
Therapeutic Antibodies for the Treatment of Respiratory Tract Infections—Current Overview and Perspectives

 , and
, and
Abstract
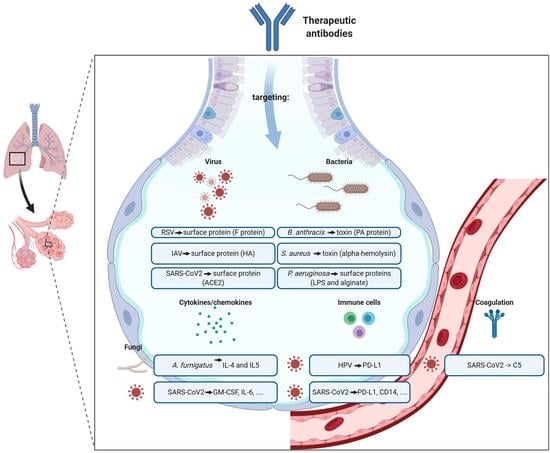
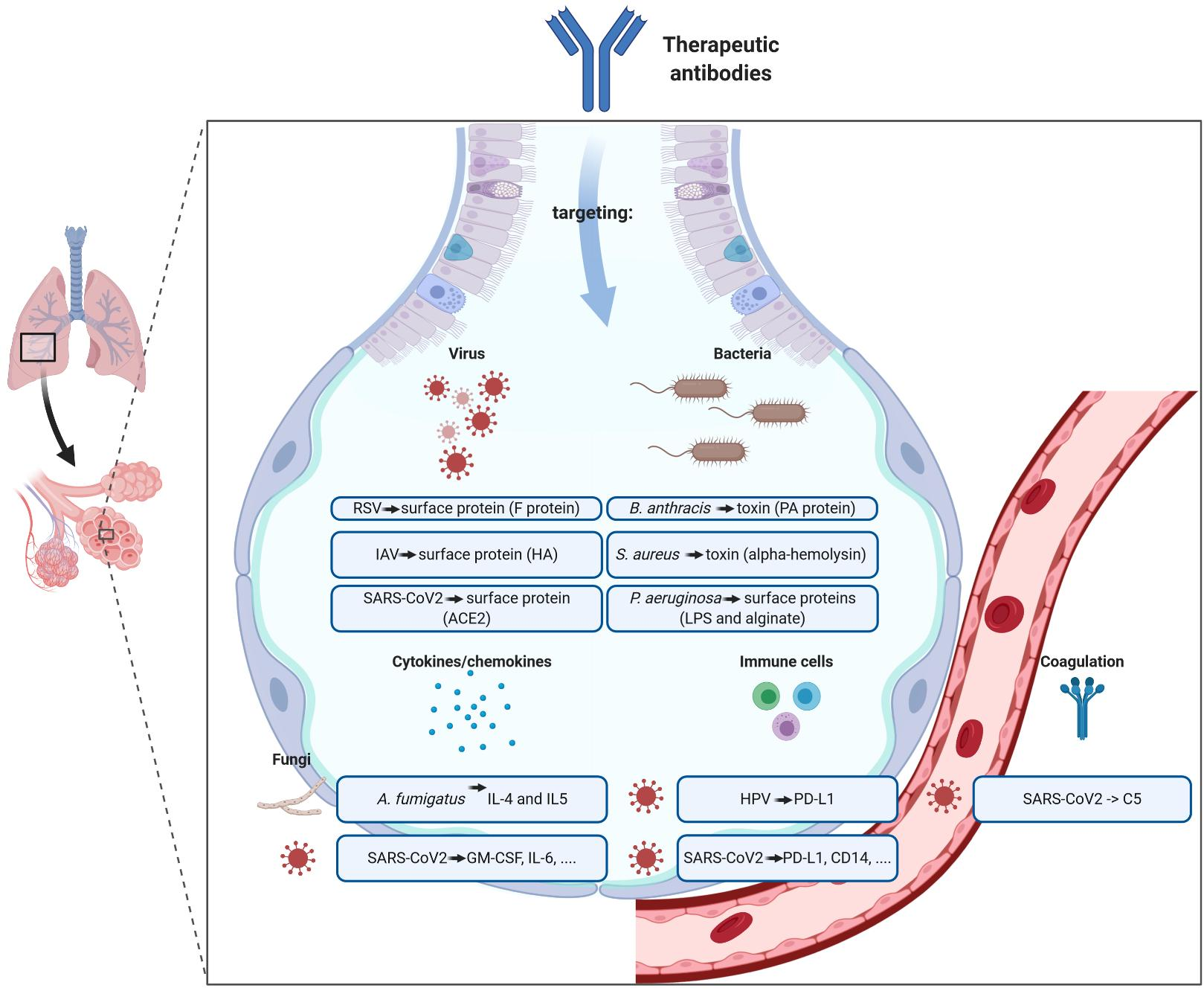
:
1. Introduction
2. Methods
3. Result
3.1. Therapeutic Antibodies—Main Features
3.2. Therapeutic Antibodies against Virus Diseases
3.2.1. Respiratory Syncytial Virus (RSV)
3.2.2. Human Papillomavirus (HPV)
3.2.3. Influenza Virus
3.2.4. The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Pandemic
3.3. Therapeutic Abs against Bacterial Diseases
3.3.1. Bacillus anthracis
3.3.2. Staphylococcus aureus
3.3.3. Pseudomonas aeruginosa
3.4. Therapeutic Abs against Fungal Diseases
3.5. Pharmacokinetics of Anti-Rti Abs
3.5.1. Administration Route
3.5.2. Endogenous (Non-Specific) Catabolism
3.5.3. Target-Mediated (Specific) Elimination
3.5.4. Elimination Mediated by Anti-Drug Antibody
3.5.5. Pharmacokinetic Variability, Concentration-Response Relationship and Optimal Dosing
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Foundation, W.L. Acute Respiratory Infections Atlas; World Lung Foundation: New York, NY, USA, 2010. [Google Scholar]
- Schluger, N.W.; Koppaka, R. Lung disease in a global context. A call for public health action. Ann. Am. Thorac. Soc. 2014, 11, 407–416. [Google Scholar] [CrossRef]
- Dagan, R.; Bhutta, Z.A.; de Quadros, C.A.; Garau, J.; Klugman, K.P.; Khuri-Bulos, N.; Levine, O.; Saha, S.K.; Sow, S.; Were, F.; et al. The remaining challenge of pneumonia: The leading killer of children. Pediatr. Infect. Dis. J. 2011, 30, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000-15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Salazar, G.; Zhang, N.; Fu, T.M.; An, Z. Antibody therapies for the prevention and treatment of viral infections. NPJ Vaccines 2017, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenech, M.; Sempere, J.; de Miguel, S.; Yuste, J. Combination of Antibodies and Antibiotics as a Promising Strategy against Multidrug-Resistant Pathogens of the Respiratory Tract. Front. Immunol. 2018, 9, 2700. [Google Scholar] [CrossRef] [Green Version]
- Desoubeaux, G.; Pelegrin, M. [Monoclonal antibodies in infectious diseases: New partners in the therapeutic arsenal]. Med. Sci. 2019, 35, 1008–1013. [Google Scholar] [CrossRef]
- Desoubeaux, G.; Reichert, J.M.; Sleeman, M.; Reckamp, K.L.; Ryffel, B.; Adamczewski, J.P.; Sweeney, T.D.; Vanbever, R.; Diot, P.; Owen, C.A.; et al. Therapeutic monoclonal antibodies for respiratory diseases: Current challenges and perspectives, March 31—April 1, 2016, Tours, France. mAbs 2016, 8, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Desoubeaux, G.; Daguet, A.; Watier, H. Therapeutic antibodies and infectious diseases, Tours, France, November 20–22, 2012. mAbs 2013, 5, 626–632. [Google Scholar] [CrossRef] [Green Version]
- Cecil, R.L.; Larsen, N.P. Clinical and Bacteriologic Study of One Thousand Cases of Lobar Pneumonia with Special Reference to the Therapeutic Value of Pneumococcus Antibody Solution: Preliminary Report. JAMA 1922, 79, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Pelegrin, M.; Naranjo-Gomez, M.; Piechaczyk, M. Antiviral Monoclonal Antibodies: Can They Be More Than Simple Neutralizing Agents? Trends Microbiol. 2015, 23, 653–665. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Secher, T.; Guilleminault, L.; Reckamp, K.; Amanam, I.; Plantier, L.; Heuze-Vourc’h, N. Therapeutic antibodies: A new era in the treatment of respiratory diseases? Pharmacol. Ther. 2018, 189, 149–172. [Google Scholar] [CrossRef]
