





Abstract
 HSS has yet to be observed in the gas phase in the interstellar medium (ISM). HSS has been observed in cometary material and in high abundance. However, its agglomeration to such bodies or dispersal from them has not been observed. Similarly, HSO and HOS have not been observed in the ISM, either, even though models support their formation from reactions of known sulfur monoxide and hydrogen molecules, among other pathways. Consequently, this work provides high-level, quantum chemical rovibrational spectroscopic constants and vibrational frequencies in order to assist in interstellar searches for these radical molecules. Furthermore, the HSO−HOS isomerization energy is determined to be 3.63 kcal mol−1, in line with previous work, and the dipole moment of HOS is 36% larger at 3.87 D than HSO, making the less stable isomer more rotationally intense. Finally, the S−S bond strength in HSS is shown to be relatively weak at 30% of the typical disulfide bond energy. Consequently, HSS may degrade into SH and sulfur atoms, making any ISM abundance of HSS likely fairly low, as recent interstellar surveys have observed.
HSS has yet to be observed in the gas phase in the interstellar medium (ISM). HSS has been observed in cometary material and in high abundance. However, its agglomeration to such bodies or dispersal from them has not been observed. Similarly, HSO and HOS have not been observed in the ISM, either, even though models support their formation from reactions of known sulfur monoxide and hydrogen molecules, among other pathways. Consequently, this work provides high-level, quantum chemical rovibrational spectroscopic constants and vibrational frequencies in order to assist in interstellar searches for these radical molecules. Furthermore, the HSO−HOS isomerization energy is determined to be 3.63 kcal mol−1, in line with previous work, and the dipole moment of HOS is 36% larger at 3.87 D than HSO, making the less stable isomer more rotationally intense. Finally, the S−S bond strength in HSS is shown to be relatively weak at 30% of the typical disulfide bond energy. Consequently, HSS may degrade into SH and sulfur atoms, making any ISM abundance of HSS likely fairly low, as recent interstellar surveys have observed.
Export citation and abstract BibTeX RIS
1. Introduction
Sulfur is a poor man's carbon. It bonds well to itself and other p-block atoms, but sulfur typically only forms two bonds in such stable arrangements. Allotropes of sulfur stretch from six to twenty atoms in a ring, but side chains and other functional groups are not favored in any way for connection to these rings. As a result, sulfur chemistry is significantly more limited than the more common second-row carbon. Even so, sulfur is one of the eleven elements that appear on the astrochemist's periodic table (McCall 2006), and is present in many known interstellar molecules (Penzias et al. 1971; Gottlieb & Ball 1973; Fock & McAllister 1982; McCarthy & Thaddeus 2001; Tercero et al. 2010; Neufeld et al. 2012; Gratier et al. 2016). It is necessary to life as we know it, gives rise to novel chemistry in extreme environments like hot springs and black smokers on Earth, heavily influences the atmospheric chemistry of Venus (Krasnopolsky 2007, 2012) as well as the surface chemistry of Io, and is present in various terrestrial pollutants (Tchir & Spratley 1975; Hawkins & Downs 1984). Consequently, sulfur cannot be neglected for astrochemical analysis, and an understanding for its importance in the chemistry of space has been continually growing since various molecular sulfur detections began in the 1970s (Morris et al. 1975; Henkel et al. 1983; Johansson et al. 1984; Ziurys et al. 1984; Cernicharo et al. 1987, 2000; Kaifu et al. 1987; Saito et al. 1987; Turner 1987, 1992; Kahane et al. 1988; Martín-Pintado et al. 1992; Boyle et al. 1994; Turner et al. 1998; Schilke et al. 2001, 2003; Mauersberger et al. 2004; Sakai et al. 2007; Finney et al. 2016).
Even with these various applications and numerous previous astrochemical studies, the simplest of triatomic disulfide systems,  HSS/HS2/S2H, still has plenty of unknowns. For instance, a recent survey of the low-mass warm core IRAS 16293-2422 returned a non-detection for this radical and inconclusive results about its possible interstellar abundance (Martín-Doménech et al. 2016). This is perplexing since HSS is believed to exist in relatively high abundances in icy grains and may be even be the most common form of sulfur in such environnments, even more than H2S based on observations of the Hale-Bopp comet (Bockelée-Morvan et al. 1995). However, gas phase, interstellar detection remains elusive.
HSS/HS2/S2H, still has plenty of unknowns. For instance, a recent survey of the low-mass warm core IRAS 16293-2422 returned a non-detection for this radical and inconclusive results about its possible interstellar abundance (Martín-Doménech et al. 2016). This is perplexing since HSS is believed to exist in relatively high abundances in icy grains and may be even be the most common form of sulfur in such environnments, even more than H2S based on observations of the Hale-Bopp comet (Bockelée-Morvan et al. 1995). However, gas phase, interstellar detection remains elusive.
Part of the problem with detecting HSS may come from the lack of conclusive laboratory data as to the rotational and spectroscopic constants for this radical. The aforementioned work searching for HSS in IRAS 16293-2422 (Martín-Doménech et al. 2016) relied upon low-resolution data obtained several instrumental generations ago (Yamamoto & Saito 1994). The accuracy of these experimental measurements has not been fully verified, and errors could still be on the order of tens of MHz. Such errors compound with increasing J values as the rotational ladder is climbed for the prediction of rotational transitions that can be observed in the dedicated frequency windows. Consequently, the relationship of condensed- to gas-phase HSS is still a mystery. Even though HSS is known to exist beyond the Earth's veil, the hydrogenated disulfide radical has not been conclusively observed beyond our solar system and never diffusely in the gas phase.
Vibrational spectra have been experimentally determined for this system. Most notably the  S−S stretch has been observed in the laboratory with high-resolution techniques at 596.28 cm−1 (Ashworth & Fink 2007). While rotational spectroscopy has long been the best means of detecting new interstellar molecules from the ground, the next generation of air- and space-based telescopes focus on infrared wavelengths. The flexibility of the Stratospheric Observatory for Infrared Astronomy and the upcoming James Webb Space Telescope will open new frontiers of astronomical vibrational spectroscopy. Even so, only lower-resolution values for the other fundamental frequencies of HSS are reported in this same study by Ashworth & Fink (2007). There exist previous low-resolution gas phase frequencies for the other fundamentals (Holstein et al. 1985) and data collected from HSS suspended in Ar matrices (Isoniemi et al. 1999), but errors for such measurements can vary wildly, making gas phase correlation of the fundamentals difficult.
