel eye phenotypes when in combination with loss of tcf7l1a function.
In summary, our work shows that zebrafish eye development is robust to the effects of a mutation in tcf7l1a due to compensatory growth mechanisms that may link eye size and neurogenesis. Our study adds to a growing body of research revealing a variety of mechanisms by which the developing embryo can cope with the effects of deleterious genetic mutations.
Results
The tcf7l1am881/m881 mutation is fully penetrant but maternal-zygotic mutants show no overt eye phenotype and are viable
The headless (hdl)m881 mutation in tcf7l1a (tcf7l1a-/- from here onwards) was identified because embryos lacking maternal and zygotic (MZ) gene function lacked eyes (Kim et al., 2000). However, no overt defects were observed in zygotic (Z) tcf7l1a-/- mutants, due to functional redundancy with the paralogous tcf7l1b gene (Dorsky et al., 2003). In our facility, MZtcf7l1a-/- embryos initially showed a variable eye phenotype, ranging from eyeless, to small and overtly normal eyes, with proportions that varied in clutches from different pairs of fish (not shown). We hypothesised that genetic background effects could be responsible for either enhancing or suppressing the eyeless phenotype. To test this idea, we outcrossed tcf7l1a-/- fish to ekkwill (EKW) or AB wildtype fish and identified tcf7l1+/- carriers by PCR genotyping. After three generations of outcrossing to EKW or AB fish, we incrossed tcf7l1+/- carriers to grow Ztcf7l1a-/- adults. All MZtcf7l1a-/- embryos coming from six pairings of Ztcf7l1a-/- mutant fish developed eyes only slightly reduced in size compared to eyes of wildtype embryos of the same EKW or AB strain (100%, n > 100; Figure 1A,B).
Figure 1 with 2 supplements see all
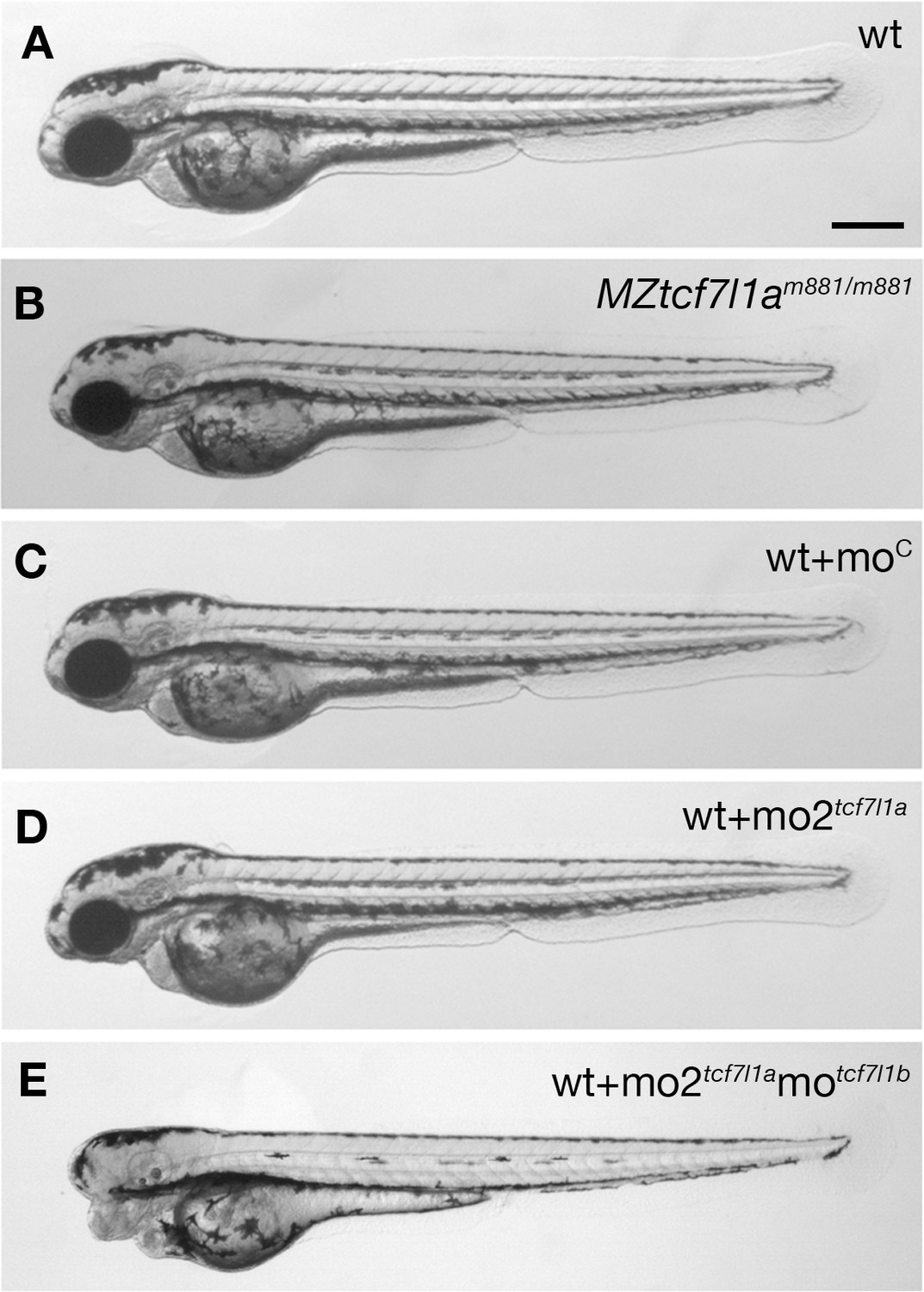
Tcf7l1a maternal zygotic (MZ) mutants and tcf7l1a morphants have no overt eye phenotype.
Lateral views of typical wildtype (A) MZtcf7l1a-/- (B) wildtype injected with control morpholino (C) tcf7l1a morphant (D) and tcf7l1a/tcf7l1b double morphant (E) embryos at 2 days post fertilisation. All conditions n > 100 and over three independent experiments except when specified. Dorsal up, anterior to the left. Scale Bar = 250 µm.
The tcf7l1am881 mutation creates a splice acceptor site in intron 7, which leads to a seven nucleotide insertion in tcf7l1a mRNA that gives rise to a truncated protein due to a premature termination codon (Kim et al., 2000). Given that the wildtype splice site in intron seven is still present in tcf7l1a mutants, we assessed whether the lack of phenotype in MZtcf7l1a-/- mutants could be due to incomplete molecular penetrance as a result of expression of mRNA from both wildtype and mutant splice sites. The chromatogram sequence of the RT-PCR product amplifying exons 7 and 8 in wildtype, mutant and heterozygous embryos showed that only wildtype tcf7l1a mRNA was detected in wildtype embryos and only mutant mRNA containing the seven nucleotide insertion was observed in mutants, while heterozygous embryos produced both wildtype and mutant mRNAs (Figure 1—figure supplement 1; Kim et al., 2000). This suggests that the mutant splice site is the only one used in tcf7l1a-/- embryos. In addition, while overexpression of wildtype tcf7l1a mRNA rescued eye formation in embryos in which both tcf7l1a and tcf7l1b are knocked down, tcf7l1am881 mutant mRNA did not, confirming that protein arising from the tcf7l1am881 allele is not functional (not shown; Kim et al., 2000). These observations suggest that the m881 allele is indeed a null mutation and that tcf7l1a is not essential for eye formation.
Supporting a requirement for tcf7l1a to form eyes, antisense morpholino knockdown of tcf7l1a (mo1tcf7l1a) leads to eyeless embryos (Dorsky et al., 2003) comparable to the originally described headless MZtcf7l1a-/- mutant phenotype (Kim et al., 2000). However, the target site for the morpholino used in that study shows considerable sequence homology to the translation start ATG region of other tcf gene family members (56–76%; Figure 1—figure supplement 2A). This suggests that the mo1tcf7l1a phenotype may be due to the morpholino knocking down expression of other tcf genes, as has been described for other morpholinos targeting paralogous genes (Kamachi et al., 2008). Indeed, injection of a different tcf7l1a morpholino (mo2tcf7l1a) with low homology to other tcf genes (36–45%, Figure 1—figure supplement 2B) did not lead to an eyeless phenotype (0.4 pMol/embryo, 100%, n > 100; Figure 1C,D). tcf7l1b morpholino injection on its own showed no overt phenotype (Dorsky et al., 2003) but co-injection of mo2tcf7l1a and motcf7l1b gave rise to eyeless embryos (each at 0.2 pMol/embryo, 78.26%, n = 92, over three experiments; Figure 1E and see Dorsky et al., 2003). This suggests that even though mo2tcf7l1a injection alone resulted in no phenotype, the morpholino does knockdown tcf7l1a.
Together, these results suggest that even though tcf7l1a-/- is a fully penetrant null mutation, lack of maternal and zygotic tcf7l1a function alone does not lead to loss of eyes in all genetic backgrounds.
tcf7l1a loss of function is not compensated by upregulation of other tcf genes
Ztcf7l1a-/- and MZtcf7l1a-/- embryos develop eyes, whereas embryos lacking both Ztcf7l1a and Ztcf7l1b do not (Dorsky et al., 2003). Thus, we hypothesised that enhanced expression of the paralogous tcf7l1b, or other lef/tcf genes may compensate for the absence of tcf7l1a function, as shown for other mutations (El-Brolosy et al., 2018; Rossi et al., 2015). To test this idea, we assessed the expression of all lef/tcf genes by RT-qPCR in sibling wildtype and Ztcf7l1a-/- mutant embryos at the stage when the eye field has been specified (10 hr post-fertilisation; hpf).
Expression levels of lef/tcf genes did not increase in Ztcf7l1a-/- mutant embryos suggesting that there is no compensatory upregulation (Figure 2A, Supplementary file 1A). As previously shown, tcf7l1a undergoes nonsense-mediated decay in mutants resulting in reduced expression levels (Kim et al., 2000; Figure 2A; Supplementary file 1A). lef1 and tcf7 levels did not change significantly in mutants and tcf7l1b (tcf3b) and tcf7l2 (tcf4) expression was actually reduced to 63 ± 6% and 62 ± 8% respectively of wildtype levels (Figure 2A; Supplementary file 1A). The otx1b and otx2 genes, which are expressed in the anterior neural plate, also showed slightly reduced expression (otx1b, reduced to 81 ± 11% and otx2, 79 ± 10%) suggesting that the anterior neural plate may be slightly reduced in size in tcf7l1a mutants. Indeed, the domain of the neural plate encompassed by expression of emx3 around the anterior margin of the neural plate up to the mesencephalic marker pax2a (Figure 2D,E) was reduced to 76% of wildtype size in tcf7l1a mutants (n = 11, p=0.0041, Figure 2B; Supplementary file 1B). This indicates that a reduction in the size of the prospective forebrain of Ztcf7l1a-/- embryos may contribute to the reduced levels of expression of tcf7l1b, tcf7l2 and otx genes.