- Glanville, J.; Zhai, W.; Berka, J.; Telman, D.; Huerta, G.; Mehta, G.R.; Ni, I.; Mei, L.; Sundar, P.D.; Day, G.M.; et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA 2009, 106, 20216–20221. [Google Scholar] [CrossRef] [Green Version]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Hey, A. History and Practice: Antibodies in Infectious Diseases. Microbiol. Spectr. 2015, 3, AID-0026-2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Irani, V.; Guy, A.J.; Andrew, D.; Beeson, J.G.; Ramsland, P.A.; Richards, J.S. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol. Immunol. 2015, 67, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Bensalem, A.; Ternant, D. Pharmacokinetic Variability of Therapeutic Antibodies in Humans: A Comprehensive Review of Population Pharmacokinetic Modeling Publications. Clin. Pharmacokinet. 2020, 59, 857–874. [Google Scholar] [CrossRef]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, Detection, and New Options for Prevention and Treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Hotard, A.L.; Currier, M.G.; Lee, S.; Stobart, C.C.; Moore, M.L. Respiratory Syncytial Virus Attachment Glycoprotein Contribution to Infection Depends on the Specific Fusion Protein. J. Virol. 2016, 90, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, A.; Chen, Z.; Cox, K.S.; Su, H.P.; Callahan, C.; Fridman, A.; Zhang, L.; Patel, S.B.; Cejas, P.J.; Swoyer, R.; et al. A potent broadly neutralizing human RSV antibody targets conserved site IV of the fusion glycoprotein. Nat. Commun. 2019, 10, 4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Academy of Pediatrics Subcommittee on Diagnosis and Management of Bronchiolitis. Diagnosis and management of bronchiolitis. Pediatrics 2006, 118, 1774–1793. [Google Scholar] [CrossRef] [Green Version]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Domachowske, J.B.; Khan, A.A.; Esser, M.T.; Jensen, K.; Takas, T.; Villafana, T.; Dubovsky, F.; Griffin, M.P. Safety, Tolerability and Pharmacokinetics of MEDI8897, an Extended Half-life Single-dose Respiratory Syncytial Virus Prefusion F-targeting Monoclonal Antibody Administered as a Single Dose to Healthy Preterm Infants. Pediatric Infect. Dis. J. 2018, 37, 886–892. [Google Scholar] [CrossRef]
- Maas, B.; Aliprantis, A.; Wolford, D.; Fayad, G.; Vora, K.; Geng, D.; Ma, H.; Caro, L. RSV Monoclonal Antibody (MK-1654) Phase 1 Pharmacokinetics (PK) in Healthy Adults and Population PK Modeling to Support Pediatric Development. Open Forum Infect. Dis. 2018, 5, S424–S425. [Google Scholar] [CrossRef] [Green Version]
- Fortes, H.R.; von Ranke, F.M.; Escuissato, D.L.; Araujo Neto, C.A.; Zanetti, G.; Hochhegger, B.; Souza, C.A.; Marchiori, E. Recurrent respiratory papillomatosis: A state-of-the-art review. Respir. Med. 2017, 126, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Bishop, J.A.; Roden, R.B.S.; Allen, C.T.; Best, S.R.A. The PD-1 and PD-L1 pathway in recurrent respiratory papillomatosis. Laryngoscope 2018, 128, E27–E32. [Google Scholar] [CrossRef]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-Dependent Cellular Cytotoxicity Activity of a Novel Anti-PD-L1 Antibody Avelumab (MSB0010718C) on Human Tumor Cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.T.; Lee, S.; Norberg, S.M.; Kovalovsky, D.; Ye, H.; Clavijo, P.E.; Hu-Lieskovan, S.; Schlegel, R.; Schlom, J.; Strauss, J.; et al. Safety and clinical activity of PD-L1 blockade in patients with aggressive recurrent respiratory papillomatosis. J. Immunother. Cancer 2019, 7, 119. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- World Health Organization. Global Influenza Strategy 2019–2030. Available online: https://www.who.int/influenza/global_influenza_strategy_2019_2030/en/ (accessed on 30 October 2020).