S−S stretch has been observed in the laboratory with high-resolution techniques at 596.28 cm−1 (Ashworth & Fink 2007). While rotational spectroscopy has long been the best means of detecting new interstellar molecules from the ground, the next generation of air- and space-based telescopes focus on infrared wavelengths. The flexibility of the Stratospheric Observatory for Infrared Astronomy and the upcoming James Webb Space Telescope will open new frontiers of astronomical vibrational spectroscopy. Even so, only lower-resolution values for the other fundamental frequencies of HSS are reported in this same study by Ashworth & Fink (2007). There exist previous low-resolution gas phase frequencies for the other fundamentals (Holstein et al. 1985) and data collected from HSS suspended in Ar matrices (Isoniemi et al. 1999), but errors for such measurements can vary wildly, making gas phase correlation of the fundamentals difficult.
Thus far, the most complete set of rovibrational data for  HSS comes from high-level quantum chemical computations (Peterson et al. 2008). Modern quantum chemical techniques utilizing coupled cluster theory (Crawford & Schaefer 2000, pp. 33–136; Helgaker et al. 2004; Shavitt & Bartlett 2009) at the singles, doubles, and perturbative triples [CCSD(T)] level (Raghavachari et al. 1989) have shown that making proper corrections for complete basis set (CBS) limit extrapolation (C), core correlation (cC), scalar relativity (R), and higher-order electron correlation (E) can create highly accurate potential energy surfaces (PESs) (Huang & Lee 2008, 2009; Huang et al. 2011) where rovibrational analyses, typically from second-order perturbation theory (Mills 1972, pp. 115–140; Watson 1977, pp. 1–89; Papousek & Aliev 1982), can produce comparison to experiment to better than 5.0 cm−1 for vibrational frequencies and 30 MHz for rotational constants (Huang et al. 2011; Fortenberry et al. 2012b, 2014a, 2015; Huang et al. 2013a; Zhao et al. 2014; Kitchens & Fortenberry 2016). Such a CcCRE PES computed for HSS has produced useful spectroscopic data (Peterson et al. 2008) to aid in the interstellar search for this simple, yet hidden disulfide radical (Martín-Doménech et al. 2016). However, the spectral data set is still somewhat incomplete, and additional corroboration for the known theoretical or even experimental data would give astronomers more trust in utilizing theoretical data for comparison to interstellar spectra. Such theoretically derived spectroscopic constants have been utilized in the detection of C5N− (Cernicharo et al. 2008), and forced the community to provide more accurate experiments and theoretical results in the detection of C3H+ (Pety et al. 2012; Fortenberry et al. 2013; Huang et al. 2013b; Botschwina et al. 2014; Brünken et al. 2014; McGuire et al. 2014; Fortenberry 2017). In any case, this work will utilize such theoretical approaches to provide the necessary rovibrational data for the HSS radical.
HSS comes from high-level quantum chemical computations (Peterson et al. 2008). Modern quantum chemical techniques utilizing coupled cluster theory (Crawford & Schaefer 2000, pp. 33–136; Helgaker et al. 2004; Shavitt & Bartlett 2009) at the singles, doubles, and perturbative triples [CCSD(T)] level (Raghavachari et al. 1989) have shown that making proper corrections for complete basis set (CBS) limit extrapolation (C), core correlation (cC), scalar relativity (R), and higher-order electron correlation (E) can create highly accurate potential energy surfaces (PESs) (Huang & Lee 2008, 2009; Huang et al. 2011) where rovibrational analyses, typically from second-order perturbation theory (Mills 1972, pp. 115–140; Watson 1977, pp. 1–89; Papousek & Aliev 1982), can produce comparison to experiment to better than 5.0 cm−1 for vibrational frequencies and 30 MHz for rotational constants (Huang et al. 2011; Fortenberry et al. 2012b, 2014a, 2015; Huang et al. 2013a; Zhao et al. 2014; Kitchens & Fortenberry 2016). Such a CcCRE PES computed for HSS has produced useful spectroscopic data (Peterson et al. 2008) to aid in the interstellar search for this simple, yet hidden disulfide radical (Martín-Doménech et al. 2016). However, the spectral data set is still somewhat incomplete, and additional corroboration for the known theoretical or even experimental data would give astronomers more trust in utilizing theoretical data for comparison to interstellar spectra. Such theoretically derived spectroscopic constants have been utilized in the detection of C5N− (Cernicharo et al. 2008), and forced the community to provide more accurate experiments and theoretical results in the detection of C3H+ (Pety et al. 2012; Fortenberry et al. 2013; Huang et al. 2013b; Botschwina et al. 2014; Brünken et al. 2014; McGuire et al. 2014; Fortenberry 2017). In any case, this work will utilize such theoretical approaches to provide the necessary rovibrational data for the HSS radical.
Additionally, the oxygen analogues of this system will also be explored. The  HSO radical has been well-studied (Ohashi et al. 1980; Webster et al. 1982; Sears & McKellar 1983; Yoshikawa et al. 2009; Cazzoli et al. 2016), but far less is known about
HSO radical has been well-studied (Ohashi et al. 1980; Webster et al. 1982; Sears & McKellar 1983; Yoshikawa et al. 2009; Cazzoli et al. 2016), but far less is known about  HOS, even less than
HOS, even less than  HSS. The search for HSO has recently offered some frustrating results with coincidences for rotational lines of HSO observed in some regions and spectral windows but not in others (Cazzoli et al. 2016). Consequently, it is hypothesized that, while interstellar reaction models expect some presence of HSO, its actual observable abundance will be quite low. The observations appear to bear this out (Cazzoli et al. 2016) even though sulfur monoxide was one of the first sulfurous molecules observed in the interstellar medium (ISM; Gottlieb & Ball 1973). In any case, the known experimental data for HSO will serve as a barometer for the quality of the CcCR computations utilized in this work to predict the rotational constants and vibrational frequencies for HSS and HOS. Finally, HSO isomerizes to HOS preferentially when compared to hydride dissociation (Martinez-Nunez et al. 2002), and is known to be 4.2 kcal mol−1 lower in energy than HOS (Wilson & Dunning 2004). As a result, if HSO has any abundance in the ISM, HOS likely will, as well, and its spectroscopic values can be refined beyond those available in the current literature (Denis 2009; Ovsyannikov et al. 2013).
HSS. The search for HSO has recently offered some frustrating results with coincidences for rotational lines of HSO observed in some regions and spectral windows but not in others (Cazzoli et al. 2016). Consequently, it is hypothesized that, while interstellar reaction models expect some presence of HSO, its actual observable abundance will be quite low. The observations appear to bear this out (Cazzoli et al. 2016) even though sulfur monoxide was one of the first sulfurous molecules observed in the interstellar medium (ISM; Gottlieb & Ball 1973). In any case, the known experimental data for HSO will serve as a barometer for the quality of the CcCR computations utilized in this work to predict the rotational constants and vibrational frequencies for HSS and HOS. Finally, HSO isomerizes to HOS preferentially when compared to hydride dissociation (Martinez-Nunez et al. 2002), and is known to be 4.2 kcal mol−1 lower in energy than HOS (Wilson & Dunning 2004). As a result, if HSO has any abundance in the ISM, HOS likely will, as well, and its spectroscopic values can be refined beyond those available in the current literature (Denis 2009; Ovsyannikov et al. 2013).