Figure 2 with 3 supplements see all
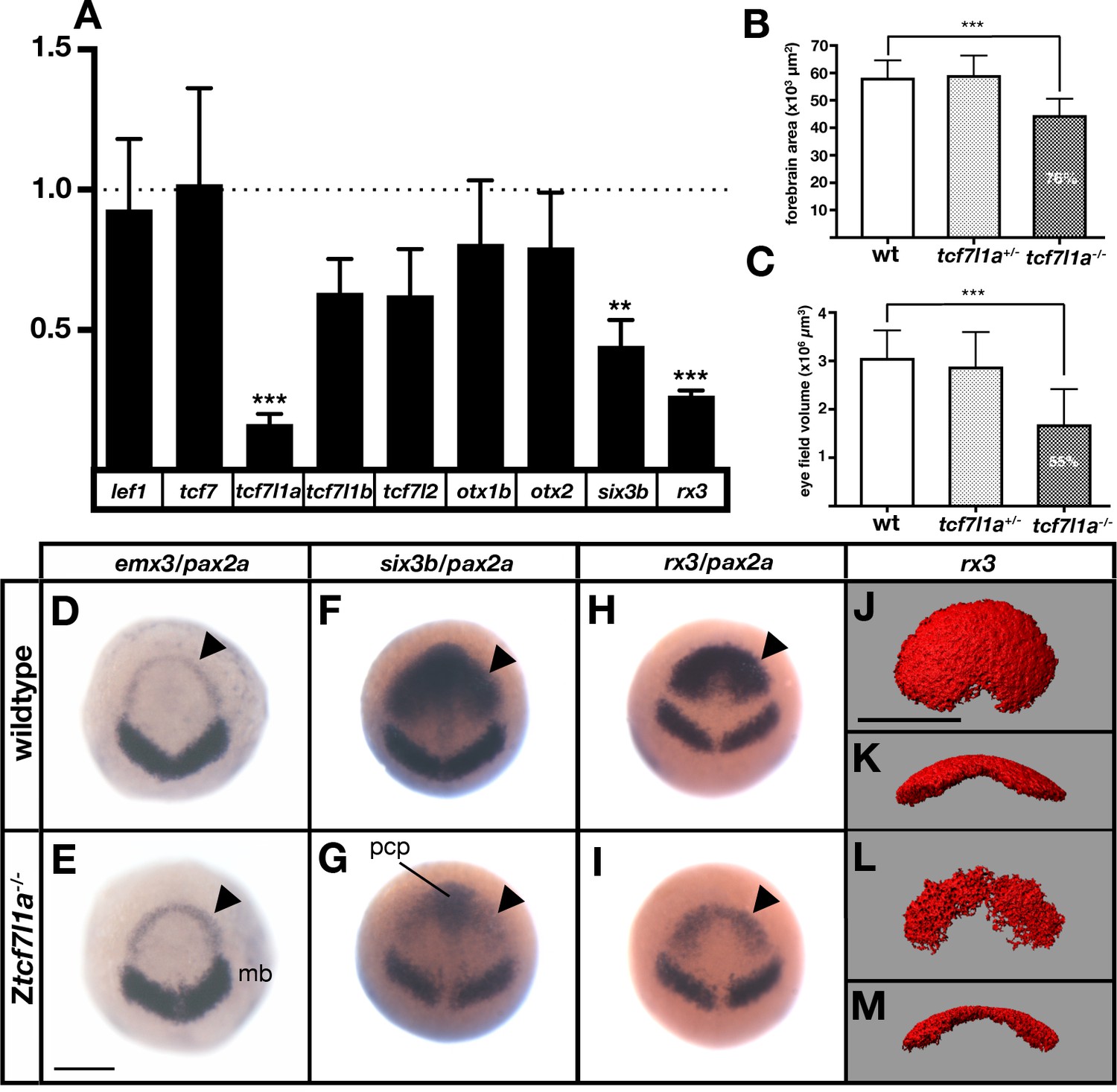
The prospective forebrain and eye field domains of the neural plate are reduced in Ztcf7l1a-/- mutants.
(A) Graph showing RT-qPCR quantification of the mRNA levels of lef1, tcf7, tcf7l1a, tcf7l1b, tcf7l2, otx1b, otx2, six3b and rx3 in Ztcf7l1a-/- mutants relative to wildtype embryos at 10hpf. Biological and technical triplicates, two independent experiments. (B, C) Quantification of the forebrain domain of the anterior neural plate (B) enclosed by emx3 up to pax2a (D, E) expression by in situ hybridisation (reduction to an average of 76%, n = 11, one experiment, data in Supplementary file 1B), and eye field volume (C) by rx3 fluorescent in situ hybridisation confocal volume reconstruction (J–M) (reduction to an average of 55%, n = 10, one experiment, data in Supplementary file 1C). (D–I) Expression of emx3 (arrowhead)/pax2a (D, E), six3b (arrowhead)/pax2a (F, G) and rx3 (arrowhead)/pax2a (H, I) in wildtype (D, F, H) and Ztcf7l1a-/- (E, G, I) embryos detected by in situ hybridisation at 10hpf. Reduction of six3b and rx3 expression 100%, n > 40, three experiments. (J–M) Confocal volume reconstruction of rx3 fluorescent in situ hybridisation in wildtype (J, K) and Ztcf7l1a-/- (L, M) mutants at 10hpf. (J, L) Dorsal view, anterior to top, and (K, M) transverse view from posterior, dorsal up. Abbreviations: mb, midbrain; pcp, prechordal plate Scale Bars = 250 µm.
Overall, these results suggest that tcf genes do not show compensatory regulation in response to loss of tcf7l1a function.
Optic vesicles evaginate and form eyes in MZtcf7l1a-/- mutants despite a much-reduced eye field
More remarkable than the modest changes in tcf and otx gene expression was the finding that RT-qPCR showed very reduced expression of eye field genes in Ztcf7l1a-/- mutant embryos (Figure 2A; rx3 reduced to 26 ± 1%, p=0.0002 and six3b reduced to 44 ± 5%, p=0.0091 of wildtype levels). Consequently, the presence of overtly normal looking eyes in both Ztcf7l1a-/- and MZtcf7l1a-/- embryos is surprising given that rx3-/- mutant embryos lack eyes due to impaired specification/evagination of the optic vesicles (Loosli et al., 2003; Stigloher et al., 2006). We confirmed that expression of six3b and rx3 is reduced in the anterior neural plate by in situ hybridisation in Ztcf7l1a-/- and tcf7l1a morphant embryos (100%,n > 40; Figure 2F–I; Figure 2—figure supplement 1A,B; similar changes seen in MZtcf7l1a-/- mutants, data not shown). The expression of six3b was reduced in the eye field but not in the prechordal plate of Ztcf7l1a-/- mutants, likely explaining why RT-qPCR showed a greater reduction in rx3 than six3b expression (Figure 2F–I; Supplementary file 1A). Analysis of eye field volume by fluorescent in situ hybridisation of rx3 revealed a reduction to 54.7% of wildtype size (n = 10, Figure 2C,J–M; Supplementary file 1C) and intensity of expression within the reduced eye field also appeared reduced (Figure 2H,I).
Further in situ hybridasation analysis suggests that it is the caudal region of the eye field that is most affected in Ztcf7l1a-/- mutants. emx3 expression directly rostral to the eye field was slightly broader in Ztcf7l1a-/- mutants than wildtypes but expression did not encroach into the reduced eye field (Figure 2D,E; Figure 2—figure supplement 2A,B, n = 5 each condition). Conversely, expression of the prospective diencephalic marker barhl2 caudal to the reduced eye field was expanded rostrally at 10hpf (Figure 2—figure supplement 2C,D n = 5 each condition) and even more evidently at 9hpf (Figure 2—figure supplement 2E,F, 13/13 Ztcf7l1a-/-). These observations suggest a caudalisation of the anterior neural plate in Ztcf7l1a-/- mutants leading to reduced eye field specification consistent with phenotypes observed in conditions in which Wnt pathway repression is reduced (Heisenberg et al., 2001; van de Water et al., 2001).
RNAseq analysis of wildtype, Ztcf7l1a-/- and Ztcf7l1a-/-/Ztcf7l1b+/- embryos at 8.5hpf (80–90% epiboly stage), when the eye field is first specified confirmed and extended RT-qPCR and in situ hybridisation analyses (Supplementary file 1D). In Ztcf7l1a-/-, hesx1 (Kazanskaya et al., 1997), rx3, tcf7l1a, and fezf2 (Sun et al., 2006) which are expressed in the prospective forebrain/eyefield were downregulated, whereas her5 and irx1b which are expressed more caudally in the neural plate (Müller et al., 1996, Wang et al., 2001b) were upregulated consistent with mild caudalisation of the neural plate. Additionally, in Ztcf7l1a-/-/tcf7l1b+/- embryos, which to not form eyes, the expression of tcf7l1b was enhanced by about 40%, suggesting transcriptional compensation but evidently this was not sufficient to rescue eye formation.
Despite the small size of eye field in tcf7l1a-/- mutants, optic vesicles appear to evaginate normally. Time lapse analysis of optic vesicle evagination using the Tg(rx3:GFP)zf460Tg transgene to visualise eye field cells (Brown et al., 2010) showed that optic vesicle morphogenesis in Ztcf7l1a-/- embryos proceeded as in heterozygous sibling embryos (Figure 2—figure supplement 3A,B; tcf7l1a+/-, n = 6 and Ztcf7l1a-/- n = 6; Video 1 and Video 2).
Video 1
Time lapse movies of eye vesicle evagination in and Ztcf7l1a+/- siblings.
Confocal time lapse movies (1 frame every 5 min) of Ztcf7l1a+/- sibling (S1, n = 5) expressing the Tg(rx3:GFP)zf460Tg transgene. First frame taken at 11hpf; membrane RFP in red counterstain.
Video 2
Time lapse movies of eye vesicle evagination in Ztcf7l1a-/- mutants.
Confocal time lapse movies (1 frame every 5 min) of Ztcf7l1a-/- mutants (S2, n = 5) expressing the Tg(rx3:GFP)zf460Tg transgene. First frame taken at 11hpf; membrane RFP in red counterstain.
Tcf7l1a functions cell-autonomously to promote eye field specification
Although Tcfs regulate the balance between activation and repression of the Wnt/βCatenin pathway during anterior neural plate regionalisation (Kim et al., 2000; Dorsky et al., 2003), it is unclear if Tcf function is required for cells to adopt the eye field fate. To address this, we determined whether Tcf7l1a function is required cell-autonomously during eye formation by transplanting wildtype and MZtcf7l1a-/- GFP labelled (GFP+) cells into wildtype and tcf7l1a mutant hosts and analysing the expression of rx3 when eye specification has occurred (10hpf, 100% epiboly; Figure 3).
Figure 3
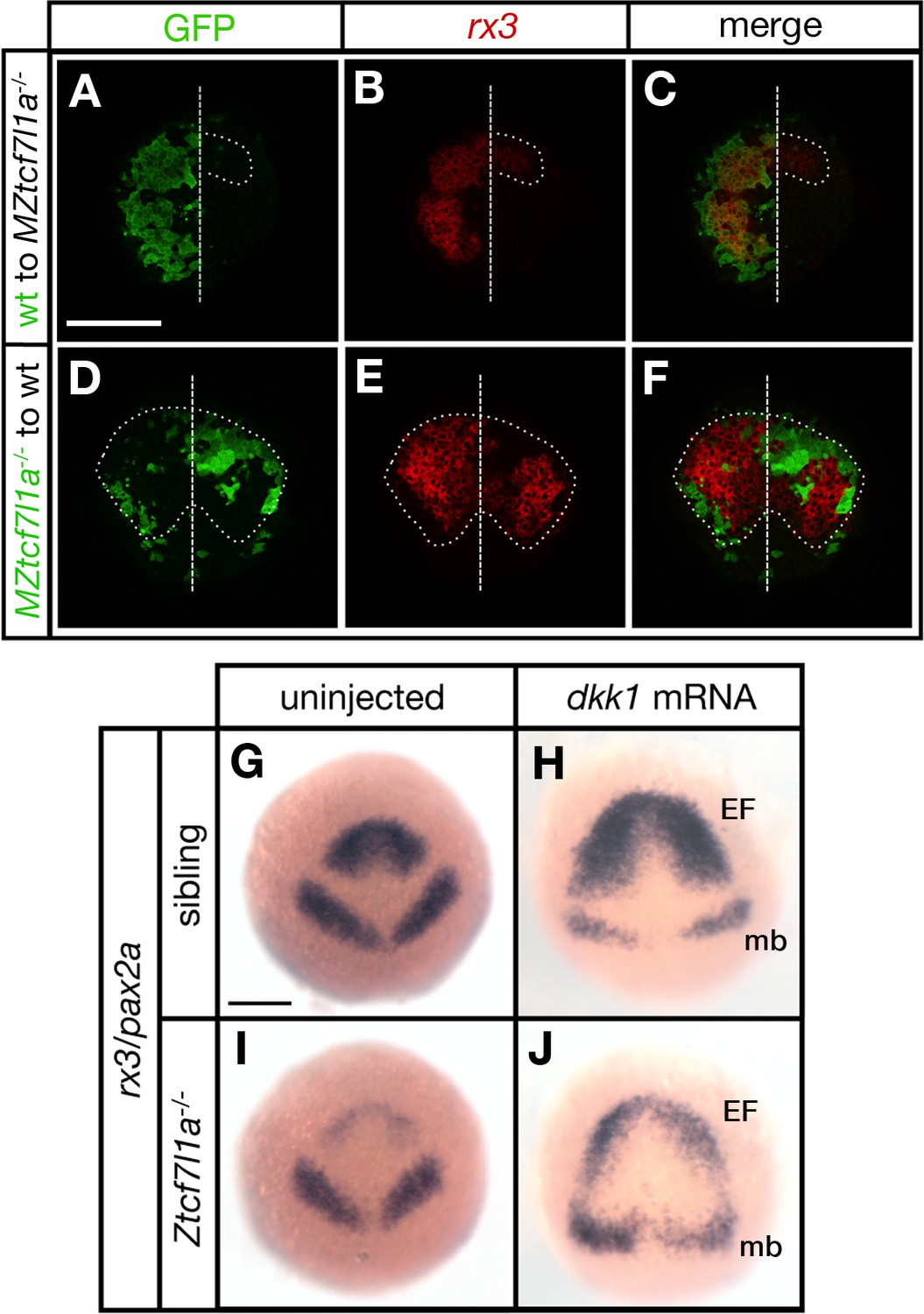
Tcf7l1a cell autonomously promotes rx3 expression in the eye field.