- Zhou, F.; Trieu, M.C.; Davies, R.; Cox, R.J. Improving influenza vaccines: Challenges to effective implementation. Curr. Opin. Immunol. 2018, 53, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Toots, M.; Plemper, R.K. Next-generation direct-acting influenza therapeutics. Transl. Res. J. Lab. Clin. Med. 2020, 220, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Wollacott, A.M.; Boni, M.F.; Szretter, K.J.; Sloan, S.E.; Yousofshahi, M.; Viswanathan, K.; Bedard, S.; Hay, C.A.; Smith, P.F.; Shriver, Z.; et al. Safety and Upper Respiratory Pharmacokinetics of the Hemagglutinin Stalk-Binding Antibody VIS410 Support Treatment and Prophylaxis Based on Population Modeling of Seasonal Influenza A Outbreaks. EBioMedicine 2016, 5, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, E.; Sloan, S.; Narayan, K.; Hay, C.A.; Smith, P.; Engler, F.; Jeeninga, R.; Smits, S.; Trevejo, J.; Shriver, Z.; et al. Safety and efficacy of monoclonal antibody VIS410 in adults with uncomplicated influenza A infection: Results from a randomized, double-blind, phase-2, placebo-controlled study. EBioMedicine 2019, 40, 574–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedeyn, K.; Saelens, X. New antibody-based prevention and treatment options for influenza. Antivir. Res. 2019, 170, 104562. [Google Scholar] [CrossRef]
- Oldach, D.; Narayan, K.; Schaefers, K.; Sloan, S.; Smith, P.; Bliss, R.; Arbrough, J.; Shriver, Z. A global, randomised, dou-ble-blind, placebo-controlled study evaluating safety and efficacy of vis410 in combination with oseltamivir versus oselta-mivir alone in hospitalized adults with influenza a requiring oxygen. In Option X for the Control of Influenza; International Society for Influenza and other Respiratory Virus Diseases (ISIRV): Singapore, 2019; Volume 11754. [Google Scholar]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351. [Google Scholar] [CrossRef]
- Polidoro, R.B.; Hagan, R.S.; de Santis Santiago, R.; Schmidt, N.W. Overview: Systemic Inflammatory Response Derived from Lung Injury Caused by SARS-CoV-2 Infection Explains Severe Outcomes in COVID-19. Front. Immunol. 2020, 11, 1626. [Google Scholar] [CrossRef]
- Sariol, A.; Perlman, S. Lessons for COVID-19 Immunity from Other Coronavirus Infections. Immunity 2020. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045 e1039. [Google Scholar] [CrossRef]
- Lega, S.; Naviglio, S.; Volpi, S.; Tommasini, A. Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19. Vaccines 2020, 8, 224. [Google Scholar] [CrossRef]
- Poduri, R.; Joshi, G.; Jagadeesh, G. Drugs targeting various stages of the SARS-CoV-2 life cycle: Exploring promising drugs for the treatment of Covid-19. Cell. Signal. 2020. [Google Scholar] [CrossRef]
- Guarali, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020, 2, 474–484. [Google Scholar] [CrossRef]
- Gritti, G.; Raimondi, F.; Ripamonti, D.; Riva, I.; Landi, F.; Alborghetti, L.; Frigeni, M.; Damiani, M.; Micò, C.; Fagiuoli, S.; et al. IL-6 signalling pathway inactivation with siltuximab in patients with COVID-19 respiratory failure: An observational cohort study. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Emergency Use Authorization. Available online: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#covid19euas (accessed on 12 January 2021).