2. Computational Details
The PES utilized is a specialized fourth-order Taylor series expansion for the potential piece of the Watson internuclear Hamiltonian. This quartic force field (QFF), as it is called, is first defined from a reference geometry. Utilizing restricted-open-shell Hartree–Fock reference wavefunctions (Gauss et al. 1991; Lauderdale et al. 1991; Watts et al. 1993) for CCSD(T), the geometry is computed with the aug-cc-pV5Z basis set (Dunning 1989; Kendall et al. 1992) with the necessary, additional tight d functions for sulfur (Wilson & Dunning 2004) as well as the Martin–Taylor (MT) core correlating basis set (Martin & Taylor 1994) inclusive and exclusive of core electrons correlated. The difference in the MT bond lengths and bond angle are added to the CCSD(T)/aug-cc-pV5Z geometry as has been done for many related systems (Fortenberry et al. 2014b; Fortenberry & Francisco 2015a, 2015b; Fortenberry & Lukemire 2015; Fortenberry & Thackston 2015; Finney et al. 2016; Kitchens & Fortenberry 2016).
From this reference geometry, the CcCR QFF is computed by first displacing the bond lengths by 0.005 Å and the bond angle by 0.005 radians. Coordinate one corresponds to the hydride stretch with coordinates two and three corresponding to the heavy atom stretch and the bend, respectively. At each of the 129 QFF points, CCSD(T)/aug-cc-pVTZ, aug-cc-pVQZ, and aug-cc-pV5Z energies are extrapolated to the CBS limit via a three-point formula (Martin & Lee 1996). To this CCSD(T)/CBS energy, the difference between the MT energies both with and without core electrons included as well as Douglas−Kroll scalar relativistic corrections (Douglas & Kroll 1974; de Jong et al. 2001) are added to produce the CcCR QFF. All computations make use of the MOLPRO 2010.1 quantum chemistry program (Werner et al. 2010).
A fitting of the QFF via a least-squares algorithm gives the equilibrium geometry, and refitting the surface produces zero gradients and the force constants utilized to construct the potential. Transforming the symmetry-internal force constants to Cartesian coordinates via the INTDER 2005 (Allen et al. 2005) program gives flexibly defined force constants for vibrational second-order perturbation theory (VPT2) or rotational perturbation theory (Mills 1972, pp. 115–140; Watson 1977, pp. 1–89; Papousek & Aliev 1982) encoded within the SPECTRO program (Gaw et al. 1991, pp. 170–185) to produce the fundamental vibrational frequencies and the spectroscopic constants for each of these three sulfurous radicals. HSO has a  Fermi resonance polyad as well as a
Fermi resonance polyad as well as a  /
/ Darling–Denison resonance and C-type Coriolis resonance for these same modes. HSS has a
Darling–Denison resonance and C-type Coriolis resonance for these same modes. HSS has a  /
/ Darling–Denison resonance. CCSD(T)/aug-cc-pV5Z dipole moment computations from the equilibrium geometry are also undertaken, and double-harmonic intensities from MP2/6-31+G* within the Gaussian09 program (Mller & Plesset 1934; Hehre et al. 1972; Frisch et al. 2009) produce the fundamental vibrational intensities. Such intensities are known to corroborate well with CCSD(T)/aug-cc-pVTZ intensities (Yu et al. 2015; Finney et al. 2016).
Darling–Denison resonance. CCSD(T)/aug-cc-pV5Z dipole moment computations from the equilibrium geometry are also undertaken, and double-harmonic intensities from MP2/6-31+G* within the Gaussian09 program (Mller & Plesset 1934; Hehre et al. 1972; Frisch et al. 2009) produce the fundamental vibrational intensities. Such intensities are known to corroborate well with CCSD(T)/aug-cc-pVTZ intensities (Yu et al. 2015; Finney et al. 2016).
3. Results and Discussion
3.1. HSO
The force constants computed from the fitting of the CcCR QFF PES points are given in Table 1. Badger's rule gives a general rule of thumb that the harmonic, diagonal force constants are proportional to the bond strength of the given mode. Hence, the F11 force constant for HSO corresponds to the H–S stretch. It is within 0.1 mdyn Å−2 of the same value previously computed with a similar approach for HSO+ (Kitchens & Fortenberry 2016). The F22 S–O bond in HSO is weaker than the same value for HSO+, but in the range of what is expected for O–X single bonds.
Table 1. The HSO CcCR QFF Force Constantsa (in mdyn Å−n radm)b
| F11 | 3.482792 | F221 | 0.6909 | F1111 | 87.56 | F3222 | −64.59 |
| F21 | 0.325388 | F222 | −78.6649 | F2111 | 0.96 | F3311 | −2.55 |
| F22 | 7.913832 | F311 | −0.0230 | F2211 | 1.97 | F3321 | 3.00 |
| F31 | −0.114004 | F321 | −0.3804 | F2221 | −43.38 | F3322 | 10.61 |
| F32 | 0.458784 | F322 | 1.7697 | F2222 | −261.26 | F3331 | 0.79 |
| F33 | 1.153105 | F331 | −0.3742 | F3111 | 0.44 | F3332 | 2.22 |
| F111 | −19.5719 | F332 | −1.2340 | F3211 | 0.70 | F3333 | −0.24 |
| F211 | 0.8217 | F333 | −0.8757 | F3221 | 10.21 | ⋯ | ⋯ |
Notes.
aFollowing the numbering from the internal coordinates defined in-text from the equilibrium geometry. b1 mdyn = 10−8 N; n and m are exponents corresponding to the number of units from the type of modes present in the specific force constant.Download table as: ASCIITypeset image
These force constants are then utilized to compute the geometrical, vibrational, and rotational values produced in Table 2. Comparison with experiment (Ohashi et al. 1980; Yoshikawa et al. 2009) shows that the CcCR HSO values agree well with experiment for the rotational constants, quartic and sextic distortion constants, and the subsequent geometry. The vibrationally averaged rotational constants vary by less than 0.004 cm−1 (∼100 MHz) for the B and C constants, similar to values computed previously (Fortenberry et al. 2011, 2012a, 2012b, 2013, 2014b).
Table 2.