(A–F) Dorsal views of confocal images of rx3 mRNA expression (red) detected by fluorescent in situ hybridisation at 10hpf in the anterior neural plates of chimeric embryos containing transplants of (A–C) wildtype (GFP+) donor cells in MZtcf7l1a-/- host embryos (100%, n = 13), and (D–F) MZtcf7l1a-/- (GFP+) donor cells in wildtype host embryos (100%, n = 9). Dotted line outlines eye fields; note in A-C that rx3 expression extends considerably caudal to the reduced mutant eye field on the side of the neural plate containing wild-type cells. Dashed line marks the embryo midline. (G–J) In situ hybridisation of rx3 and pax2a in sibling (G, H) and Ztcf7l1a-/- (I, J) 9hpf embryos, uninjected (G, I) or injected with 50 pg of dkk1 mRNA (H, J). Abbreviations; EF, eyefield; mb, midbrain Scale Bars = 200 µm.
Transplants of wildtype cells to MZtcf7l1a-/- mutant embryos led to the recovery of rx3 expression exclusively restricted to the wildtype GFP+ cell clones (13/13 transplants, Figure 3A–C). However, the border of the GFP+ wildtype clones showed less rx3 expression, suggesting that cells at the edge of the graft are subject to cell non-autonomous signalling effects from cells surrounding the clone. Conversely, MZtcf7l1a-/- mutant GFP+ cells expressed much lower levels of rx3 than wildtype neighbours when positioned in the eye field of wildtype embryos (9/9 transplants, Figure 3D–F). The reduction in rx3 expression was limited to the MZtcf7l1a-/- GFP+ mutant cells. Control transplants of cells from wildtype donor embryos to wildtype hosts showed no effect on rx3 expression (not shown). Consistent with a cell autonomous role for Tcf7l1a in eye formation, overexpression of the Wnt inhibitor Dkk1 (Hashimoto et al., 2000) expanded the anterior neural plate in both wildtype and tcf7l1a mutants, but rx3 expression and eye field size remained much smaller in the enlarged anterior plate of tcf7l1a mutants (Figure 3G–J).
All together, these results support a cell-autonomous role for Tcf7l1a in promoting eye field specification.
Eye size in Ztcf7l1a-/- embryos recovers with growth kinetics similar to wildtype embryos
Despite a much-reduced eye field, eyes in Ztcf7l1a-/- fry and adults seem indistinguishable from those in wildtype siblings. Indeed, optokinetic responses of Ztcf7l1a-/- and wildtype 5dpf larvae showed no significant differences at any of the four tested spatial frequencies (Figure 4—figure supplement 1, Supplementary file 1E), suggesting that by this stage, Ztcf7l1a-/- eyes are functional and have visual acuity comparable to that of wildtype siblings. Consequently, although Ztcf7l1a-/- embryos show a robust and severe neural plate patterning phenotype, eye formation recovers over time. To explore how this recovery happens, we measured eye size in Ztcf7l1a-/- embryos from 24 to 96hpf (Figure 4A,C–L; Supplementary file 1F), estimating eye volumes from retinal profiles (see Materials and methods).
Figure 4 with 4 supplements see all

Eye size recovers in Ztcf7l1a-/- mutant and eye vesicle-cell removed embryos.
(A) Growth kinetics of the eye in wildtype (blue line) and Ztcf7l1a-/- (red line) embryos at stages indicated (data in Supplementary file 1F, one experiment, 24hpf, wt n = 12, Ztcf7l1a-/- n = 14; 28hpf, wt n = 15, Ztcf7l1a-/- n = 12; 32hpf, wt n = 13, Ztcf7l1a-/- n = 15; 36hpf, wt n = 16, Ztcf7l1a-/- n = 14; 48hpf, wt n = 11, Ztcf7l1a-/- n = 19; 60hpf, wt n = 11, Ztcf7l1a-/- n = 14; 72hpf, wt n = 13, Ztcf7l1a-/- n = 19; 96hpf, wt n = 13, Ztcf7l1a-/- n = 15). (B) Plot showing the ratio of Ztcf7l1a-/- to wildtype eye volume from data in (A). (C–L) Lateral views (dorsal up, anterior to left) of wildtype (C–G) and Ztcf7l1a-/- (H–L) eyes at stages indicated above panels. (M–O) Eye development following partial ablation of the optic vesicle in wildtype embryos at five somite stage. (M) Coronal confocal section of evaginating optic vesicles (red) in a wildtype Tg(rx3:RFP) five somite stage embryo. Dashed line indicates the approximate extent of ablations performed. 36hpf (N) and 4dpf (O) eyes in embryos in which cells were unilaterally removed from one optic vesicle (from n = 20). Asterisk indicates the eye that develops from the partially ablated optic vesicle. ZO1, zona ocludens 1. Scale bars = 200 µm.
At 24hpf, eye volume in mutants was about 57% of the estimated volume of wildtype eyes at the same stage (Figure 4A,C,H; Supplementary file 1F). However, by 48hpf mutant eyes were about 85% of the size of eyes in wildtype/heterozygous siblings (Figure 4A,G,L; Supplementary file 1F). Ztcf7l1a-/- eye size did not further recover beyond this time point and up to 5dpf (Figure 4B). Eye growth in both wildtypes and Ztcf7l1a-/- mutants showed similar growth kinetics (Figure 4A). This suggests that even though Ztcf7l1a-/-eyes are smaller, they follow a comparable developmental time-course as wildtype eyes in the early growth phase between 24 and 36hpf but with about 8 hr delay (for instance, a 32hpf Ztcf7l1a-/- eye is about the same size as a wild-type 24hpf eye).
The temporal shift in eye growth in Ztcf7l1a-/- mutants is not explained by an overall developmental delay as the position of the posterior lateral line primordium (pLLP) was similar to wildtype at all stages tested (Figure 4—figure supplement 2, Supplementary file 1G). Volumes of eye cells in Ztcf7l1a-/- mutants and siblings were not significantly different at 24 or 36hpf and consequently cell size changes likely play no role in eye size compensation in Ztcf7l1a mutants (Figure 4—figure supplement 3, Supplementary file 1H).
Eye size recovers after physical removal of optic vesicle cells
To assess if size recovery is a general feature of eye development, we physically removed optic vesicle cells in wildtype embryos and assessed the effect on eye growth. Cells were aspirated from one of the two nascent optic vesicles at 12hpf (six somite stage), leaving approximately the medial half of the vesicle intact (Figure 4M). At 36hpf, there was still a clear size difference between the experimental and control eyes (Figure 4N, Figure 4—figure supplement 4, Supplementary file 1I). However, by 4dpf we observed little or no size difference between control and experimental eyes (three independent experiments, n=20/20, Figure 4O, Figure 4—figure supplement 4, Supplementary file 1I). Three eyes from partially ablated optic vesicles which were ~90% the size of their control contralateral eyes at 30hpf, recovered to 100% by 3dpf (Figure 4—figure supplement 4K, Supplementary file 1I) and five eyes which were between ~65 and~75% of control size at 30hpf recovered to 90% by 3dpf with little or no further recovery by 4dpf (Figure 4—figure supplement 4A–I,K, Supplementary file 1I). Consequently, the forming eye can effectively recover from either genetic or physical reduction in the size of the eye field/evaginating optic vesicle.
Neurogenesis is delayed in tcf7l1a mutant eyes
The observation that wildtype and Ztcf7l1a-/- mutant eyes display similar, but temporally offset, growth kinetics led us to speculate that that retinal neurogenesis might be delayed in Ztcf7l1a-/- eyes to extend the period of proliferative growth prior to retinal precursors undergoing neurogenic divisions.
In the zebrafish eye, neurogenesis can be visualised by tracking expression of atoh7 (ath5) in retinal neurons starting in the ventronasal retina at ~28 hpf and spreading clockwise across the central retina until it reaches the ventrotemporal side (Masai et al., 2000; Hu and Easter, 1999; Figure 5A–E,Q, Supplementary file 1J). Although atoh7 was induced at a similar time in Ztcf7l1a-/- as in wildtype eyes, the subsequent progression of expression was delayed (Figure 5F–J,Q; Supplementary file 1J). Classifying the expression of atoh7 in six categories according to its progression across the neural retina (see legend to Figure 5) revealed that atoh7 expression in mutant retinas was slow to spread and remained restricted to the ventro-nasal or nasal retina for longer (Figure 5Q, Supplementary file 1J). Indeed, between 36 and 40hpf, Ztcf7l1a-/- retinas expressed atoh7 exclusively in the nasal half of the retina (Figure 5H,Q), a phenotype we did not see at any stage in sibling embryo eyes. These data indicate that progression of atoh7 expression and neurogenesis is delayed by about 8–12 hr in Ztcf7l1a-/- retinas compared to siblings, a timeframe comparable to the delays seen in optic vesicle growth. In line with our results in Ztcf7l1a-/- embryos, eye vesicle ablated wildtype retinas also showed delayed neurogenesis compared to control non-ablated contralateral eyes at 36hpf (Figure 5K,L; 6/6 ablated eyes, two independent experiments).
Figure 5

Neurogenesis is delayed in small tcf7l1a-/- eyes and accelerated in large eyes following hsp70:dkk1 overexpression.
(A–P) Lateral views of eyes showing atoh7 fluorescent in situ hybridisation in typical wildtype (A–E, M, O), Ztcf7l1a-/- (F–J), wildtype left-side optic vesicle-ablated (K, L); from n = 5 embryos) and Tg(HS:dkk1)w32 (N, P) embryos at stages indicated. (M–P) Wildtype (M, O) and heterozygous sibling Tg(HS:dkk1)w32 embryos (N, P) heat-shocked at 6hpf (M, N); from n = 7/9 embryos) or 24hpf (O, P); from n = 10/10 embryos) for 45’ at 37°C and grown to 28hpf. Anterior is to the left except in (K) in which anterior is to the right. Arrows indicate ventro-nasal retina; arrowheads indicate dorso-temporal retina; dashed line approximate the nasal-temporal division; dashed circle marks lens position. Abbreviations: n, nasal, t, temporal. Scale bar = 100 µm. (Q) Histogram showing the spatial distribution of atoh7 expression in sibling and Ztcf7l1a-/- retinas at the indicated hours post-fertilisation (data in Supplementary file 1F). VN, ventro nasal expression; VN+, ventro-nasal expression plus a few scattered cells; N+, nasal expression plus scattered cells covering the whole retina; NR, nasal retina expression; WR, whole retina expression; PR, expression localised to the peripheral retina. Numbers in bars represent the number of embryos scored for the particular category of atoh7 expression. (R) Plot showing the growth kinetics of the eye in wildtype (blue line) and Tg(HS:dkk1)w32 (red line) embryos at times indicated (data in Supplementary file 1K).
Our results suggest that retinal precursors in Ztcf7l1a-/- eyes remain proliferative at stages when precursors in wildtype eyes are already producing neurons.
Larger eyes undergo premature neurogenesis
Our results are consistent with the idea that neurogenesis may be triggered when the optic vesicle reaches a critical size. To explore this possibility, we generated embryos with larger optic vesicles by overexpressing the Wnt antagonist Dkk1 (Hashimoto et al., 2000). Heatshocking tg(hsp70:dkk1-GFP)w32 transgenic embryos at 6hpf led to eyes ~ 34% bigger than control heat-shocked embryos by 28hpf (Figure 5M,N,R, n = 12; Supplementary file 1K). After 36hpf, wildtype eyes gradually caught up in size as growth slowed in eyes in dkk1-overexpressed embryos (Figure 5R; Supplementary file 1K).
Neurogenesis was prematurely triggered by 28hpf in the eyes of dkk1 overexpressing embryos, with many more cells expressing atoh7 compared to eyes in heat-shocked control embryos (Figure 5M,N, n = 7 out of 9 embryos). This result is unlikely to be due to a direct effect of dkk1 overexpression on neurogenesis as premature neurogenesis was not triggered in tg(hsp70:dkk1-GFP)w32 retinas heat-shocked at 24hpf (Figure 5O,P, n = 10, 100%). These results further support a link between eye size and the onset of neurogenesis and the size self-regulating ability of the forming eye.
tcf7l1a mutant eyes have more proliferating cells
The delayed production of neurons in tcf7l1a mutant eyes suggests that more retina progenotor cells (RPCs) may continue proliferating and contribute to growth compensation. To address this possibility, we counted mitotic phosphohistone3 (PH3) positive cells in wildtype and tcf7l1a mutant embryos carrying the Tg(atoh7:GAP-RFP)cu2Tg transgene that is expressed from the last mitotic event prior to neuronal birth (Zolessi et al., 2006).