- Dasaraju, P.V.; Liu, C. Infections of the respiratory system. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Gavelston: Gavelston, TX, USA, 1996. [Google Scholar]
- GBD Lower Respiratory Infections Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, G. The definition and classification of pneumonia. Pneumonia 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T.; Opal, S.M. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 2009, 374, 1543–1556. [Google Scholar] [CrossRef]
- Hendricks, K.; Vieira, A.R.; Marston, C.K. Anthrax. Available online: https://wwwnc.cdc.gov (accessed on 30 October 2020).
- Frankel, A.E.; Kuo, S.R.; Dostal, D.; Watson, L.; Duesbery, N.S.; Cheng, C.P.; Cheng, H.J.; Leppla, S.H. Pathophysiology of anthrax. Front. Biosci. 2009, 14, 4516–4524. [Google Scholar] [CrossRef] [Green Version]
- Casadevall, A. Antibodies for defense against biological attack. Nat. Biotechnol. 2002, 20, 114. [Google Scholar] [CrossRef]
- Migone, T.S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, B.J.; Shadiack, A.M.; Carpenter, S.; Sanford, D.; Henning, L.N.; Gonzales, N.; O’Connor, E.; Casey, L.S.; Serbina, N.V. Obiltoxaximab Prevents Disseminated Bacillus anthracis Infection and Improves Survival during Pre- and Postexposure Prophylaxis in Animal Models of Inhalational Anthrax. Antimicrob. Agents Chemother. 2016, 60, 5796–5805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, C.F.; Leach, T.S.; King, A.; Guttendorf, R. Safety, Pharmacokinetics, and Immunogenicity of Obiltoxaximab After Intramuscular Administration to Healthy Humans. Clin. Pharmacol. Drug Dev. 2018, 7, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grgurich, P.E.; Hudcova, J.; Lei, Y.; Sarwar, A.; Craven, D.E. Management and prevention of ventilator-associated pneumonia caused by multidrug-resistant pathogens. Expert Rev. Respir. Med. 2012, 6, 533–555. [Google Scholar] [CrossRef]
- Reyes-Robles, T.; Torres, V.J. Staphylococcus aureus Pore-Forming Toxins. Clin. Microbiol. Rev. 2017, 409, 121–144. [Google Scholar] [CrossRef]
- Spaan, A.N.; van Strijp, J.A.G.; Torres, V.J. Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 2017, 15, 435–447. [Google Scholar] [CrossRef]
- Diep, B.A.; Hilliard, J.J.; Le, V.T.; Tkaczyk, C.; Le, H.N.; Tran, V.G.; Rao, R.L.; Dip, E.C.; Pereira-Franchi, E.P.; Cha, P.; et al. Targeting Alpha Toxin To Mitigate Its Lethal Toxicity in Ferret and Rabbit Models of Staphylococcus aureus Necrotizing Pneumonia. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Cohen, T.S.; Shi, Y.; Datta, V.; Hilliard, J.J.; Tkaczyk, C.; Suzich, J.; Stover, C.K.; Sellman, B.R. MEDI4893* Promotes Survival and Extends the Antibiotic Treatment Window in a Staphylococcus aureus Immunocompromised Pneumonia Model. Antimicrob. Agents Chemother. 2015, 59, 4526–4532. [Google Scholar] [CrossRef] [Green Version]
- Hilliard, J.J.; Datta, V.; Tkaczyk, C.; Hamilton, M.; Sadowska, A.; Jones-Nelson, O.; O’Day, T.; Weiss, W.J.; Szarka, S.; Nguyen, V.; et al. Anti-alpha-toxin monoclonal antibody and antibiotic combination therapy improves disease outcome and accelerates healing in a Staphylococcus aureus dermonecrosis model. Antimicrob. Agents Chemother. 2015, 59, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, B.; Sanchez, M.G.; Eggimann, P.; Dequin, P.; Laterre, P.; Huberlant, V.; Escudero, D.; Boulain, T.; Bretonniere, C.; Pugin, J.; et al. Suvratoxumab Reduces Staphylococcus Aureus Pneumonia in High-Risk ICU Patients: Results of the SAATELLITE Study. In Proceedings of the American Thoracic Society 2019 International Conference, Dallas, TX, USA, 17–22 May 2019. [Google Scholar]
- Drumm, J.E.; Mielach, A.; Aridis Pharmaceuticals, Inc. Presents Positive Phase 2a Safety and Efficacy Data of Salvecin™ (AR-301) in Patients with Severe Pneumonia Caused by Staphylococcus aureus During the 2017 ASM Microbe Congress. Newswire, P., Ed.; 2017. Available online: https://www.prnewswire.com/ (accessed on 13 February 2021).