The  HSO/DSO/H34SO CcCR QFF Equilibrium and Zero-point (
HSO/DSO/H34SO CcCR QFF Equilibrium and Zero-point ( ) Geometries, Spectroscopic Constants, and Vibrational Frequenciesa
) Geometries, Spectroscopic Constants, and Vibrational Frequenciesa
| HSO | Experimentb | DSO | H34SO | |
|---|---|---|---|---|
| r0(H–S) Å | 1.383240 | 1.389c | 1.378734 | 1.383227 |
| r0(S–O) Å | 1.495692 | 1.494c | 1.495926 | 1.495615 |
| ∠(H–S–O) | 104.828 | 106.6c | 104.724 | 104.831 |
| A0 cm−1 | 10.010706 | 9.9897076 | 5.300349 | 9.981891 |
| B0 cm−1 | 0.687070 | 0.6838992 | 0.664435 | 0.674999 |
| C0 cm−1 | 0.641108 | 0.6382982 | 0.588392 | 0.630529 |
| A1 cm−1 | 9.694658 | ⋯ | 5.187261 | 9.966354 |
| B1 cm−1 | 0.688651 | ⋯ | 0.664843 | 0.676623 |
| C1 cm−1 | 0.641542 | ⋯ | 0.587650 | 0.630968 |
| A2 cm−1 | 10.155715 | ⋯ | 5.385994 | 10.113151 |
| B2 cm−1 | 0.683909 | ⋯ | 0.665743 | 0.671254 |
| C2 cm−1 | 0.634151 | ⋯ | 0.585640 | 0.623271 |
| A3 cm−1 | 10.038461 | ⋯ | 5.279072 | 10.022816 |
| B3 cm−1 | 0.680674 | ⋯ | 0.655305 | 0.669482 |
| C3 cm−1 | 0.636600 | ⋯ | 0.581295 | 0.626540 |
| DJ kHz | 27.355 | 29.4675 | 22.493 | 26.493 |
| DJK kHz | 890.163 | 904.18 | 673.742 | 866.130 |
| DK MHz | 25.887 | 26.24 | 7.474 | 25.665 |
| d1 kHz | −1.794 | −2.02504 | −2.808 | −1.708 |
| d2 Hz | −0.293 | −0.35187 | −0.786 | −0.276 |
| HJ mHz | −87.204 | −28.42 | −55.728 | −83.841 |
| HJK Hz | 1.120 | 2.779 | 0.946 | 1.112 |
| HKJ Hz | 148.220 | 144.7 | 55.016 | 145.774 |
| HK kHz | 6.428 | ⋯ | 1.244 | 6.295 |
| h1 mHz | −5.496 | ⋯ | −5.318 | −5.269 |
| h2 mHz | 2.464 | ⋯ | 7.418 | 2.274 |
| h3 mHz | 0.746 | 1.10 | 3.165 | 0.681 |
| re(H–S) Å | 1.367 166 | ⋯ | ⋯ | ⋯ |
| re(S–O) Å | 1.489 038 | ⋯ | ⋯ | ⋯ |
| ∠(H–S–O) | 104.740 | ⋯ | ⋯ | ⋯ |
| Ae cm−1 | 10.082349 | ⋯ | 5.324710 | 10.053566 |
| Be cm−1 | 0.691001 | ⋯ | 0.668161 | 0.678844 |
| Ce cm−1 | 0.646681 | ⋯ | 0.593666 | 0.635906 |
| μ D | 2.84 | 2.20d | ⋯ | ⋯ |
 H–S stretch H–S stretch |
2462.3 (94) | ⋯ | 1769.6 | 2460.0 |
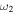 bend bend |
1049.9 (1) | ⋯ | 792.2 | 1043.9 |
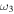 S–O stretch S–O stretch |
1131.2 (52) | ⋯ | 1111.7 | 1124.9 |
| Harmonic 0-Pt | 2321.7 | ⋯ | 1836.8 | 2314.4 |
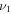 H–S stretch H–S stretch |
2335.6 | ⋯ | 1707.4 | 2333.4 |
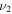 bend bend |
1013.9 | 1011 | 779.4 | 1005.8 |
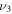 S–O stretch S–O stretch |
1070.6 | 1084 | 1036.6 | 1068.0 |
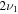
|
4548.9 | ⋯ | 3339.6 | 4545.3 |
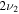
|
2015.7 | 2004 | 1551.1 | 1998.6 |

|
2097.2 | ⋯ | 1992.0 | 2099.2 |
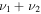
|
3336.8 | ⋯ | 2456.6 | 3329.4 |
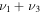
|
3408.0 | ⋯ | 2749.7 | 3401.2 |
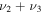
|
2048.9 | 2078 | 1816.8 | 2033.4 |
| 0-Pt | 2284.4 | ⋯ | 1811.3 | 2277.4 |
Notes.
aDouble harmonic MP2/6-31+G* intensities in parentheses. bExperimental spectroscopic constants from Cazzoli et al. (2016) and vibrational data from Yoshikawa et al. (2009). cStructural data from Ohashi et al. (1980). dHSO dipole moment from Webster et al. (1982).Download table as: ASCIITypeset image
Additionally, the  bending frequency computed here to be 1013.9 cm−1 is 2.9 cm−1 above the experimental result of 1011 cm−1 (Yoshikawa et al. 2009) and an improvement over a recently computed value using a near-global PES (Ovsyannikov et al. 2013). The
bending frequency computed here to be 1013.9 cm−1 is 2.9 cm−1 above the experimental result of 1011 cm−1 (Yoshikawa et al. 2009) and an improvement over a recently computed value using a near-global PES (Ovsyannikov et al. 2013). The  S–O stretch, on the other hand, is off from experiment (1084 cm−1) here by 13.4 cm−1, while the previous PES is much closer to experiment at 1087.79 cm−1. However, the most intense of the fundamental vibrational frequencies will be the H–S stretch at 2335.7 cm−1 which is nearly 80 cm−1 less than that computed previously (Ovsyannikov et al. 2013). No previously computed CcCR fundamental vibrational frequencies have ever produced errors of this magnitude, leading us to put forth that the H–S stretch is at a lower frequency than was previously believed. The
S–O stretch, on the other hand, is off from experiment (1084 cm−1) here by 13.4 cm−1, while the previous PES is much closer to experiment at 1087.79 cm−1. However, the most intense of the fundamental vibrational frequencies will be the H–S stretch at 2335.7 cm−1 which is nearly 80 cm−1 less than that computed previously (Ovsyannikov et al. 2013). No previously computed CcCR fundamental vibrational frequencies have ever produced errors of this magnitude, leading us to put forth that the H–S stretch is at a lower frequency than was previously believed. The  combination band is experimentally determined to lie at 2078 cm−1 while the previous theory places this value at 2109.92 cm−1. The VPT2 CcCR results are 30 cm−1 below while the previous theory is 40 cm−1 above, giving a 70 cm−1 difference between the theoretical results. However, combination bands are known to produce compounding errors in the CcCR VPT2 approach (Huang et al. 2013c), further indicating that the
combination band is experimentally determined to lie at 2078 cm−1 while the previous theory places this value at 2109.92 cm−1. The VPT2 CcCR results are 30 cm−1 below while the previous theory is 40 cm−1 above, giving a 70 cm−1 difference between the theoretical results. However, combination bands are known to produce compounding errors in the CcCR VPT2 approach (Huang et al. 2013c), further indicating that the  stretch should likely be in less error than the previous theory. Hence, a new prediction for the placement of the H–S stretch puts forth that this fundamental is likely to be observed at a slightly more redshifted frequency in the gas phase.
stretch should likely be in less error than the previous theory. Hence, a new prediction for the placement of the H–S stretch puts forth that this fundamental is likely to be observed at a slightly more redshifted frequency in the gas phase.