The proportion of PH3-positive (PH3+) cells standardised to the total number of cells in confocal sections at 36hpf was about 20% higher in Ztcf7l1a mutants than wildtypes (Figure 6A,B,C, Supplementary file 1L, wildtype, n = 7; Ztcf7l1a-/-, n = 8, p=0.021, unpaired t-test). The proportion of PH3+ cells was similar between nasal and temporal retina and similar between wildtypes and Ztcf7l1a mutants (Figure 6—figure supplement 1, Supplementary file 1L). Furthermore, the percentage of PH3+ RPCs co-expressing atoh7:GAP-RFP was 3.6 times higher in wildtypes compared to tcf7l1a mutants (Figure 6A,B,D, Supplementary file 1L, wildtype, 35.21 ± 3.5, n = 7, Ztcf7l1a-/-, 9.67 ± 1.58, n = 8, unpaired t-test p<0.0001). Wildtype nasal RPCs were about three times more likely to be atoh7:GAP-RFP positive than nasal RPCs in mutants (Figure 6D, Supplementary file 1L, wildtype, 45.91 ± 3.71, n = 7, Ztcf7l1a-/-, 15.44 ± 2.25, n = 8, unpaired t-test p<0.0001), and PH3+ RPCs in the temporal retina of tcf7l1a mutants almost never showed atoh7:GAP-RFP co-expression (Figure 6D, Supplementary file 1L, wildtype, 20.97 ± 3.67, n = 7, Ztcf7l1a-/-, 0.48 ± 0.48, n = 8, unpaired t-test p<0.0001).
Figure 6 with 1 supplement see all
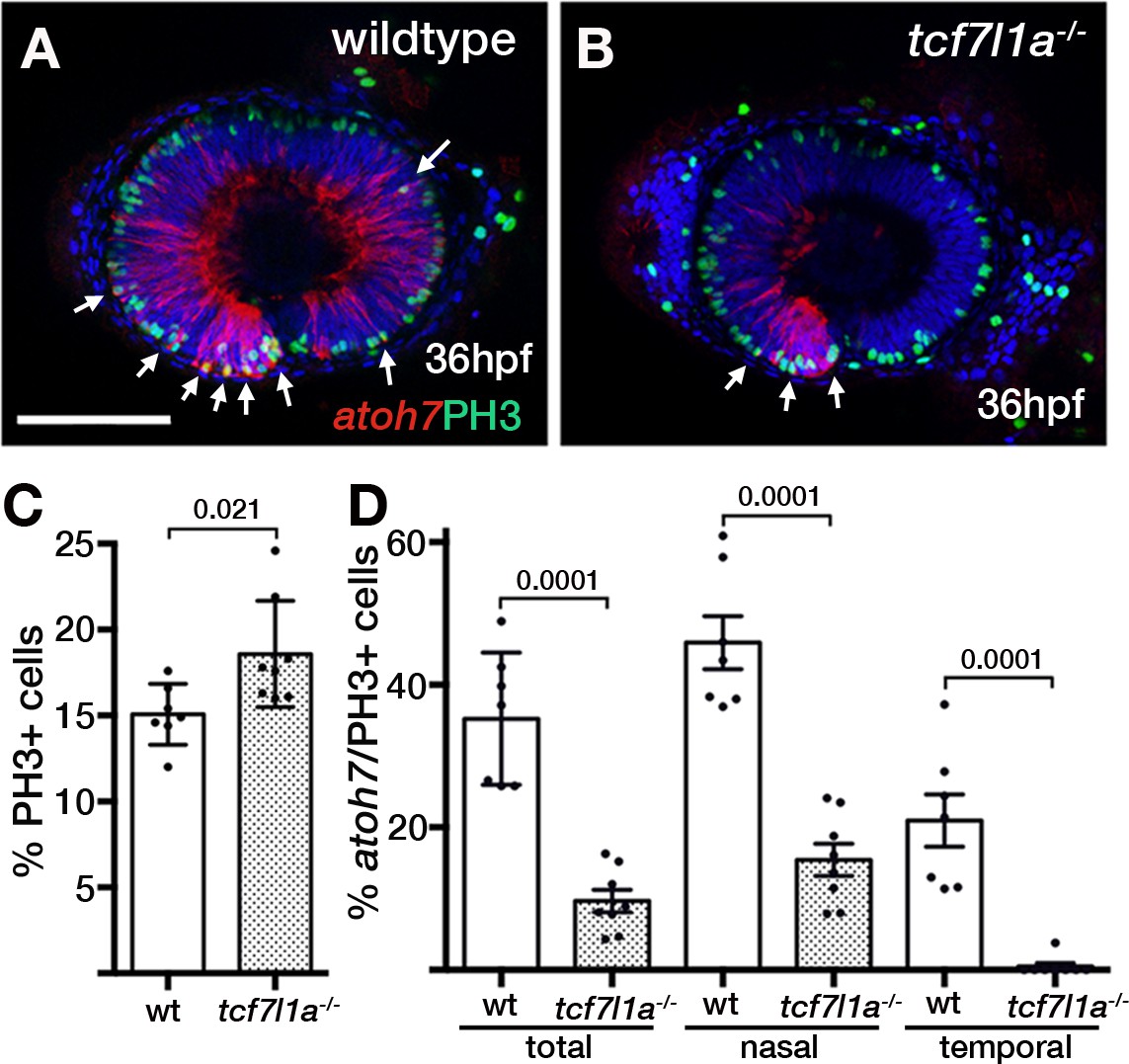
Ztcf7l1a mutants show more retinal progenitor cells undergoing proliferation.
(A–B) Immunostaining detecting phosphohistone3 (PH3, green) and RFP (Tg(atoh7:GAP-RFP)cu2Tg-, red) in wildtype (A) and Ztcf7l1a-/- (B) eyes at 36hpf . Arrows indicate selected double PH3/RFP positive cells. n, nasal; t, temporal. Scale bar = 100 µm. (C–D) Plot showing the percentage of PH3-positive cells (C) data in Supplementary file 1L) and double PH3/RFP-positive cells (D), data in Supplementary file 1L). Single experiment, wildtype n = 7, Ztcf7l1a-/- n = 8, figures over the bars show p-values from unpaired t-tests.
These results suggest that at a stage when many wildtype RPCs are undergoing neurogenic divisions, more RPCs in tcf7l1a mutants are still proliferating, likely contributing to compensatory growth of the mutant eye.
An ENU modifier mutagenesis screen in tcf7l1a mutant background reveals two groups of genetic modifiers
Although eye formation can recover in tcf7l1a-/- mutants despite a much smaller eye field, we speculated that eye development in these embryos might be sensitised to showing the effects of additional mutations. To test this, we performed an ENU mutagenesis screen on fish carrying the tcf7l1a mutation (Figure 7A).
Figure 7 with 1 supplement see all

Ztcf7l1a-/- mutants lacking Hesx1 function fail to form eyes.
(A) Schematic of the genetic strategy to isolate mutations that modify the tcf7l1a-/- mutant phenotype. (B–C) U910 modifier of the tcf7l1a-/- mutant phenotype. Lateral views of homozygous U910 embryos that are heterozygous (B) or homozygous (C) for the tcf7l1a mutation. (D) Representation of SSLP segregation linkage analysis mapping of U910 modifier of tcf7l1a to a 1.46 megabase (Mb) interval on chromosome 11 (Ch11; rec, recombinants). (E) RT-PCR for hesx1 spanning exons 1–2 (top panel), exons 2–3 (middle panel) and β-actin (bottom panel) on wildtype (lanes 1 and 2) and U910-/- (lanes 3 and 4) embryo cDNA from 1 cell stage (lanes 1 and 3) and 10hpf (lanes 2 and 4). Single experiment. (F–I) hesx1 in situ hybridisation on wildtype (F–H) and U910-/- (I) embryos at epiboly (ep) stages indicated. Dorsal views, anterior up. (J, K) Lateral views of hesx1-/- (Δex1/2)/tcf7l1a+/- (J) and hesx1-/- (Δex1/2)/Ztcf7l1a-/- (K) embryos. Four independent experiments, n = 53. Scale bars = 200 µm.
Homozygous Ztcf7l1a mutant adult male fish (F0 founders) were treated with four rounds of ENU (van Eeden et al., 1999) and then crossed with Ztcf7l1a-/- adult females to generate F1 families (Figure 7A). However, possibly because of cellular stress or the synergistic cumulative effect of many mutations induced by ENU, we observed many eyeless F1 embryos. To circumvent this problem, we injected 10 pg/embryo of zebrafish tcf7l1a mRNA to rescue any Tcf-dependent eyeless phenotypes in the F1 embryos (Figure 7A). Adult F1 fish were outcrossed to EKW wildtype fish. All F2 fish were tcf7l1a+/- and half carried unknown mutations (m) in heterozygosity (Figure 7A). To screen, we randomly crossed F2 pairs from each family aiming for at least 6 clutches of over 100 embryos. The probability of finding double Ztcf7l1a-/-/m-/- embryos for independently segregating mutations is 1/16, hence we would expect to find ~6 double mutants in 100 embryos. Here, we describe examples of synthetic lethal mutations that lead to microphthalmia/anophthalmia (U910; Figure 7B-K) or eyes that fail to grow (U762, U768; Figures 8 and 9).
Figure 8 with 3 supplements see all

Loss of tcf7l1a modifies the cct5u762 mutant eye phenotype.
(A–J) Lateral views of wildtype (A, F), Ztcf7l1a-/- mutant (B, G) cct5U762/u762 mutants (C, H), double cct5U762/u762/Ztcf7l1a-/- mutants (D, I) and double cct5U762/u762/Ztcf7l1a-/- mutants injected with 0.8 pmol of cct3 morpholino (E, J) at indicated stages. Scale bar = 100 µm. Full data in Supplementary file 10O, single experiment, 36hpf, wt n = 4, Ztcf7l1a-/- n = 9, cct5-/- n = 8, cct5/Ztcf7l1a-/- n = 3; 52hpf, wt n = 8, Ztcf7l1a-/- n = 8, cct5-/- n = 4, cct5/Ztcf7l1a-/- n = 3; 52hpf + 2 pmol tp53 morpholino, wt n = 6, Ztcf7l1a-/- n = 13, cct5-/- n = 13, cct5/Ztcf7l1a-/- n = 12; 52hpf + 0.8 pmol cct3 morpholino, wt n = 12, Ztcf7l1a-/- n = 12. (K) Eye volume quantification at the indicated timepoints and conditions shown in A–J) (data in Supplementary file 1O). Unpaired t-test. (L–O) Immunostaining detecting phosphohistone3 (PH3, green) in wildtype (L), Ztcf7l1a-/- (M), cct5-/- (N), cct5-/-/Ztcf7l1a-/- (O) eyes at 32hpf. (P) Plot showing the percentage of PH3 positive cells in the eyes shown in L–O) (data in Supplementary file 1Q) Single experiment, wildtype n = 10, Ztcf7l1a-/- n = 10, cct5-/- n = 9, cct5/Ztcf7l1a-/-n = 10, unpaired t-tests.