- Sadikot, R.T.; Blackwell, T.S.; Christman, J.W.; Prince, A.S. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 1209–1223. [Google Scholar] [CrossRef] [Green Version]
- Que, Y.A.; Lazar, H.; Wolff, M.; Francois, B.; Laterre, P.F.; Mercier, E.; Garbino, J.; Pagani, J.L.; Revelly, J.P.; Mus, E.; et al. Assessment of panobacumab as adjunctive immunotherapy for the treatment of nosocomial Pseudomonas aeruginosa pneumonia. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1861–1867. [Google Scholar] [CrossRef] [Green Version]
- Ryder, C.; Byrd, M.; Wozniak, D.J. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Current Opin. Microbiol. 2007, 10, 644–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberger, P.A.; Bush, R.K.; Demain, J.G.; Luong, A.; Slavin, R.G.; Knutsen, A.P. Allergic bronchopulmonary aspergillosis. J. Allergy Clin. Immunol. Pract. 2014, 2, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Soeda, S.; Kono, Y.; Tsuzuki, R.; Yamawaki, S.; Katsube, O.; To, M.; To, Y. Allergic bronchopulmonary aspergillosis successfully treated with benralizumab. J. Allergy Clin. Immunol. Pract. 2019, 7, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Yu, X.Q.; François, B.; Eggimann, P.; Huberlant, V.; Dequin, P.F.; Valia, J.C.; Ali, S.O.; Jensen, K.; Colbert, S.; et al. Interim pharmacokinetic analysis from the SAATELLITE Phase 2 Clinical Trial of Suvratoxumab (MEDI4893), an extended half-life monoclonal antibody against Staphylococcus aureus alpha toxin. In Proceedings of the 28th ECCMID, Madrid, Spain, 21–24 April 2018. [Google Scholar]
- Griffin, M.P.; Khan, A.A.; Esser, M.T.; Jensen, K.; Takas, T.; Kankam, M.K.; Villafana, T.; Dubovsky, F. Safety, Tolerability, and Pharmacokinetics of MEDI8897, the Respiratory Syncytial Virus Prefusion F-Targeting Monoclonal Antibody with an Extended Half-Life, in Healthy Adults. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Ternant, D.; Azzopardi, N.; Raoul, W.; Bejan-Angoulvant, T.; Paintaud, G. Influence of Antigen Mass on the Pharmacokinetics of Therapeutic Antibodies in Humans. Clin. Pharmacokinet. 2019, 58, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Robbie, G.J.; Zhao, L.; Mondick, J.; Losonsky, G.; Roskos, L.K. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob. Agents Chemother. 2012, 56, 4927–4936. [Google Scholar] [CrossRef] [Green Version]
- Mould, D.R.; Upton, R.N. Basic concepts in population modeling, simulation, and model-based drug development-part 2: Introduction to pharmacokinetic modeling methods. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e38. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Wellcome Trust UK Department of Health: London, UK, 2016. [Google Scholar]
- Alonzo, F., 3rd; Benson, M.A.; Chen, J.; Novick, R.P.; Shopsin, B.; Torres, V.J. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 2012, 83, 423–435. [Google Scholar] [CrossRef] [Green Version]
- DiGiandomenico, A.; Sellman, B.R. Antibacterial monoclonal antibodies: The next generation? Curr. Opin. Microbiol. 2015, 27, 78–85. [Google Scholar] [CrossRef]
- Buyel, J.F.; Twyman, R.M.; Fischer, R. Very-large-scale production of antibodies in plants: The biologization of manufacturing. Biotechnol. Adv. 2017, 35, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, E.; Friede, M.; Sheikh, M.; Torvaldsen, S. Therapeutic antibodies for infectious diseases. Bull. World Health Organ. 2017, 95, 235–237. [Google Scholar] [CrossRef]