Beyond the  fundamental vibrational frequency, new values are produced for the overtones and combination bands as well as vibrationally averaged rotational constants for the fundamental vibrational frequencies for
fundamental vibrational frequency, new values are produced for the overtones and combination bands as well as vibrationally averaged rotational constants for the fundamental vibrational frequencies for  HSO as well as the DSO and H34SO isotopologues. The principal rotational constant is nearly halved in DSO compared to HSO, and the D–S stretching and bending fundamental frequencies are significantly reduced upon deuteration. Inclusion of 34S changes the observables little, but enough to be noted with high-resolution instruments. Consequently, searches for new rotational transitions corresponding to the vibrationally excited states of HSO or the DSO/H34SO isotopologues can now be undertaken both in the laboratory and the ISM. The newest generation of radio telescopes like the Atacama Large Millimeter Array may allow for the detection of previously hypothesized, but unobserved, interstellar molecules like HSO. The additional vibrationally excited rotations may also be accessed in circumstellar envelopes or supernova remnants, giving vital clues as to the progression of those regions (Cazzoli et al. 2016). Consequently, these data provided here, shown to be accurate, will aid in such searches.
HSO as well as the DSO and H34SO isotopologues. The principal rotational constant is nearly halved in DSO compared to HSO, and the D–S stretching and bending fundamental frequencies are significantly reduced upon deuteration. Inclusion of 34S changes the observables little, but enough to be noted with high-resolution instruments. Consequently, searches for new rotational transitions corresponding to the vibrationally excited states of HSO or the DSO/H34SO isotopologues can now be undertaken both in the laboratory and the ISM. The newest generation of radio telescopes like the Atacama Large Millimeter Array may allow for the detection of previously hypothesized, but unobserved, interstellar molecules like HSO. The additional vibrationally excited rotations may also be accessed in circumstellar envelopes or supernova remnants, giving vital clues as to the progression of those regions (Cazzoli et al. 2016). Consequently, these data provided here, shown to be accurate, will aid in such searches.
3.2. HOS
With accuracies for fundamentals of HSO in the ∼10 cm−1 range and known accuracies of similar systems comparable or lower, prediction for the HOS spectroscopic values should produce reliable results. First, the force constants for HOS are given in Table 3 showing that the H–O bond in F11 is more than twice as strong as the H–S stretch in HSO with the S–O bond significantly weaker. Here, this differs from HOS+ where the S–O bond did not change by nearly the same amount upon isomerization (Kitchens & Fortenberry 2016). The hydroxyl radical is a fairly stable system so such a bond weakening in the neutral form is not unexpected.
Table 3. The HOS CcCR QFF Force Constants (in mdyn Å−n radm)
| F11 | 8.004 919 | F221 | −0.6042 | F1111 | 349.89 | F3222 | 1.57 |
| F21 | −0.019 912 | F222 | −27.7643 | F2111 | 1.92 | F3311 | 0.62 |
| F22 | 4.958 901 | F311 | −0.2784 | F2211 | −2.98 | F3321 | −0.24 |
| F31 | 0.033 171 | F321 | −0.0070 | F2221 | 3.35 | F3322 | 1.05 |
| F32 | 0.469 582 | F322 | −1.2591 | F2222 | 130.45 | F3331 | 1.00 |
| F33 | 0.721 186 | F331 | −0.0798 | F3111 | −1.98 | F3332 | −0.13 |
| F111 | −57.1424 | F332 | −0.7779 | F3211 | 0.27 | F3333 | −1.11 |
| F211 | 0.7229 | F333 | −0.6161 | F3221 | −0.07 | ⋯ | ⋯ |
Download table as: ASCIITypeset image
There exist very little reliable experimental data for HOS with some of the best computations for the spectral features of this radical coming in the past decade. The current work, given in Table 4, is a likely improvement on these earlier results (Denis 2009), but the differences in the values are not so great that they arouse questions in either approach. Consequently, the A rotational constant for HOS is more than double the value in HSO with the B and C constants reduced by over 15%. Hence, HOS is more prolate in its character than its mercapto isomer.
Table 4.
The  HOS/DOS/HO34S CcCR QFF Equilibrium and Zero-point Geometries, Spectroscopic Constants, and Vibrational Frequenciesa
HOS/DOS/HO34S CcCR QFF Equilibrium and Zero-point Geometries, Spectroscopic Constants, and Vibrational Frequenciesa
| HOS | Previous Theoryb | DOS | HO34S | |
|---|---|---|---|---|
| r0(H–O) Å | 0.973775 | ⋯ | 0.971071 | 0.973768 |
| r0(O−S) Å | 1.634668 | ⋯ | 1.634463 | 1.634615 |
| ∠(H–O−S) | 108.334 | ⋯ | 108.219 | 108.335 |
| A0 cm−1 | 21.880094 | 21.6218 | 11.892 954 | 21.878356 |
| B0 cm−1 | 0.555046 | 0.5460 | 0.521 434 | 0.543775 |
| C0 cm−1 | 0.540276 | 0.5315 | 0.498 317 | 0.529590 |
| A1 cm−1 | 21.055060 | ⋯ | 11.574 432 | 21.053304 |
| B1 cm−1 | 0.554927 | ⋯ | 0.520 918 | 0.543624 |
| C1 cm−1 | 0.539679 | ⋯ | 0.497 350 | 0.529016 |
| A2 cm−1 | 22.917953 | ⋯ | 12.330 344 | 22.917002 |
| B2 cm−1 | 0.555014 | ⋯ | 0.521 752 | 0.543728 |
| C2 cm−1 | 0.538874 | ⋯ | 0.499 751 | 0.528219 |
| A3 cm−1 | 21.836301 | ⋯ | 11.857 272 | 21.833508 |
| B3 cm−1 | 0.558530 | ⋯ | 0.517 533 | 0.539389 |
| C3 cm−1 | 0.535707 | ⋯ | 0.491 708 | 0.525160 |
| DJ kHz | 26.129 | ⋯ | 21.432 | 25.110 |
| DJK MHz | 1.455 | ⋯ | 0.833 | 1.407 |
| DK MHz | 189.956 | ⋯ | 62.132 | 189.865 |
| d1 kHz | −0.588 | ⋯ | −0.918 | −0.554 |
| d2 kHz | −0.046 | ⋯ | −0.112 | −0.043 |
| HJ mHz | −96.074 | ⋯ | −53.535 | −89.852 |
| HJK Hz | 2.473 | ⋯ | 1.534 | 2.288 |