Figure 9
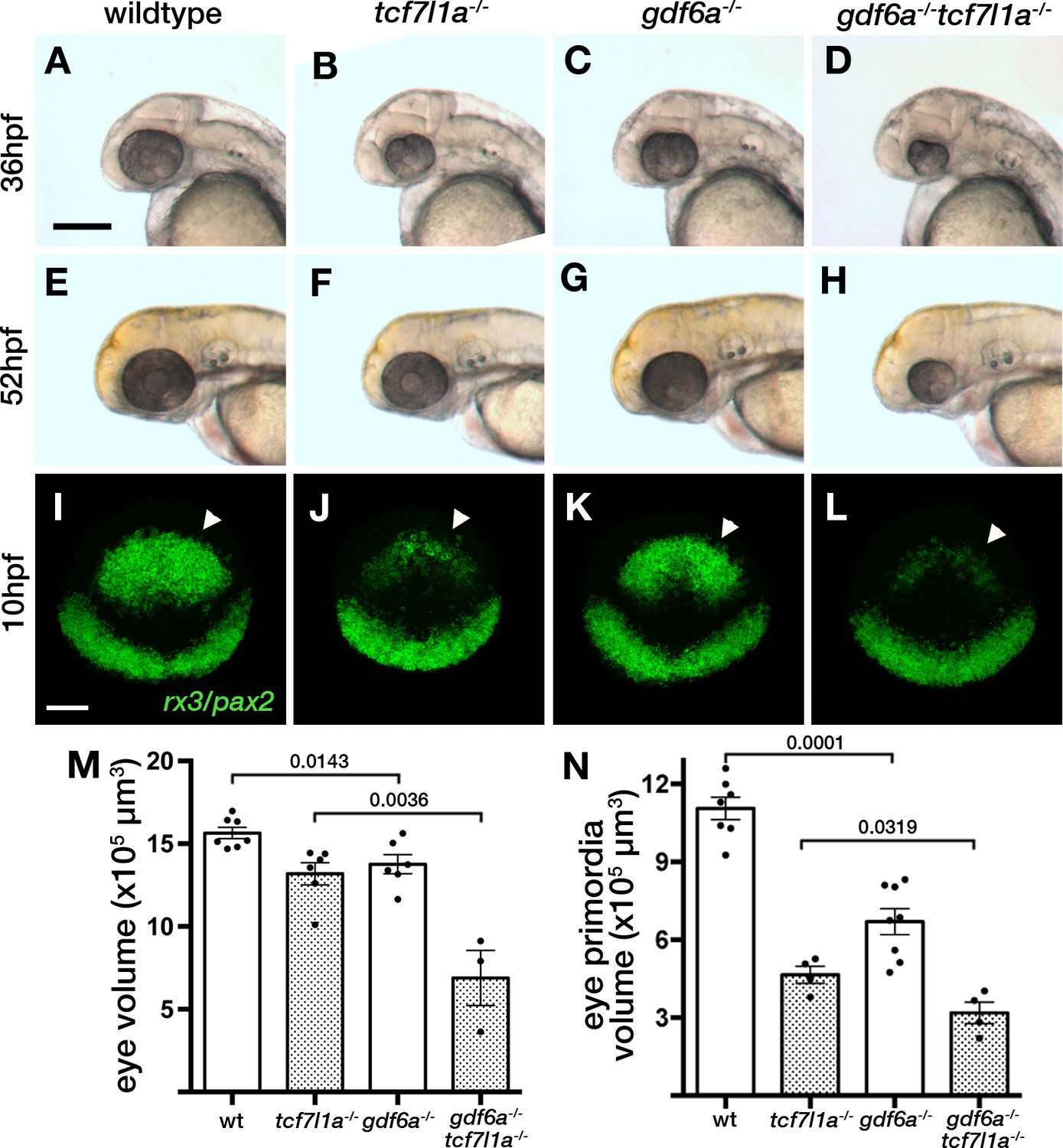
Loss of tcf7l1a modifies the gdf6aU768/U768mutant eye phenotype.
(A–H) Lateral views of eyes in wildtype (A, E), Ztcf7l1a-/- (B, F), gdf6aU768/U768 (C, G) and double gdf6aU768/U768/Ztcf7l1a-/- (D, H) embryos at 36hpf (A–D) and 52hpf (E–H). Dorsal up, anterior to left. Arrows indicate the lens. Scale bar = 200 µm. (I) Whole mount fluorescent in situ hybridisation for rx3 and pax2a in wildtype (I), Ztcf7l1a-/- (J), gdf6aU768/U768(K) and double gdf6aU768/U768/Ztcf7l1a-/- (L) embryos at 10hpf. Dorsal view, anterior up. Arrows point to rx3 eye field expression. Scale bar = 100 µm. (M) Eye volume quantification in wildtype (n = 7), Ztcf7l1a-/- (n = 6), gdf6a-/-(n = 6) and gdf6aU768/U768/Ztcf7l1a-/- double mutant siblings (n = 3) at 36hpf (data in Supplementary file 1R). Single experiment, unpaired t-test. (N) Eye field volume quantification from rx3 fluorescent in situ hybridisation shown in I-L in wildtype (n = 7), Ztcf7l1a-/- (n = 4), gdf6a-/-(n = 8) and gdf6aU768/U768/Ztcf7l1a-/- double mutant siblings (n = 4) at 10hpf (data in Supplementary file 1S). Single experiment, unpaired t-test.
A deletion in the hesx1 locus is a modifier of the tcf7l1a-/- phenotype that leads to loss of eyes
tcf7l1a-/- embryos homozygous for the U910 mutation were eyeless (Figure 7B,C) whereas homozygous U910 mutants with one or no mutant tcf7l1a alleles showed no eye phenotype. U910 was mapped by SSLP segregation analysis (Kelly et al., 2000) to a 1.46 Mb interval between 41.36 Mb (four recombinants/276meioses) and 42.82 Mb (two recombinants/276meioses) on chromosome 11 in GRCz10 assembly (Figure 6D, Supplementary file 1M). Within this interval is hesx1, which morpholino knock-down experiments had previously suggested to genetically interact with tcf7l1a (Andoniadou et al., 2011). Primers for hesx1 cDNA failed to amplify in U910/Ztcf7l1a-/- eyeless embryo cDNA samples. Using a primer set that spans the hesx1 locus, we found that all U910/Ztcf7l1a-/- eyeless embryos had a ~ 2700 bp deletion that covers hesx1 exons 1 and 2 (hesx1Δex1/2; Figure 7—figure supplement 1); this was unexpected as deletions are not normally induced by ENU (see below). Sequencing of the hesx1 locus revealed that there is a polyA stretch of approximately 80 nucleotides followed by a 33 AT microsatellite repeat on the 3’ end of intron two that may have generated a chromosomal instability that led to the deletion of exons 1 and 2 (Figure 7—figure supplement 1). As a consequence of the deletion, hesx1 mRNA was not detected by RT-PCR or in situ hybridisation in U910 homozygous embryos (Figure 6E,H,I). We further confirmed that only U910-F2 embryos that were homozygous for both the tcf7l1a mutation and hesx1U910/U910 were eyeless (Figure 7B,C).
As ENU usually generates point mutations, we speculated that the deletion in hesx1Δex1/2 was not caused by our mutagenesis but was already present in one or more fish used to generate the mutant lines. Indeed, we found the same deletion in wildtype fish not used in the mutagenesis project. To confirm that the eyeless phenotype in hesx1U910/U910/Ztcf7l1a-/- double mutants is not caused by another mutation induced by ENU, we crossed Ztcf7l1a-/- fish to one such wildtype TL fish carrying hesx1Δex1/2. Incrossing of hesx1Δex1/2/Δex1/2/tcf7l1a+/- adult fish led to embryos with a very small rudiment of eye pigment with no detectable lens (Figure 7J,K). Genotyping of eyeless and sibling embryos confirmed that only double homozygosity for hesx1Δex1/2/Ztcf7l1a-/- led to the eyeless embryo phenotype (Four independent experiments, n = 53, Supplementary file 1N).
The interaction between hesx1 and tcf7l1a mutations strikingly illustrates how the developing eye can fully cope with loss of function of either gene alone but fails to form in absence of both gene activities. Additional eyeless families that do not carry the hesx1 deletion were identified but they remain to be validated and mutations cloned.
cct5 and gdf6a mutations compromise the ability of tcf7l1a mutant eyes to undergo compensatory growth
U762 mutant eyes showed no significant size difference compared to wildtypes at 36hpf (Figure 8A,C,K, Supplementary file 1O); neither did Ztcf7l1a-/- compared to double U762/Ztcf7l1a-/- eyes (Figure 8B,D,K, Supplementary file 1O). However, by 52hpf U762 mutants showed slightly reduced eye size and this phenotype was considerably more severe in embryos additionally homozygous for the Ztcf7l1a mutation (Figure 8F–I,K, Supplementary file 1O).
The U762 mutation was mapped by SSLP segregation analysis to a 1.69 Mb interval between 15.50 Mb and 17.19 Mb on chromosome 24 (Figure 8—figure supplement 1A) and through sequencing candidate genes in this interval (Figure 8—figure supplement 1A; Supplementary file 1P), we identified a mutation in the splice donor of cct5 (chaperonin containing TCP-1 epsilon) intron 4 (GT >GC, Figure 8—figure supplement 1B). The mutation leads to the usage of an alternative splice donor in the 3’ most end of cct5 exon 4, which induces a two nucleotide deletion in the mRNA (Figure 8—figure supplement 1C). This deletion changes the reading frame of the protein after amino acid 176, encoding a 29aa nonsense stretch followed by a stop codon (Figure 8—figure supplement 1C,D). The mutation also induces nonsense-mediated decay of the mRNA (not shown). U762 and cct5hi2972bTg mutations failed to complement (not shown) supporting the conclusion that the cct5 mutation in U762 is responsible for the tcf7l1a modifier phenotype. Cct5 is one of the eight subunits of the chaperonin TRiC/TCP-1 protein chaperone complex, which assists the folding of actin, tubulin and many proteins involved in cell cycle regulation (Sternlicht et al., 1993; Dekker et al., 2008; Yam et al., 2008).
To assess if the phenotype in double cct5U762/tcf7l1a mutants is likely due to TRiC/TCP-1 chaperone activity or an independent function of Cct5 we knocked down cct3, another member of the chaperonin complex, in tcf7l1a mutants. Morpholino knockdown of cct3 abrogated eye growth in Ztcf7l1a mutants as in cct5U762/Ztcf7l1a mutants (Figure 8K, last two bars; Supplementary file 1O), suggesting that the genetic interaction is between TRiC/TCP-1 and Tcf7l1a function.
Compared to single cct5U762 or tcf7l1a mutants, double cct5U762/Ztcf7l1a homozygous mutant eyes did not grow beyond 36hpf (Figure 8 compare D, I to B. G, Figure 8K, dotted line, Supplementary file 1O). As described for other cct gene mutants (Matsuda and Mishina, 2004), we observed dying cells in 36hpf and 48hpf cct5U762and cct5U762/Ztcf7l1a mutant eyes and tecta (Figure 8—figure supplement 2C), whereas dying cells were rarely detected in these regions at these times in wildtype or Ztcf7l1a-/- mutant siblings (Figure 8—figure supplement 2). To assess if apoptosis contributes to the lack of compensatory eye growth in cct5U762/Ztcf7l1a mutants, we inhibited cell death by knocking down tp53 (Figure 8E,J,K, Supplementary file 1O). Double cct5U762/Ztcf7l1a mutants with abrogated Tp53 function showed little or no apoptosis in the eye or tectum (Figure 8, compare J to I, arrows; Figure 8—figure supplement 2E,J,O) but still showed eye size reduced similarly to cct5/Ztcf7l1a mutants at 36hpf and 52hpf (Figure 8E,J,K; Supplementary file 1O).
As Cct5 is implicated in the folding of cell cycle related proteins, we assessed the presence of proliferative RPCs and neurons in cct5U762/Ztcf7l1a mutants (Figure 8L–P, Supplementary file 1Q). cct5U762 mutants showed no significant difference in PH3 +cells compared to wildtype siblings (Figure 8L,N,P, Supplementary file 1Q, n = 9), whereas double cct5U762/Ztcf7l1a-/- mutants showed a 48% increase compared to single Ztcf7l1a mutants (Figure 8M,O,P, Supplementary file 1Q, n = 10, p<0.0051, unpaired t-test, two experiments). By 48hpf, wildtype eyes show strong atoh7:GFP expression in the retinal ganglion cell layer (Figure 8—figure supplement 3A, n = 4, arrow head; Masai et al., 2003), whereas although cct5U762/U762 eye cells do express GFP, lamination of neurons is abnormal (Figure 8—figure supplement 3B, n = 5). In cct5U762/U762/Ztcf7l1a-/- eyes, GFP-expressing neurons are almost completely restricted to the ventro-nasal retina (Figure 8—figure supplement 3C, n = 6).
Homozygous U768 mutants show slightly smaller (Figure 9A,C,M, Supplementary file 1R, n = 6, p=0.0143, unpaired t-test) and misshapen eyes; this mutation was mapped to gdf6a (Valdivia et al., 2016). Eyes in gdf6aU768/U768/Ztcf7l1a-/- embryos at 36hpf were reduced to 52% the size of eyes in Ztcf7l1a-/- mutants (Figure 9B,D,M, Supplementary file 1R, n = 3, p=0.0036, unpaired t-test). Unlike Ztcf7l1a-/- mutants in which eye size recovered, eyes in gdf6aU768/U768/Ztcf7l1a-/- embryos remained smaller than in single mutants or wildtypes at 52hpf (Figure 9E–H). This suggests that the ability to compensate eye size is compromised in absence of both gdf6a and tcf7l1a function.
The smaller eye in gdf6aU768/U768/Ztcf7l1a-/- mutants at 36hpf suggested that early eye development and maybe eye field specification in these mutants is compromised. Volumetric analysis of rx3 expression by fluorescent in situ hybridisation showed that gdf6a-/- eye fields were reduced to 63% of wildtype size at 10hpf (Figure 9I,K,N, arrowheads; Supplementary file 1S, n = 8, p=0.0001, unpaired t-test). Moreover, gdf6aU768/U768/Ztcf7l1a-/- eye fields were about ~68% of the size of Ztcf7l1a-/- mutants (Figure 9I–L,N, arrowheads; Supplementary file 1S, n = 4, p=0.0143, unpaired t-test).