- Simon, M.S.; Sfeir, M.M.; Calfee, D.P.; Satlin, M.J. Cost-effectiveness of ceftazidime-avibactam for treatment of carbapenem-resistant Enterobacteriaceae bacteremia and pneumonia. Antimicrob. Agents Chemother. 2019, 63, e00897-19. [Google Scholar] [CrossRef] [PubMed]
- Secher, T.; Dalonneau, E.; Ferreira, M.; Parent, C.; Azzopardi, N.; Paintaud, G.; Si-Tahar, M.; Heuze-Vourc’h, N. In a murine model of acute lung infection, airway administration of a therapeutic antibody confers greater protection than parenteral administration. J. Control. Release 2019, 303, 24–33. [Google Scholar] [CrossRef] [PubMed]


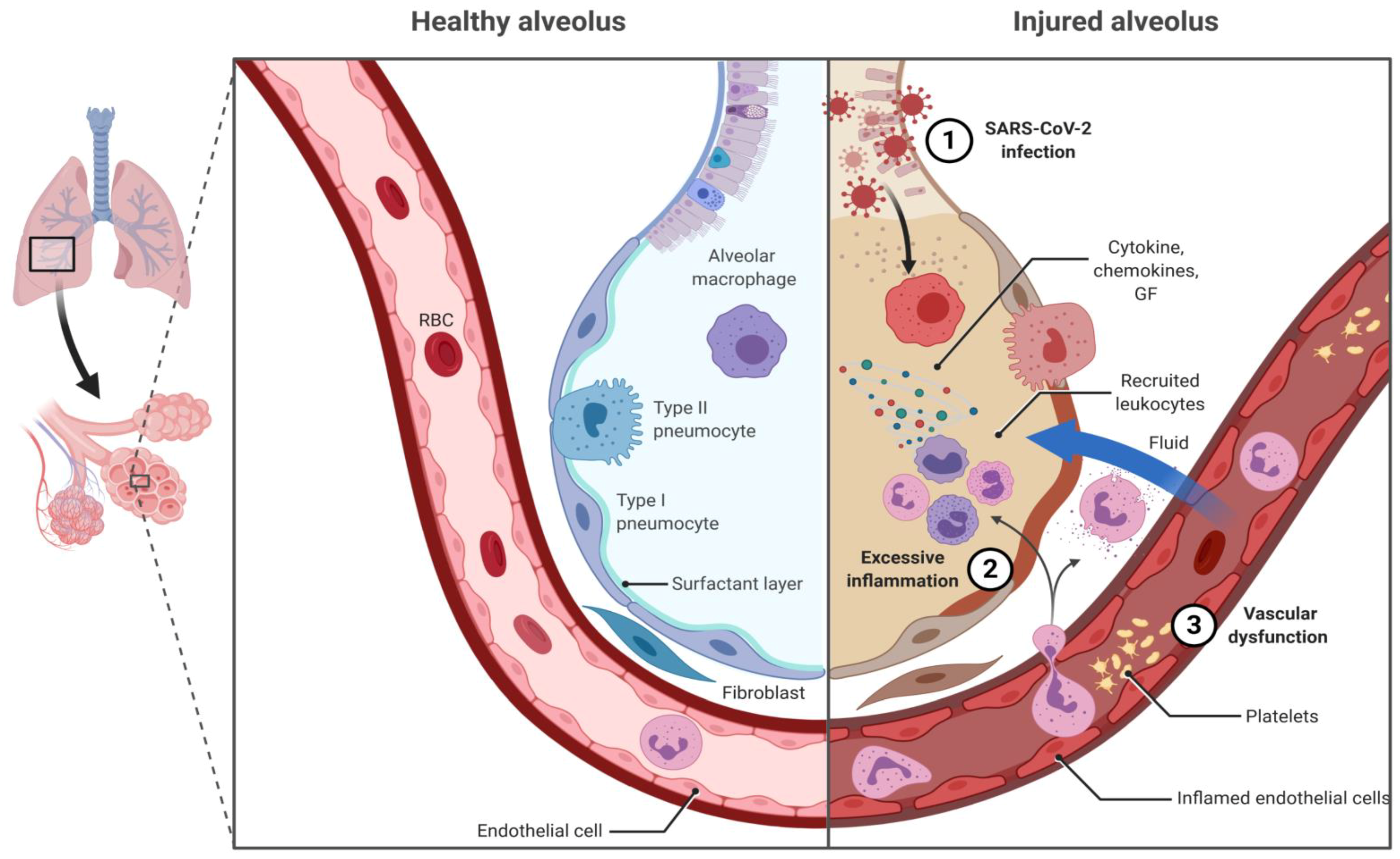
{kind=link}
{kind=link}
| Indication | Generic Name | Sponsoring Company | Target | Development Stage |
|---|---|---|---|---|
| Respiratory syncytial virus | Palivizumab (synagis) | AstraZeneca | F-protein | Approved in 1998 |
| Bacillus anthracis | Raxibacumab (abthrax) | GSK | PA | Approved in 2013 |
| Obiltoxaximab (anthim) | Elusys Therapeutics | PA | Approved in 2016 |
| Indication. | Generic Name | Sponsoring Company | Target | Development Stage | NCT | Completion Date | Status |
|---|---|---|---|---|---|---|---|
| Respiratory syncytial virus | MEDI-8897 (nirsevimab) | MedImmune | F-protein | Phase 2/3 | NCT03959488, NCT03979313 | 2021–2023 | Recruiting |
| MK-1654 | Merck | F-protein | Phase 2 | NCT03524118 | 2022 | Active | |
| Influenza A virus | VIR-2482 | Vir Biotechnology | HA | Phase 2 | NCT04033406 | 2022 | Recruiting |
| CT-P27 (antibody mixture) | Celltrion | HA | Phase 2 | NCT03511066 | 2018 | Active on Celltrion website | |
| VIS-410 | Visterra | HA | Phase 2 | NCT03040141, NCT02989194, NCT02468115 | 2018 | Active on Visterra website | |
| HPV-associated Recurrent Respiratory Papilloma | Avelumab | Merck | PD-L1 | Phase 2 | NCT02859454 | 2019 | Active |
| Pembrolizumab | Merck | PD-1 | Phase 2 | NCT02632344 | 2020 | Active, not yet recruiting |
| Strategy | Generic Name | Sponsoring Company | Target | Development Stage | NCT (Status) | Completion Date |
|---|---|---|---|---|---|---|
| ARDS-related target | IC14 | Implicit Bioscience | CD14 | Phase 2 | NCT04391309 (Active, not recruiting) | 2021 |
| Itolizumab | Biocon | CD6 | Phase 2 | NCT04475588 (Completed) | 2020 | |
| Leronlimab | CytoDyn | CCR5 | Phase 2 | NCT04343651 (Active, not recruiting), NCT04347239 (Recruiting) | 2021 | |
| CPI-006 | Corvus Pharmaceuticals | CD73 | Phase 1 | NCT04464395 (Recruiting) | 2021 | |
| Meplazumab | Jiangsu Pacific Meinuoke Bio Pharmaceutical | CD147 | Phase 1 | NCT04369586 (Recruiting) | 2020 | |
| Phase 1/2 | NCT04275245 (Recruiting) | 2020 | ||||
| Phase 2/3 | NCT04586153 (Not yet recruiting) | 2021 | ||||
| Lanadelumab | Takeda | Kallikrein | Phase 1/2 | NCT04422509 (Recruiting) | 2021 | |
| Phase 1 | NCT04460105 (Withdrawn) | 2020 | ||||
| Otilimab | GlaxoSmithKline | GM-CSF | Phase 2 | NCT04376684 (Recruiting) | 2020 | |
| Lenzilumab | Humanigen | GM-CSF | Phase 2 | NCT04583969 (Recruiting) | 2021 | |
| Phase 3 | NCT04351152 (Recruiting), NCT04534725 (Not yet recruiting) | 2020 | ||||
| Gimsilumab | Kinevant Sciences Roivant Sciences | GM-CSF | Phase 2 | NCT04351243 (Active, not recruiting) | 2020/2021 | |
| TJ003234 | I-Mab Biopharma | GM-CSF | Phase 2/3 | NCT04341116 (Recruiting) | 2021 | |
| Mavrilimumab | Kiniksa pharmaceuticals | GM-CSF-R | Phase 2 | NCT04397497 (Not yet recruiting), NCT04463004 (Recruiting), NCT04399980 (Active, not recruiting), NCT04492514 (Recruiting) | 2020–2021 | |
| Phase 2/3 | NCT04447469 (Recruiting) | 2021 | ||||