| HKJ kHz | 1.145 | ⋯ | 1.899 | 1.108 |
| HK kHz | 275.237 | ⋯ | 54.122 | 274.847 |
| h1 mHz | 0.572 | ⋯ | 1.529 | 0.532 |
| h2 mHz | 0.466 | ⋯ | 1.203 | 0.420 |
| h3 mHz | 0.063 | ⋯ | 0.235 | 0.056 |
| re(H–O) Å | 0.963028 | 0.9629 | ⋯ | ⋯ |
| re(O−S) Å | 1.629057 | 1.6302 | ⋯ | ⋯ |
| ∠(H–O−S) | 108.140 | 107.97 | ⋯ | ⋯ |
| Ae cm−1 | 21.795577 | 21.5522 | 11.851360 | 21.739982 |
| Be cm−1 | 0.557429 | 0.5484 | 0.523511 | 0.546098 |
| Ce cm−1 | 0.543528 | 0.5348 | 0.501365 | 0.532749 |
| μ D | 3.87 | ⋯ | ⋯ | ⋯ |
 H–O stretch H–O stretch |
3786.0 (118) | 3766.3 | 2757.5 | 3786.0 |
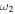 bend bend |
1188.8 (53) | 1184.9 | 871.1 | 1188.1 |
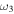 O−S stretch O−S stretch |
858.8 (89) | 843.8 | 855.7 | 850.3 |
| Harmonic 0-Pt | 2916.8 | ⋯ | 2242.2 | 2912.2 |
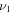 H–O stretch H–O stretch |
3592.3 | 3577.3 | 2655.1 | 3592.3 |
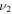 bend bend |
1157.7 | 1150.2 | 854.4 | 1157.0 |
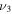 O−S stretch O−S stretch |
846.3 | 830.2 | 843.7 | 838.0 |
|
7001.3 | ⋯ | 5213.1 | 7001.3 |
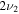
|
2296.2 | ⋯ | 1698.6 | 2294.8 |
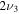
|
1682.7 | ⋯ | 1677.7 | 1666.3 |
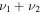
|
4730.5 | ⋯ | 3500.3 | 4729.8 |

|
4437.4 | ⋯ | 3497.5 | 4429.2 |
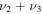
|
1999.7 | ⋯ | 1694.7 | 1666.3 |
| 0-Pt | 2879.7 | ⋯ | 2221.8 | 2875.1 |
Notes.
aDouble harmonic MP2/6-31+G* intensities in parentheses. bCCSD(T)/aug-cc-pV(T+d)Z or "Recommended" values from Denis (2009).Download table as: ASCIITypeset image
All of the intensities for fundamental vibrational frequencies of  HOS in Table 4 are non-negligible with the H–O stretch coming in as the most intense of the group. The
HOS in Table 4 are non-negligible with the H–O stretch coming in as the most intense of the group. The  H–O stretching frequency itself is in the range one would expect for such a motion at 3592.3 cm−1. The bend is higher in frequency for HOS at 1157.7 cm−1 than for HSO at 1013.9 cm−1, but the heavy atom stretch is significantly reduced to 846.3 cm−1 when the heavier sulfur atom is placed on the terminus of the molecule. The rotational and vibrational spectroscopic values for the DOS and HO34S isotopologues are also given in Table 4 for subsequent analysis of this system.
H–O stretching frequency itself is in the range one would expect for such a motion at 3592.3 cm−1. The bend is higher in frequency for HOS at 1157.7 cm−1 than for HSO at 1013.9 cm−1, but the heavy atom stretch is significantly reduced to 846.3 cm−1 when the heavier sulfur atom is placed on the terminus of the molecule. The rotational and vibrational spectroscopic values for the DOS and HO34S isotopologues are also given in Table 4 for subsequent analysis of this system.
Utilizing both the minima and the anharmonic zero point vibrational energies (0-Pt) for the HSO and HOS isomers in the fitted CcCR QFF PES, the isomerization energy is computed here to be 3.64 kcal mol−1 (1273 cm−1) with HSO being the more stable isomer. The present result is in line with, but slightly below, that from earlier CCSD(T)/CBS vibrationally averaged computations at 4.2 kcal mol−1 (Wilson & Dunning 2004). Hence, if H2 is reacted with the known silicon monoxide interstellar molecule in some fashion to create HSO and a hydrogen atom (Cazzoli et al. 2016), some HOS will likely be produced as well, since H2 has an equal probability to interact first with the oxygen or the sulfur atom. The 3.87 D dipole moment computed herein is quite large indicating that HOS may be more observable than HSO if their concentrations are as equal as their relative energies suggest. Consequently, the vibrational and rotational spectroscopic data produced in this work will aid in classification of these molecules with the newest generation of space telescopes or laboratory techniques.
3.3. HSS
The  HSS force constants are given in Table 5. These values highlight that the H–S bond in this radical is slightly stronger than that in HSO. The S−S bond is weaker in the smaller F22 force constant than either of the S–O bonds in the oxygenated systems. Consequently, the dissociation of the S−S bond may be more favored than any hydrogen migration, leading directly to some contribution of the known abundances for the mercapto radical (Neufeld et al. 2012). This is further corroborated in that the breaking of the S−S bond by CCSD(T)/aug-cc-pV5Z computations puts the disulfide bond dissociation energy at a relatively small 76.0 kcal mol−1. The standard disulfide bond dissociation energy is quite strong at ∼250 kcal mol−1 indicating that the addition of the hydrogen atom significantly weakens the S−S bond in HSS.
HSS force constants are given in Table 5. These values highlight that the H–S bond in this radical is slightly stronger than that in HSO. The S−S bond is weaker in the smaller F22 force constant than either of the S–O bonds in the oxygenated systems. Consequently, the dissociation of the S−S bond may be more favored than any hydrogen migration, leading directly to some contribution of the known abundances for the mercapto radical (Neufeld et al. 2012). This is further corroborated in that the breaking of the S−S bond by CCSD(T)/aug-cc-pV5Z computations puts the disulfide bond dissociation energy at a relatively small 76.0 kcal mol−1. The standard disulfide bond dissociation energy is quite strong at ∼250 kcal mol−1 indicating that the addition of the hydrogen atom significantly weakens the S−S bond in HSS.