Altogether, analysis of the interacting mutations reveals that although abrogation of Tcf7l1a function alone has little effect on formation of eyes, it can lead to complete loss of eye formation or more severe eye phenotypes in combination with additional mutations. Consequently, although eye development is sufficiently robust to cope with loss of Tcf7l1a, mutant embryos are sensitised to the effects of additional mutations.
Discussion
In this study, we show that although Tcf7l1a is required for cells to adopt eye field identity and express rx3, tcf7l1a mutants form normal, functional eyes. This finding reveals a remarkable ability of the eye to develop normally from an eye field that is half the size of that in the wildtype condition. Tcf function in tcf7l1a mutants is not genetically compensated by upregulation of other tcf genes nor by other genetic mechanisms that restore neural plate regionalisation and eye field formation. Instead, we find that tcf7l1a mutant optic vesicles maintain more proliferative RPCs and delay neurogenesis enabling size recovery. We observe a similar effect when optic vesicle cells are physically ablated. In contrast, neurogenesis is prematurely induced in larger optic vesicles, likely depleting progenitors and slowing growth. Our results suggest that size-dependent regulation of the balance between proliferation and differentiation may buffer the developing eye against initial differences in cell number. Although the developing eye can cope with loss of Tcf7l1a function, we speculated that embryos lacking Tcf7l1a would not be robust to the consequences of additional mutations affecting eye formation. In support of this, we identify mutations in three other genes that give synthetically enhanced eye phenotypes when combined with the tcf7l1a mutation. This approach facilitates identification of genes that participate in genetic networks that make developing eyes robust to mutations that compromise eye field specification and optic vesicle growth.
The tcf7l1a mutation is fully penetrant with no apparent genetic compensation during neural plate patterning
Tcf7l1 is a core Wnt/β-catenin pathway transcription factor that can activate or repress genes dependent upon the status of the Wnt signalling cascade (Cadigan and Waterman, 2012). Homozygous tcf7l1 mutant mice present severe mesodermal and ectodermal patterning defects (Merrill et al., 2004), but the duplication of tcf7l1 into tcf7l1a and tcf7l1b in zebrafish has led to functional redundancy (Dorsky et al., 2003).
Although Ztcf7l1a embryos have a severe eye field specification phenotype, they still develop normal eyes. We confirmed that the tcf7l1am881 mutant allele is null, generates no wildtype transcript and that morpholino knock-down specifically of Tcf7l1a does not give an eyeless phenotype. Hence, the originally described MZtcf7l1a eyeless phenotype (Kim et al., 2000) may have been due to genetic background effects modifying the outcome of the tcf7l1am881 allele. The fact that we were able to recover an eyeless modifier of the tcf7l1a phenotype in our own mutagenesis pilot screen lends support to this idea.
At the stage of eye specification, we did not find genetic compensation in tcf7l1a mutants by other tcf genes. Even though tcf7l1a mutants develop eyes, they do so from an eye field that is ~50% smaller than wild-type. Although we did not find evidence for genetic compensation, and despite tcf7l1 being duplicated in fish, the fact that neither gene has been lost due to genetic drift suggests that having both genes may confer enhanced fitness and robustness to zebrafish. As an example, paralogous Lefty proteins make Nodal signalling more stable to noise and perturbations during early embryogenesis (Rogers et al., 2017).
Tcf7l1a is cell-autonomously required for the expression of rx3 and consequently is a bona fide eye field gene regulatory network transcription factor that functions upstream to rx3. tcf7l1a is expressed very early in the anterior neural plate and so may work alongside otx, sox, six and pax genes to regionalise the eye-forming region of the neural plate (Beccari et al., 2013; Zuber et al., 2003). Considering that it is the repressor activity of Tcf7l1a that promotes eye formation (Kim et al., 2000), the most likely role for Tcf7l1a is to repress transcription of a gene that suppresses eye field formation.
Compensatory tissue growth confers robustness to eye development
We show that despite the small eye field in tcf7l1a mutants, the optic vesicles evaginate and undergo overtly normal morphogenesis. Although tcf7l1a mutant eye vesicles are still much smaller than wild-type at 24hpf, we found that their eye growth kinetics and cell volumes are similar. This suggests that the mechanisms that regulate overall growth of the retina in both conditions are comparable albeit delayed in the tcf7l1a mutant retina.
Although atoh7 expression is initiated in the ventronasal retina in tcf7l1a mutants at the same stage as in wild-type eyes, the wave of atoh7 expression that spreads across the retina is delayed by approximately 8–12 hr in mutants. atoh7 is required for the first wave of neurogenesis in the retina (Brown et al., 2001; Kay et al., 2001; Wang et al., 2001a) and thus, the delay we see in tcf7l1a mutants suggests that RPCs continue proliferating in mutants at stages when they are already generating neurons in wild-type eyes. Indeed, we show that the tcf7l1a mutant eye has more mitotic RPCs, fewer of which are undergoing neurogenic divisions. This suggests that the extended period of proliferative growth due to delayed neurogenesis enables the forming eye to continue growing and recover its size. We observed a similar phenomenon of delayed neurogenesis and prolonged growth when cells were removed from one optic vesicle. Conversely, atoh7 spreads precociously in experimentally enlarged optic vesicles. The premature neurogenesis of RPCs in these conditions may contribute to eyes achieving a final size similar to wild-type. Altogether, our data suggest that the timing of the spread of neurogenesis across the retina may be coupled to size of the eye, thereby providing a mechanism to buffer eye size. It is intriguing that the compensatory changes in growth seen in tcf7l1a mutant and optic-vesicle ablated eyes seem to occur prior to the establishment of the ciliary marginal zone, which accounts for the vast majority of eye growth (Fischer et al., 2013).
Our results support classical embryology experiments from Ross Harrison, Victor Twitty and others (Harrison, 1929; Twitty and Schwind, 1931; Twitty and Elliott, 1934). These investigators showed that when eye primordia from small-eyed salamander species (A. punctatum) were transplanted to larger-eyed salamanders (A. tigrinum) or vice-versa, the eye derived from the grafted tissue formed an eye of a size corresponding to the donor salamander species. Species-specific size differences are also observed in self-organising in vitro cultured eye organoids derived from mouse or human embryonic stem cells (Nakano et al., 2012). Our work, together with the experiments in salamanders and organoids, suggests that the developing eye has intrinsic size-determining mechanisms.
Size regulatory mechanisms have been previously described in other species and perhaps most extensively studied in the fly wing imaginal disc (Potter and Xu, 2001). Indeed, many models have been put forward to explain imaginal disk size control (Eder et al., 2017; Irvine and Shraiman, 2017; Vollmer et al., 2017). It is evident that the final size of paired structures within individuals is remarkably similar supporting the idea that the mechanisms that control the size of such organs/tissues are highly robust.
Addressing the robustness of eye formation through a forward mutagenesis screen in fish carrying the tcf7l1a mutation
Our results indicate that tcf7l1a mutant eyes are sensitised to the effects of additional mutations. Indeed, a homozygous deletion of the two first exons of hesx1 leads to eyeless embryos when in combination with tcf7l1a. This result also confirms our previous observations suggesting a genetic interaction between hesx1 and tcf7l1a based upon morpholino knock-down experiments (Andoniadou et al., 2007). Furthermore, both hesx1 and tcf7l1a are expressed in the anterior neural plate including the eye field, and as observed in tcf7l1a zebrafish mutants, hesx1 mutant mice also show a posteriorised forebrain (Andoniadou et al., 2007; Martinez-Barbera et al., 2000). These and our results suggest that Tcf7l1a and Hesx1 have similar, overlapping functions in the anterior neural plate such that the eyeless phenotype is expressed in zebrafish only when both genes are abrogated. Mutations in hesx1 lead to anophthalmia, microphthalmia, septo-optic dysplasia (SOD) and pituitary defects in humans and mice (Dattani et al., 1998; Gaston-Massuet et al., 2008; Martinez-Barbera et al., 2000; Thomas et al., 2001). Interaction of hesx1 mutations with other genetic lesions may also occur in patients carrying Hesx1 mutations, as the phenotypes in these individuals show variable expressivity (McCabe et al., 2011). In these patients, tcf7l1a should be considered as a candidate modifier for hesx1-related genetic conditions.
Gdf6a is a TGFβ pathway member (David and Massagué, 2018) that when mutated in zebrafish results in small mis-patterned eyes, neurogenesis defects and retino-tectal axonal projection errors (Gosse and Baier, 2009; French et al., 2009). In humans, mutations in GDF6 have been identified in anophthalmic, microphthalmic and colobomatous patients (Asai-Coakwell et al., 2009) as well as in some cases of Leber congenital Amaerurosis (Asai-Coakwell et al., 2013). Double gdf6aU768/tcf7l1a mutant eye fields are smaller than both single mutants and their eyes fail to recover their size at later stages. This suggests that gdf6a/tcf7l1a double mutant eye fields have more severe specification defects compared to either individual mutant, and that double mutant optic vesicles lack the compensatory growth seen in tcf7l1a mutants. Given the phenotypes we describe, it is perhaps surprising that gdf6a appears not to be expressed in the eye field (Rissi et al., 1995), although it is expressed in neighbouring tissues and also prior to eye specification (Sidi et al., 2003). Consequently, we presume that Gdf6 acting prior to eye field formation or arising from outside of the eye field impacts eye field specification. It is also intriguing that gdf6a mutants show premature expression of atoh7 and neurogenesis (Valdivia et al., 2016). If this phenotype is epistatic to the compensatory growth mechanisms, then this may contribute to the lack of growth in double mutant eyes.
Mutations in cct5 in combination with tcf7l1a also led to phenotypes in which eye size failed to recover. cct5 codes for the epsilon subunit of the TCP-1 Ring Complex (TRiC) chaperonin that is composed of eight different subunits that form a ring, the final complex organised as a stacked ring in a barrel conformation (Yébenes et al., 2011). In vitro studies indicate TRiC chaperonin mediates actin and tubulin folding (Sternlicht et al., 1993); however, it also assists in the folding of cell cycle-related and other proteins (Dekker et al., 2008; Yam et al., 2008). A mutation in cct2 has been found in a family with Leber congenital ameurosis retinal phenotype (Minegishi et al., 2016; Minegishi et al., 2018) and mutations in cct4 and cct5 have been related to sensory neuropathy (Pereira et al., 2017; Lee et al., 2003; Hsu et al., 2004; Bouhouche et al., 2006). Similar to our cct5 mutant, cct1, cct2, cct3, cct4 and cct8 mutant zebrafish show retinal degeneration (Berger et al., 2018; Matsuda and Mishina, 2004; Minegishi et al., 2018), suggesting that the cct5/tcf7l1a double mutant phenotype is due to abrogation of TRiC chaperonin function, a conclusion supported by cct3 knockdown in tcf7l1a mutants. Double cct5/tcf7l1a homozygous mutant eyes degenerate prematurely and to a greater extent than cct5 single mutants, and neurogenesis is also severely compromised. However, the lack of compensatory growth is not solely due to cell death as blocking apoptosis in cct5/tcf7l1a mutants fails to restore eye size. Our results show that the consequence of cct5 loss of function is exacerbated by the lack of tcf7l1a function, although it is currently unclear how such an interaction might occur. However, this genetic interaction does highlight that in some conditions a gene of pleiotropic function, like cct5, can lead to a specific phenotype in the eye.
Surprisingly many eyeless embryos were observed in the F1 clutches of embryos used to establish homozygous tcf7l1a fish carrying new mutations. These phenotypes were suppressed by providing exogenous wildtype tcf7l1a. It is not unusual to see mutant phenotypes in F1 embryos in ENU screens and we suspect that heavy mutational load may impact developmental processes such that phenotypic penetrance is enhanced due to cellular or tissue level stress. Additionally, we now think it likely that the hesx1 mutant allele was in the background of some parent fish and this may also have contributed to enhancing the homozygous tcf7l1a phenotype in combination with the many newly induced ENU mutations.
Anophthalmia and microphthalmia are generally associated with eye field specification defects (Reis and Semina, 2015), but given that normal eyes can still develop from a much reduced eye field, further analysis of the genetic and developmental mechanisms that lead to small or absent eyes is warranted. Our isolation and identification of modifiers of tcf7l1a highlights the utility of genetic modifier screens to identify candidate genes underlying congenital abnormalities of eye formation. Indeed, given that Tcf7l1a itself can now be classified as a bona fide gene in the eye transcription factor regulatory network, it should be considered when screening patients with inherited morphological defects in eye formation.