| Pamrevlumab | FibroGen | CTGF | Phase 2 | NCT04432298 (Recruiting) | 2020/2021 | |
| SNDX-6352 (axatilimab) | Syndax Pharmaceuticals | CSF-1R | Phase 2 | NCT04415073 (Suspended) | 2020 | |
| Phase 2 | NCT04344782 (Active, Not yet recruiting), NCT04275414 (Completed) | 2020 | ||||
| BDB-001 | Staidson Biopharmaceuticals | PD-L1 | Phase 2/3 | NCT04449588 (Recruiting) | 2021/2022 | |
| Nivolumab | Bristol-Myers Squibb | PD1 | Phase 2 | NCT04413838 (Active, Not yet recruiting) | 2021 | |
| Phase 2 | NCT04343144 (Active, Not yet recruiting) | 2020 | ||||
| Phase 2 | NCT04356508 (Not yet recruiting) | 2021 | ||||
| Phase 2 | NCT04356508S | 2020/2021 | ||||
| Crizanlizumab | Novartis | P-selectin | Phase 2 | NCT04435184 (Recruiting) NCT03474965 (Recruiting) | 2020 2024 | |
| CSL312 (garadacimab) | CSL Behring | Factor XIIa | Phase 2 | NCT04409509 (Recruiting) | 2020 | |
| LY3127804 | Eli Lilly | Angiopoietin 2 | Phase 2 | NCT04342897 (Terminated) | 2020 | |
| Ravulizumab | Alexion pharmaceuticals | C5 | Phase 3 | NCT04369469 (Recruiting) NCT04570397 (Not yet recruiting) | 2020/2021 | |
| Phase 4 | NCT04390464 (Recruiting) | 2021/2022 | ||||
| Secukinumab | Novartis | IL-17A | Phase 2 | NCT04403243 (Recruiting) | 2020 | |
| Canakinumab | Novartis | IL-1ß | Phase 2 | NCT04365153 (Active, Not yet recruiting) | 2020 | |
| Phase 3 | NCT04362813 (Active, not recruiting) NCT04510493 (Recruiting) | 2020 2023 | ||||
| Clazakizumab | Vitaeris | IL-6 | Phase 2 | NCT04381052 (Not yet recruiting), NCT04343989 (Recruiting), NCT04363502 (Recruiting), NCT04494724 (Recruiting), NCT04348500 (Not yet recruiting) NCT04659772 (Enrolling by invitation) | 2020/2021 | |
| NCT04348500 (Active, not recruiting) | 2021 | |||||
| Sirukumab | Janssen | IL-6 | Phase 2 | NCT04380961 (Recruiting) | 2021 | |
| Sarilumab | Regeneron Pharmaceuticals | IL-6 | Phase 1 | NCT04386239 (Active, Not yet recruiting) | 2020 | |
| Phase 2 | NCT04357860 (Not yet recruiting), NCT04357808 (Active, not recruiting), NCT04359901 (Recruiting), NCT04661527 (Recruiting) | 2020/2023 | ||||
| Phase 2/3 | NCT04315298 (Completed), NCT04324073 (Active, not recruiting), NCT04341870 (Suspended) | 2021 | ||||
| Phase 3 | NCT04327388 (Completed), NCT04345289 (Recruiting) | 2020/2021 | ||||
| Olokizumab | R-Pharm International | IL-6 | Phase 2/3 | NCT04380519 (Completed), NCT04452474 (Not yet recruiting) | 2020–2021 | |
| Tozilizumab + Sarilumab | Roche, Sanofi | IL-6 | Phase 2 | NCT04322773 (Terminated) | 2021 | |
| Siltuximab | Janssen | IL-6 | Phase 2 | NCT04329650 (Recruiting) | 2020 | |
| Tocilizumab | Roche | IL-6R | Phase 1 | NCT04560205 (Recruiting) | 2020 | |
| Phase 2 | NCT04445272 (Recruiting), NCT04479358 (Recruiting), NCT04412291 (Recruiting), NCT04331795 (Completed), NCT04377659 (Active, not recruiting), NCT04363736 (Completed), NCT04435717 (Recruiting), NCT04377503 (Active, Not yet recruiting), NCT04339712 (Recruiting), NCT04370834 (Suspended), NCT04315480 (Active, not recruiting), NCT04331808 (Active, not recruiting), NCT04317092 (Active, Not yet recruiting) NCT04363853 (Active, Not yet recruiting) | 2020–2022 | ||||
| Phase 2/3 | NCT04381936 | 2021/2031 | ||||
| Phase 3 | NCT04403685 (Terminated), NCT04423042 (Not yet recruiting), NCT04412772 (Recruiting), NCT04345445 (Not yet recruiting), NCT04424056 (Not yet recruiting), NCT04335071 (Terminated), NCT04361032 (Not yet recruiting), NCT04320615 (Completed), NCT04372186 (Active, not recruiting), NCT04356937 (Active, not recruiting), NCT04577534 (Recruiting) | 2020–2022 | ||||