Table 5. The HSS CcCR QFF Force Constants (in mdyn Å−n radm)
| F11 | 3.893427 | F221 | −0.6209 | F1111 | 98.68 | F3222 | 1.79 |
| F21 | 0.140979 | F222 | −18.6077 | F2111 | −0.45 | F3311 | 1.57 |
| F22 | 3.664970 | F311 | −0.1047 | F2211 | 0.15 | F3321 | 0.43 |
| F31 | −0.115820 | F321 | 0.1204 | F2221 | 1.21 | F3322 | 3.07 |
| F32 | 0.373726 | F322 | −1.0323 | F2222 | 79.47 | F3331 | −0.61 |
| F33 | 0.871176 | F331 | −0.0608 | F3111 | 0.99 | F3332 | 2.54 |
| F111 | −21.5613 | F332 | −1.1642 | F3211 | 1.07 | F3333 | 1.09 |
| F211 | 0.4949 | F333 | −0.4217 | F3221 | 0.38 | ⋯ | ⋯ |
Download table as: ASCIITypeset image
There exist some previous rotational and vibrational data for HSS from both experiment (Yamamoto & Saito 1994; Isoniemi et al. 1999; Ashworth & Fink 2007) and theory (Peterson et al. 2008), and this is compared with the CcCR QFF results in Table 6. The previous CcCRE discrete variable representation (DVR) results utilizing a similar PES to that constructed in this work, but a different method for solving the Watson Hamiltonian, are in excellent agreement with those values computed here as has been shown previously for SPSi (Finney et al. 2016). The quartic distortion constants are also nearly identical for both HSS and DSS as a prime example of the correlation between the two theoretical approaches. Furthermore, the experimental B and C rotational constants from Yamamoto & Saito (1994) are within 0.0005 cm−1 (15 MHz) of the present CcCR QFF results. Consequently, the vibrationally averaged ( ) geometry and spectroscopic constants of HSS reported here should be quite accurate, and the previously determined rotational constants should be sufficient as a basis for any interstellar searches for HSS. The 1.53 D dipole moment makes this molecule a plausible target for astronomical detection.
) geometry and spectroscopic constants of HSS reported here should be quite accurate, and the previously determined rotational constants should be sufficient as a basis for any interstellar searches for HSS. The 1.53 D dipole moment makes this molecule a plausible target for astronomical detection.
Table 6.
The  HSS/DSS and 34S Isotopologue CcCR QFF Equilibrium and Zero-point Geometries, Spectroscopic Constants, and Vibrational Frequenciesa
HSS/DSS and 34S Isotopologue CcCR QFF Equilibrium and Zero-point Geometries, Spectroscopic Constants, and Vibrational Frequenciesa
| HSS | Previous HSSb | DSS | Previous DSSb | H34SS | HS34S | H34S34S | |
|---|---|---|---|---|---|---|---|
| r0(H–S) Å | 1.362468 | ⋯ | 1.358815 | ⋯ | 1.362451 | 1.362461 | 1.362444 |
| r0(S−S) Å | 1.963115 | ⋯ | 1.963175 | ⋯ | 1.963 049 | 1.963 048 | 1.962 981 |
| ∠(H–S−S) | 101.800 | ⋯ | 101.701 | ⋯ | 101.802 | 101.800 | 101.803 |
| A0 cm−1 | 9.932542 | 9.905 999c | 5.186836 | 5.183868c | 9.908756 | 9.932003 | 9.908222 |
| B0 cm−1 | 0.267181 | 0.266 730c | 0.260474 | 0.260030c | 0.259838 | 0.259228 | 0.251880 |
| C0 cm−1 | 0.259860 | 0.259 404c | 0.247610 | 0.247194c | 0.252893 | 0.252331 | 0.245348 |
| A1 cm−1 | 9.640912 | ⋯ | 5.080 793 | ⋯ | 9.617 649 | 9.640336 | 9.617080 |
| B1 cm−1 | 0.267821 | ⋯ | 0.260 783 | ⋯ | 0.260 469 | 0.259850 | 0.252493 |
| C1 cm−1 | 0.260296 | ⋯ | 0.247 684 | ⋯ | 0.253 330 | 0.252760 | 0.245778 |
| A2 cm−1 | 10.135960 | ⋯ | 5.261 972 | ⋯ | 10.111 139 | 10.135312 | 10.110498 |
| B2 cm−1 | 0.266799 | ⋯ | 0.260 329 | ⋯ | 0.259 457 | 0.258849 | 0.251502 |
| C2 cm−1 | 0.259013 | ⋯ | 0.247 143 | ⋯ | 0.252 070 | 0.251510 | 0.244552 |
| A3 cm−1 | 9.925311 | ⋯ | 5.186 529 | ⋯ | 9.901 816 | 9.924703 | 9.901213 |
| B3 cm−1 | 0.265554 | ⋯ | 0.259 025 | ⋯ | 0.258 278 | 0.257671 | 0.250388 |
| C3 cm−1 | 0.258238 | ⋯ | 0.245 930 | ⋯ | 0.251 338 | 0.250779 | 0.243863 |
| DJ kHz | 5.765 | 5.80 | 5.075 | 5.11 | 5.470 | 5.436 | 5.149 |
| DJK MHz | 0.230 | 0.2299 | 0.182 | 0.1821 | 0.220 | 0.217 | 0.208 |
| DK MHz | 22.4606 | 22.41 | 6.357 | 6.37 | 22.294 | 22.461 | 22.295 |
| d1 kHz | −0.150 | ⋯ | −0.243 | ⋯ | −0.135 | −0.134 | −0.123 |
| d2 kHz | −0.010 | ⋯ | −0.031 | ⋯ | −0.009 | −0.009 | −0.008 |
| HJ mHz | −19.860 | ⋯ | −18.589 | ⋯ | −18.241 | −18.077 | −16.558 |
| HJK Hz | 0.191 | ⋯ | 0.191 | ⋯ | 0.178 | 0.171 | 0.159 |
| HKJ kHz | 31.390 | ⋯ | 9.799 | ⋯ | 20.591 | 29.773 | 28.996 |
| HK kHz | 53.241 | ⋯ | 928.128 | ⋯ | 52.263 | 53.149 | 52.172 |
| h1 mHz | 0.025 | ⋯ | 0.030 | ⋯ | 0.022 | 0.023 | 0.020 |
| h2 mHz | 0.042 | ⋯ | 0.135 | ⋯ | 0.037 | 0.036 | 0.031 |
| h3 mHz | 0.005 | ⋯ | 0.029 | ⋯ | 0.005 | 0.005 | 0.004 |
| re(H–S) Å | 1.349025 | 1.3482 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
| re(S−S) Å | 1.958809 | 1.9608 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
| ∠(H–S−S) | 101.571 | 101.52 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
| Ae cm−1 | 9.980263 | 9.98906 | 5.202443 | 5.20666 | 9.956587 | 9.979831 | 9.956160 |
| Be cm−1 | 0.267872 | 0.26736 | 0.261122 | 0.26064 | 0.260499 | 0.259892 | 0.252513 |
| Ce cm−1 | 0.260871 | 0.26039 | 0.248642 | 0.24822 | 0.253857 | 0.253296 | 0.246268 |
| μ D | 1.53 | 1.425 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
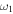 H–S stretch H–S stretch |
2602.5 (10) | 2598.3 | 1869.8 | 1866.8 | 2600.1 | 2602.5 | 2600.1 |
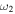 bend bend |
919.8 (3) | 919.0 | 668.0 | 667.3 | 919.1 | 919.0 | 918.4 |
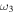 S−S stretch S−S stretch |
608.8 (14) | 605.0 | 607.3 | 603.6 | 599.7 | 599.9 | 590.7 |
| Harmonic 0-Pt | 2065.6 | ⋯ | 1572.6 | ⋯ | 2059.5 | 2060.7 | 2054.6 |
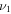 H–S stretch H–S stretch |
2485.5 | 2463d | 1809.7 | ⋯ | 2483.3 | 2485.5 | 2483.2 |
 bend bend |
910.0 | 903d | 663.1 | ⋯ | 909.4 | 909.3 | 908.7 |
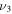 S−S stretch S−S stretch |
601.9 | 596.8e | 600.3 | 594.4e | 593.0 | 593.2 | 584.1 |
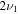
|
4856.9 | ⋯ | 3560.6 | ⋯ | 4852.7 | 4856.9 | 4852.6 |
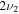
|
1816.5 | ⋯ | 1325.3 | ⋯ | 1815.4 | 1815.0 | 1813.8 |
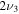
|
1198.4 | ⋯ | 1195.6 | ⋯ | 1180.7 | 1181.1 | 1163.2 |
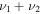
|
3387.8 | ⋯ | 2469.3 | ⋯ | 3385.1 | 3387.0 | 3384.3 |
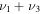
|
3089.1 | ⋯ | 2410.9 | ⋯ | 3077.9 | 3080.3 | 3069.0 |
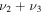
|
1507.2 | ⋯ | 1258.8 | ⋯ | 1497.7 | 1497.8 | 1488.3 |
| 0-Pt | 2045.9 | ⋯ | 1562.1 | ⋯ | 2039.8 | 2041.0 | 2035.0 |
Notes.