Materials and methods
Animal use, mutant and transgene alleles, genotyping and heat shock
Request a detailed protocolAdult zebrafish were kept under standard husbandry conditions and embryos were obtained by natural spawning. Wildtype and mutant embryos were raised at 28.5°C and staged according to Kimmel et al. (1995). To minimise variations in staging, embryos were collected every 30 min and kept separate clutches according to their time of fertilisation. Fish lines used were tcf7l1a/headless(hdl)m881 (Kim et al., 2000), cct5hi2972bTg (Amsterdam et al., 2004), cct5U762, gdf6aU768 (Valdivia et al., 2016), hesx1U910, tcf7l1bzf157Tg (Gribble et al., 2009), Tg(atoh7:GFP)rw021Tg (Masai et al., 2000), Tg(atoh7:GAP-RFP)cu2Tg (Zolessi et al., 2006), Tg(hsp70:dkk1-GFP)w32 (Stoick-Cooper et al., 2007) and Tg(rx3:GFP)zf460Tg (Brown et al., 2010). All the alleles except for cct5hi2972bTg were genotyped by KASP assays (K Biosciences, assay barcodes: 1077647141 (cct5U762), 1077647146 (gdf6aU768), 172195883 (this assay discriminates a SSLP 500 bp from the 3’ end of the deletion in hesx1U910), 1145062619 (tcf7l1am881)) using 1 µl of genomic DNA for 8 µl of reaction volume PCR as described by K Biosciences.
For heatshock (HS) gene induction, embryos from a heterozygous Tg(hsp70:dkk1-GFP)w32 to wild type cross were moved from embryo media at 28.5°C to 37°C at 6hpf or 24hpf for 45 min, and then back to 28.5°C embryo media. Three hours post HS, embryos were separated in controls (GFP-) and HS experimental (GFP+) groups, and fixed at the stages described in results.
ENU mutagenesis and mutant mapping
Request a detailed protocolHomozygous male tcf7l1am881 fish were exposed to four rounds of ENU according to van Eeden et al. (1999). Details of the mutagenesis pipeline are in the results section. Embryos from incrosses of carriers of the cct5U762 or gdf6aU768 mutations, which show a phenotype as homozygous embryos independently of mutations in tcf7l1a, were identified for the described eye phenotype at 3dpf to avoid ambiguity and false positives. For rough mapping, batches of 30 mutants and 30 siblings were fixed in methanol and genomic DNA was extracted by proteinase K protocol. This gDNA was then used for bulk segregant analysis PCR to test a library of 245 polymorphic SSLP variants spanning the whole zebrafish genome (Stickney et al., 2002). SSLP markers heterozygous in the sibling samples and homozygous in the mutant sample were confirmed on gDNA samples of 12 mutant and 12 sibling individuals. Markers that showed linkage to a locus were tested on additional mutant samples, and more SSLP markers were tested for the mapped region until a genomic interval was defined.
Homozygous tcf7l1a/hesx1U910 mutant carriers were incrossed, and eyeless embryos and siblings were fixed in methanol. Rough mapping was carried out as above but in this case sibling embryos used for bulk segregant analysis were genotyped for tcf7l1am881 and only homozygous mutants with eyes were included in the sibling pool.
mRNA synthesis, embryo microinjection and morpholinos
Request a detailed protocolmRNA for overexpression was synthesised using RNA mMessage mMachine transcription kits (Ambion). One- to two-cell stage embryos were co-injected with 10 nl of 5 pg of GFP mRNA and morpholinos or in vitro synthesised mRNA at the indicated concentrations. Only embryos with an even distribution of GFP fluorescence were used for experiments.
For cell volume analysis, one-cell stage embryos from a tcf7l1a ± incross were injected with 5 pg pCS2-GFP DNA and 10 pg lyn-cherry mRNA. The following day, embryos were sorted for GFP mosaicism in the eye and mounted in PTU/Tricaine-containing 1% low-melt agarose and were imaged at 24 and 36hpf in a Leica SP8 confocal microscope.
Morpholino sequences: mo2 tcf7l1a (5’ AGG CAT GTT GGC ACT TTA AAT G 3’), motcf7l1b (5’-CAT GTT TAA CGT TAC GGG CTT GTC T-3’; Dorsky et al., 2003) and moC (TGT TGA AAT CAG CGT GTT CAA G). tcf7l1am881/m881 embryos injected with motcf7l1b phenocopy the loss of eye phenotype seen in tcf7l1am881/m881/tcf7l1b+/zf157tg double mutants (Young and Wilson, unpublished).
RNA extraction, reverse transcription and qPCR
Request a detailed protocolTotal RNA and genomic DNA were isolated from individual embryos at 10hpf following Life Technologies Trizol protocol. cDNA was synthesised by reverse transcription using SuperscriptII (Life Technologies) with 200 ng of total RNA to a final volume of 40 µl and oligo dT for priming. The cDNA reaction was diluted 10 times and 5 µl were used in 25 µl final volume reactions using GoTaq qPCR Master mix (Biorad). Each experimental condition was processed in technical and biological triplicates. All primers used had PCR efficiencies within 90–100% range: gapdh (F-ACC CGT GCT GCT TTC TTG, R-CTG CCT TAA CCT CAC CCT TG); hprt1 (F-AAC AGT GAT CGC TCC ATT CC, R-GGA CAG ATC ATC TCC ACC AAT C); lef1(F-GCT TCA GGT ACA GGC CAG AG, R-AAA GAC GTC CGC TTT CCT CC); otx1a (F-GGT GTT TCT TGG CTT TGT GG, R-GGG CTT GCT TGA GGT ATG A); otx2 (F-TAC ACG GTC AAC GGG CTA A, R-CTC GTC TCT GGT TTC GAG GA); rx3 (F-TCC GAG TAC AGG TGT GGT TCC, R-CTC CTG TCG CCG CCA TTT A); six3b (F-TGC CAA AAA CAG GCT TCA GCA, R-CTG ACA TGG AGC GCA GAC T); tcf7l1a (F- AGC ACA CGA ACG TAT CTC CA, R-GAG TCT TTA AGA GCC GCC GA); tcf7 (F-TGC TGC CGT ATG AAC ACT TC, R-TCT CCT GCG TCT GAT GTC TG); tcf7l1b (F-GGC TAA AGT AGT GGC CGA GTG, R-CTG GCC AGC TCG TAG TAT TTG); tcfl2 (F-GCC TCC GCC TAG ATC TGA AA, R-CTT GCC TTT TTG CAG CCT CC).
Wildtype and tcf7l1am881 mutant cDNA fragments spanning the tcf7l1a exon 7/8 border for DNA sequencing were amplified with primers P2 and P3 (Kim et al., 2000).
RNA sequencing
Request a detailed protocolTotal RNA was extracted from zebrafish embryos at 80% epiboly by Trizol extraction and gDNA was genotyped for tcf7l1a to identify wildtype, heterozygous and homozygous embryos. RNA from six wild-type and six tcf7l1a-/-mutant embryos was DNase treated for 20 min at 37°C followed by addition of 1 ml 0.5M EDTA and inactivation at 75°C for 10 min to remove residual DNA. RNA was then cleaned using 2 volumes of Agencourt RNAClean XP (Beckman Coulter) beads under the standard protocol. Stranded RNA-seq libraries were constructed using the Illumina TruSeq Stranded RNA protocol with oligo dT pulldown. Libraries were pooled and sequenced on two lanes of Illumina HiSeq 2000 in 75 bp paired-end mode. Sequence data were deposited in ENA under accession PRJEB9957. FASTQ files were aligned to the GRCz11 reference genome using TopHat (v2.0.13, options: --library-type fr-firststrand, Kim et al., 2013). The data were assessed for technical quality (GC-content, insert size, proper pairs etc.) using QoRTs (Hartley and Mullikin, 2015). Counts for genes were produced using htseq-count (v0.6.0 options: --stranded=reverse, Anders et al., 2015) with the Ensembl v93 annotation as a reference. Sequence data were deposited in ENA under accession PRJEB9957. Differential gene expression was analysed using DESeq2 (Love et al., 2014).
In situ hybridisation, probe synthesis and tunel labelling
Request a detailed protocolWhole mount in situ hybridisation was performed using digoxigenin (DIG) and fluorescein (FLU)-labelled RNA probes according to standard protocols (Thisse and Thisse, 2008). Probes were synthesized using T7 or T3 RNA polymerases (Promega) according to manufacturers’ instructions and supplied with DIG or FLU labelled UTP (Roche). Probes were detected with anti-DIG-AP (1:5000, Roche), anti-FLU-AP (1:10000, Roche), or anti-DIG-POD (1:1000, Roche) antibodies and developed with NBT/BCIP mix (Roche), for regular microscopy or Fast Red (Sigma) or CY-3 tyramide (Lauter et al., 2011) substrate for confocal analysis.
For Tunel assays, embryos were processed as for in situ hybridisation up to the washing of PK stage. After this, embryos were incubated in an acetone:ethanol (2:1)–20°C prechilled solution at −20°C for 10 min. After PBS tween 0.5% washes, embryos were incubated for 1 hr in the equilibration buffer (Millipore ApopTag kit) at room temperature. The buffer was removed and 20 µl of fresh equilibration buffer was added plus 15 µl of TdT mix (12 µl reaction buffer, 6 µl TdT enzyme, 0.5 µl of 10% triton X100), and embryos incubated at 37°C over night. Samples were washed for 1 hr at 37°C and 1 hr at room temperature, and the protocol was continued as for in situ hybridisation.
Quantification of eye profile, eye volume, cell volume, PH3 +cells, and posterior lateral line primordium (pLLP) position
Request a detailed protocolThe eye profile and eye volume were calculated from confocal imaging of vsx2 in situ hybridisation stained embryos at 24hpf. The eye volume/eye profile ratio average from 10 embryos was 53.24. This ratio was used to estimate eye volume from eye profile area as the profile area to eye volume ratio is approximately constant after 24hpf (Matejčić et al., 2018). The sizes of eye profiles were quantified from lateral view images of PFA-fixed embryos by delineating the eye using Adobe Photoshop CS5 magic wand tool and measuring the area of pixels included in the delineated region. The surface area was then transformed from px2 to µm2.
For estimation of cell volume, 3d stacks were first contrast enhanced to increase the intensity of the labelled cells in the entire volume. Subsequently, a 3d median filter was applied to filter out high intensity noise. Next, a fixed threshold was applied to segment individual cells in the volume and their surface area and volume were calculated. For each imaged and analysed image stack, we also manually inspected the processed data to ensure that post processing did not result in partial segmentation of cell volumes. Cells that were undergoing division or were within ~20 µm from the dorsal or ventral surface of the imaged volume were excluded from the analysis. Image processing and analysis was carried out using ImageJ.
For PH3 quantification, embryos were oriented to yield a lateral view of the retina, and the widest plane of the retina was imaged. The z-series was defined as 2 μm above and 2 μm below the widest plane of the retina. Stacks were taken at a step size of 1 μm for a total of 5 stacks per imaged volume. The number of nuclei undergoing mitosis, marked by α-PH3, were counted. The total number of nuclei for each eye was estimated by counting the DAPI labeled nuclei in a standardized area of 40 × 40 pixels. This result was multiplied by the total area of the retina (in pixels), obtained using the freehand tool in ImageJ and the software’s measuring capabilities, and then divided by 1600. To normalize the data, the number of α-PH3 positive cells was presented as a percentage of the total number of nuclei per retina.
pLLP migration was measured by analysing the position of the posterior end of the primordium relative to the somite boundary labelled by in situ hybridisation with eya1 and xirp2a respectively.
Confocal microscopy and image analysis
Request a detailed protocolConfocal imaging was performed on a Leica TCS SP8 confocal microscope. For time lapse analyses, the stage was set in an air chamber heated to 28.5°C. Live embryos were immobilized in 1% low melting point agarose (Sigma) and 0.016% Tricaine (Sigma) to anesthetize. Image volume analysis measurement was performed on Imaris 7.7.0 and Fuji.
Cell transplantation
Request a detailed protocolWIldtype or MZtcf7l1a-/- embryos used as donors were injected with 50 pg of GFP mRNA at 1 cell stage. At 3-4hpf, blastula stage, dechorionated donor and host embryos were mounted in 3% methylcellulose in fish water supplemented with 1% v/v penicillin/streptomycin (5000 units penicillin and 5 mg streptomycin per ml) and viewed with a fixed-stage compound microscope (Nikon Optiphot). Approximately 30–40 cells were taken from the animal pole of donors and transplanted to approximately the same position in hosts by suction using an oil-filled manual injector (Sutter Instrument Company). Embryos were moved to 1% penicillin/streptomycin supplemented fish media and fixed at 10hpf.
Eye vesicle cell removal
Request a detailed protocolEmbryos were mounted in 1% low melting point agarose in Ringer’s solution supplemented with 1% v/v penicillin/streptomycin. A slice of set agarose was removed to expose one of the eyes and a drop of mineral oil (sigma) was placed over the target eye to dissolve the epidermis (Picker et al., 2009). After 2 min, the oil drop was removed and optic vesicle cells were sucked out with a capillary needle filled with mineral oil. Embryos were left to recover for half an hour before being released from the agarose.
Optokinetic response
Request a detailed protocolOptokinetic responses were examined using a custom-built rig to track horizontal eye movements (optokinetic nystagmus) in response to whole-field motion stimuli. Larvae at 4dpf were mounted in 1% low melting point agarose in fish water and analysed at 5dpf. The agarose surrounding the eyes was removed to allow normal eye movements. Sinusoidal gratings with spatial frequencies of 0.05, 0.1, 0.13 and 0.16 cycles/degree were presented on a cylindrical diffusive screen 25 mm from the centre of the fish’s head with a MicroVision SHOWWX + projector. Gratings had a constant velocity of 10 degrees/s and changed direction and/or spatial frequency every 20 s. Eye movements were tracked under infrared illumination (720 nm) at 60 Hz using a Flea3 USB machine vision camera controlled with custom-written LABVIEW software. MATLAB scripts were used to extract slow phase eye velocity from recorded eye position data (degrees per second).
Data availability
All the data used for this study was provided in the uploaded manuscript.
-
European Nucleotide ArchiveID PRJEB9957. Transcriptome_profiling_of_zebrafish_tcf3a_and_tcf3b_knockout_embryos.
References
-
Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypesHuman Molecular Genetics 18:1110–1121.https://doi.org/10.1093/hmg/ddp008
-
Contribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophiesHuman Molecular Genetics 22:1432–1442.https://doi.org/10.1093/hmg/dds560
-
The logic of gene regulatory networks in early vertebrate forebrain patterningMechanisms of Development 130:95–111.https://doi.org/10.1016/j.mod.2012.10.004
-
Math5 is required for retinal ganglion cell and optic nerve formationDevelopment 128:2497–2508.
-
Nlcam modulates midline convergence during anterior neural plate morphogenesisDevelopmental Biology 339:14–25.https://doi.org/10.1016/j.ydbio.2009.12.003
-
TCF/LEFs and wnt signaling in the nucleusCold Spring Harbor Perspectives in Biology 4:a007906.https://doi.org/10.1101/cshperspect.a007906
-
Integration of anterior neural plate patterning and morphogenesis by the wnt signaling pathwayDevelopmental Neurobiology 74:759–771.https://doi.org/10.1002/dneu.22135
-
Dynamic tissue rearrangements during vertebrate eye morphogenesis: insights from fish modelsJournal of Developmental Biology 6:4.https://doi.org/10.3390/jdb6010004
-
Contextual determinants of tgfβ action in development, immunity and cancerNature Reviews Molecular Cell Biology 19:419–435.https://doi.org/10.1038/s41580-018-0007-0
-
The interaction network of the chaperonin CCTThe EMBO Journal 27:1827–1839.https://doi.org/10.1038/emboj.2008.108
-
Two tcf3 genes cooperate to pattern the zebrafish brainDevelopment 130:1937–1947.https://doi.org/10.1242/dev.00402
-
Forces controlling organ growth and sizeMechanisms of Development 144:53–61.https://doi.org/10.1016/j.mod.2016.11.005
-
Pervasive robustness in biological systemsNature Reviews Genetics 16:483–496.https://doi.org/10.1038/nrg3949
-
The ciliary marginal zone (CMZ) in development and regeneration of the vertebrate eyeExperimental Eye Research 116:199–204.https://doi.org/10.1016/j.exer.2013.08.018
-
Correlation in the development and growth of the eye studied by means of heteroplastic transplantationWilhelm Roux' Archiv Für Entwicklungsmechanik Der Organismen 120:1–55.https://doi.org/10.1007/BF02109662
-
Zebrafish Dkk1 functions in forebrain specification and axial mesendoderm formationDevelopmental Biology 217:138–152.https://doi.org/10.1006/dbio.1999.9537
-
Retinal neurogenesis: the formation of the initial central patch of postmitotic cellsDevelopmental Biology 207:309–321.https://doi.org/10.1006/dbio.1998.9031
-
Mechanical control of growth: ideas, facts and challengesDevelopment 144:4238–4248.https://doi.org/10.1242/dev.151902
-
Genetic linkage mapping of zebrafish genes and ESTsGenome Research 10:558–567.https://doi.org/10.1101/gr.10.4.558
-
Stages of embryonic development of the zebrafishDevelopmental Dynamics 203:253–310.https://doi.org/10.1002/aja.1002030302
-
Septo-optic dysplasia and other midline defects: the role of transcription factors: hesx1 and beyondBest Practice & Research Clinical Endocrinology & Metabolism 25:115–124.https://doi.org/10.1016/j.beem.2010.06.008
-
Mutation in the zebrafish cct2 gene leads to abnormalities of cell cycle and cell death in the retina: a model of CCT2-Related leber congenital amaurosisInvestigative Opthalmology & Visual Science 59:995.https://doi.org/10.1167/iovs.17-22919
-
Transcription of a zebrafish gene of the hairy-Enhancer of split family delineates the midbrain anlage in the neural plateDevelopment Genes and Evolution 206:153–160.https://doi.org/10.1007/s004270050041
-
Implications of developmental gene regulatory networks inside and outside developmental biologyCurrent Topics in Developmental Biology 117:237–251.https://doi.org/10.1016/bs.ctdb.2015.12.014
-
Tissue micromanipulation in zebrafish embryosMethods in Molecular Biology 546:153–172.https://doi.org/10.1007/978-1-60327-977-2_11
-
Mechanisms of size controlCurrent Opinion in Genetics & Development 11:279–286.https://doi.org/10.1016/S0959-437X(00)00191-X
-
Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1European Journal of Human Genetics 22:907–915.https://doi.org/10.1038/ejhg.2013.268
-
Conserved genetic pathways associated with Microphthalmia, Anophthalmia, and colobomaBirth Defects Research Part C: Embryo Today: Reviews 105:96–113.https://doi.org/10.1002/bdrc.21097
-
Rapid mapping of zebrafish mutations with SNPs and oligonucleotide microarraysGenome Research 12:1929–1934.https://doi.org/10.1101/gr.777302
-
Identification of a large set of rare complete human knockoutsNature Genetics 47:448–452.https://doi.org/10.1038/ng.3243
-
Sp5l is a mediator of fgf signals in Anteroposterior patterning of the neuroectoderm in zebrafish embryoDevelopmental Dynamics 235:2999–3006.https://doi.org/10.1002/dvdy.20945
-
The relative growth of the amphibian eye, studied by means of transplantationJournal of Experimental Zoology 68:247–291.https://doi.org/10.1002/jez.1400680206
-
The growth of eyes and limbs transplanted heteroplastically between two species of amblystomaJournal of Experimental Zoology 59:61–86.https://doi.org/10.1002/jez.1400590105
-
Ectopic wnt signal determines the eyeless phenotype of zebrafish masterblind mutantDevelopment 128:3877–3888.
-
Developmental mutant screens in the zebrafishMethods in Cell Biology 60:21–41.https://doi.org/10.1016/S0091-679X(08)61892-0
-
Growth and size control during developmentOpen Biology 7:170190.https://doi.org/10.1098/rsob.170190
-
BookRobustness and Evolvability in Living SystemsPrinceton: Princeton University Press.
-
Requirement for math5 in the development of retinal ganglion cellsGenes & Development 15:24–29.https://doi.org/10.1101/gad.855301
-
The genetic architecture of Microphthalmia, anophthalmia and colobomaEuropean Journal of Medical Genetics 57:369–380.https://doi.org/10.1016/j.ejmg.2014.05.002
-
Early steps in the development of the forebrainDevelopmental Cell 6:167–181.https://doi.org/10.1016/S1534-5807(04)00027-9
-
Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologiesNature Structural & Molecular Biology 15:1255–1262.https://doi.org/10.1038/nsmb.1515
-
Chaperonins: two rings for foldingTrends in Biochemical Sciences 36:424–432.https://doi.org/10.1016/j.tibs.2011.05.003
Article and author information
Author details
Florencia Cavodeassi

Claudia Wierzbicki
Elizabeth Mayela Ambrosio
Elisabeth M Busch-Nentwich
Funding
Wellcome Trust (088175/Z/09/Z)
- Rodrigo M Young
- Stephen W Wilson
H2020 Marie Skłodowska-Curie Actions (Marie Curie Incoming International Fellowship)
- Rodrigo M Young
Royal Society
- Miguel L Allende
- Stephen W Wilson
Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias (15090007)
- Miguel L Allende
Wellcome Trust (WT098051, 206194)
- Elisabeth M Busch-Nentwich
Medical Research Council (MR/L003775/1)
- Gaia Gestri
- Stephen W Wilson
Wellcome Trust (104682/Z/14/Z)
- Stephen W Wilson
Wellcome Trust (089227/Z09/Z)
- Stephen W Wilson
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are grateful to Dr. Ajay Chitnis and Dr. Richard Dorsky for sharing reagents and fish lines, Dr. Bill Harris and colleagues for support and discussions, Lisa Tucker for technical assistance, the UCL fish facility for fish care, and Elke Ober participaring in the genetic screen. We would like to thank Neha Wali and the Wellcome Sanger Institute sequencing pipelines for performing sequencing. This work was generously supported by funding from a Marie Curie Incoming International Fellowship to RY, Wellcome Trust grants to SW and RY (088175, 104682/Z/14/Z), and EB (WT098051, 206194), an MRC Programme grant to SW and GG (MR/L003775/1), Royal Society International Joint Project funding to SW and MA and FONDAP (15090007) to MA.
Ethics
Animal experimentation: This study was performed in strict accordance to the recommendations of the Home Office, UK.
Version history
- Received: July 13, 2018
- Accepted: January 26, 2019
- Version of Record published: February 19, 2019 (version 1)
Copyright
© 2019, Young et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,389
- views
-
- 331
- downloads
-
- 19
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Compensatory growth renders Tcf7l1a dispensable for eye formation despite its requirement in eye field specification
eLife 8:e40093.
https://doi.org/10.7554/eLife.40093
Further reading
-
- Computational and Systems Biology
- Developmental Biology
Organisms utilize gene regulatory networks (GRN) to make fate decisions, but the regulatory mechanisms of transcription factors (TF) in GRNs are exceedingly intricate. A longstanding question in this field is how these tangled interactions synergistically contribute to decision-making procedures. To comprehensively understand the role of regulatory logic in cell fate decisions, we constructed a logic-incorporated GRN model and examined its behavior under two distinct driving forces (noise-driven and signal-driven). Under the noise-driven mode, we distilled the relationship among fate bias, regulatory logic, and noise profile. Under the signal-driven mode, we bridged regulatory logic and progression-accuracy trade-off, and uncovered distinctive trajectories of reprogramming influenced by logic motifs. In differentiation, we characterized a special logic-dependent priming stage by the solution landscape. Finally, we applied our findings to decipher three biological instances: hematopoiesis, embryogenesis, and trans-differentiation. Orthogonal to the classical analysis of expression profile, we harnessed noise patterns to construct the GRN corresponding to fate transition. Our work presents a generalizable framework for top-down fate-decision studies and a practical approach to the taxonomy of cell fate decisions.
-
- Developmental Biology
- Evolutionary Biology
Despite rapid evolution across eutherian mammals, the X-linked MIR-506 family miRNAs are located in a region flanked by two highly conserved protein-coding genes (SLITRK2 and FMR1) on the X chromosome. Intriguingly, these miRNAs are predominantly expressed in the testis, suggesting a potential role in spermatogenesis and male fertility. Here, we report that the X-linked MIR-506 family miRNAs were derived from the MER91C DNA transposons. Selective inactivation of individual miRNAs or clusters caused no discernible defects, but simultaneous ablation of five clusters containing 19 members of the MIR-506 family led to reduced male fertility in mice. Despite normal sperm counts, motility, and morphology, the KO sperm were less competitive than wild-type sperm when subjected to a polyandrous mating scheme. Transcriptomic and bioinformatic analyses revealed that these X-linked MIR-506 family miRNAs, in addition to targeting a set of conserved genes, have more targets that are critical for spermatogenesis and embryonic development during evolution. Our data suggest that the MIR-506 family miRNAs function to enhance sperm competitiveness and reproductive fitness of the male by finetuning gene expression during spermatogenesis.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}