aDouble harmonic MP2/6-31+G* intensities in parthenses. bCcCRE DVR computations from Peterson et al. (2008) unless otherwise noted. cLow-resolution gas phase results from Yamamoto & Saito (1994). dAr matrix results from Isoniemi et al. (1999). eHigh-resolution gas phase results from Ashworth & Fink (2007).Download table as: ASCIITypeset image
The only high-resolution experimental data for the vibrational frequencies of HSS are for the  S−S stretch measured to be 596.8 and 594.4 cm−1 for DSS. The CcCR frequencies of 601.9 and 600.3 cm−1 are 5.1 and 5.9 cm−1, respectively, higher than these determined values. This is in line with previous results of similar radicals (Fortenberry et al. 2011; Kitchens & Fortenberry 2016) even though this is the first time the vibrations of a third-row atom with another third-row atom have been compared between CcCR QFF VPT2 and a gas phase experiment. The other vibrational frequencies known experimentally were measured either with low-resolution techniques or in Ar matrices. Comparison of the
S−S stretch measured to be 596.8 and 594.4 cm−1 for DSS. The CcCR frequencies of 601.9 and 600.3 cm−1 are 5.1 and 5.9 cm−1, respectively, higher than these determined values. This is in line with previous results of similar radicals (Fortenberry et al. 2011; Kitchens & Fortenberry 2016) even though this is the first time the vibrations of a third-row atom with another third-row atom have been compared between CcCR QFF VPT2 and a gas phase experiment. The other vibrational frequencies known experimentally were measured either with low-resolution techniques or in Ar matrices. Comparison of the  bend is good, but the
bend is good, but the  H–S stretch differs by more than 20 cm−1. However, CcCR VPT hydride stretches have reproduced those from experiment to better than 1 cm−1 in some cases (Huang et al. 2011, 2013a; Fortenberry et al. 2012a), indicating that the higher 2485.5 cm−1 frequency computed in this work is likely closer to the experimental value for this mode than the Ar matrix data.
H–S stretch differs by more than 20 cm−1. However, CcCR VPT hydride stretches have reproduced those from experiment to better than 1 cm−1 in some cases (Huang et al. 2011, 2013a; Fortenberry et al. 2012a), indicating that the higher 2485.5 cm−1 frequency computed in this work is likely closer to the experimental value for this mode than the Ar matrix data.
Unfortunately, most of these vibrational frequencies for HSS will not have strong intensities. The movement of the sulfur atoms does not shift either the center of mass or the center of charge enough to create strong infrared activity. As a result, rotational spectroscopy is likely a better means of observing HSS than searching for the fundamental vibrational frequencies. As a final note, inclusion of the 34S isotope in the HSS radical provides a notable shift for the vibrational frequencies. Most mass-2 increases will not greatly affect the vibrational frequencies, but each sulfur atom contains nearly half of the molecular mass. Consequently, the H34SS, HS34S, and H34S34S spectrosocpic data in Table 6 will aid in high-resolution searches for these forms of the hydrogenated disulfide radical, especially in the laboratory.
4. Conclusions
The search for HSO, HOS, and HSS in the ISM is valuable in understanding the role that interstellar sulfur may play in various regions, especially protoplanetary disks. If HSS is so plentiful in cometary media (Bockelée-Morvan et al. 1995), the question is naturally asked whether this material is created in the ice mantle or deposited onto the surface. The follow-up questions related to the dispersal of HSS from such icy bodies are agnostic to its creation. Since HSS has not been observed in the gas phase, it is unclear as to how the presence of HSS in ices may seed the surrounding environment of such bodies in planet-forming regions. A more complete understanding of HSS or its oxygenated counterparts is required for such unknowns to be addressed to any extent.
HSS will likely best be observed by rotational spectroscopy, and the current literature on the experimental rotational constants (Yamamoto & Saito 1994) are in good agreement with our high-level computations and those from previous work (Peterson et al. 2008). While our methods reproduce the S−S stretch well, this and the other vibrational modes of HSS are not strong enough absorbers to escape the noise level of most infrared telescopes unless HSS is in high enough abundance. Recent observations do not support such high concentrations (Martín-Doménech et al. 2016). One hypothesis derived from this work for the lack of gas phase HSS in interstellar observations may be that it breaks down more readily than anticipated into SH and a sulfur atom since the S−S force constant is fairly weak and the bond dissociation energy for this moiety is only 76.0 kcal mol−1, only 30% of the strength of a standard disulfide bond (Fortenberry 2016).
The small isomerization energy between HSO and HOS is confirmed here at a very high level to be less than 4.0 kcal mol−1 with HSO lower in energy. Even so, the larger dipole moment of the alcohol isomer could indicate that HOS is more observable in regions where the ambient energy or photon flux is large enough to create marked abundances of HOS relative to HSO. Consequently, these two molecules will likely be observed as a pair if at all. Furthermore, relative abundances of the isomers to one another would be clear indicators of the astrophysical environment in which they are found, giving additional measures of the temperature, density, and local energetics of such astronomical regions. Finally, the 0-Pt values computed in this work will aid reaction modelers in properly describing the interstellar chemical schema that include HSS and its oxygenated counterparts.
R.C.F. gives appreciation to Georgia Southern University for providing start-up funds and to Professor T. Daniel Crawford of Virginia Tech for the use of his computing facilities for part of the data computed in this study.