| Phase 4 | NCT04377750 (Recruiting) | 2020/2021 | ||||
| Levilimab | Biocad | IL-6R | Phase 3 | NCT04397562 (Completed) | 2021 | |
| Tocilizumab + Dexamethasone | Roche | IL-6R | Phase 2 | NCT04476979 (Recruiting) | 2021 | |
| Tocilizumab + Remdesivir | Roche | IL-6R | Phase 3 | NCT04409262 (Recruiting) | 2020 | |
| Tocilizumab + Favipiravir | Roche | IL-6R | NA | NCT04310228 (Recruiting) | 2020 | |
| Tocilizumab +Siltuximab | Roche, Janssen | IL-6 + IL-6R | Phase 3 | NCT04330638 (Active, not recruiting) | 2020 | |
| Tocilizumab + Pembrolizumab | Roche, Merck | IL-6R + PD-1 | Phase 2 | NCT04335305 (Recruiting) | 2020 | |
| Tocilizumab + Hydroxychloroquine + Azithromycin | Roche | IL-6R | Phase 2 | NCT04332094 (Recruiting) | 2020 | |
| Heparin + Tocilizumab | Roche | IL-6R | Phase 3 | NCT04600141 (Not yet recruiting) | 2021 | |
| BMS-986253 | Bristol-Myers Squibb | IL-8 | Phase 2 | NCT04347226 (Recruiting) | 2021/2022 | |
| Risankizumab | Jiangsu Pacific Meinuoke Bio Pharmaceutical | IL-23 | Phase 2 | NCT04583956 (Recruiting) | 2021 | |
| Phase 2/3 | NCT04586153 (Not yet recruiting) | 2021 | ||||
| MSTT1041A (astegolimab) | Genetech | IL-33 | Phase 2 | NCT04386616 (Recruiting) | 2020 | |
| Emapalumab | Swedish Orphan BIovitrum | IFN-γ | Phase 2/3 | NCT04324021 (Terminated) | 2020 | |
| IFX1 (vilobelimab) | InflaRx GmbH | C5a | Phase2/3 | NCT04333420 (Recruiting) | 2021 | |
| Infliximab | MSD | TNF-alpha | Phase 2 | NCT04425538 (Recruiting) | 2020 | |
| Remdesivir + infliximab | MSD | TNF-alpha | Phase 3 | NCT04593940 (Recruiting) | 2021 | |
| Eculizumab | Alexion pharmaceuticals | C5 | Phase 2 | NCT04346797 (Recruiting) | 2020 | |
| AK119 | Akesobio | CD73 | Phase 1 | NCT04516564 (Recruiting) | 2021 | |
| CERC-002 | Cerecor | LIGHT | Phase 2 | NCT04412057 (Recruiting) | 2020 | |
| Glenzocimab | Acticor Biotech | Platelet glycoprotein VI | Phase 2 | NCT04659109 (Not yet recruiting) | 2021 | |
| VIB7734 | Viela Bio | ILT7 | Phase 1 | NCT04526912 (Recruiting) | 2021 | |
| NGM621 | ngmbio | C3 | Phase 1/2 | NCT04582318 (Recruiting) | 2021 | |
| EB05 | Edesa Biotech | TLR4 | Phase 2/3 | NCT04401475 (Not yet recruiting) | 2021 | |
| Avdoralimab | Innate Pharma | C5aR | Phase 2 | NCT04371367 (Recruiting) | 2020 | |
| Virus-related target | BRII-196 | Brii biosciences | S protein | Phase 1 | NCT04479631 (Active, not recruiting) | 2021 |
| BRII-198 | S protein | Phase 1 | NCT04479644 (Active, not recruiting) | 2021 | ||
| JS016 | Shanghai Junshi Bioscience Co., Ltd. | S protein | Phase 1 | NCT04441918 (Recruiting) | 2020 | |
| LY-CoV555, LY3819253 | Eli Lilly | S protein | Phase 1 | NCT04411628 (Completed), NCT04537910 (Active, not recruiting) | 2020 | |
| Phase 2 | NCT04427501 (Recruiting) | 2020 | ||||
| Phase 2/3 | NCT04518410 (Recruiting) | 2021 | ||||
| Phase 3 | NCT04497987 (Recruiting), NCT04501978 (Active, not recruiting) | 2021 | ||||
| Phase 4 | NCT04656691 (Not yet recruiting) | 2021 | ||||
| REGN-COV2 (REGN10933 + REGN10987) | Regeneron Pharmaceuticals | S protein | Phase 1 | NCT04519437 (Active, not recruiting) | 2021 | |
| Phase 1/2 | NCT04425629 (Recruiting), NCT04426695 (Recruiting) | 2020–2021 | ||||
| Phase 2 | NCT04666441 (Not yet recruiting) | 2021 | ||||
| Phase 3 | NCT04452318 (Recruiting) | 2021 | ||||
| SCTA01 | Sinocelltech | S protein | Phase 1 | NCT04483375 (Recruiting) | 2021 | |
| Phase 2/3 | NCT04644185 (Not yet recruiting) | 2021 | ||||
| STI-2020 (COVI-AMG™) | Sorrento Therapeutics | S protein | Phase 1/2 | NCT04584697 (Recruiting) | 2021 | |
| COVI-GUARD (STI-1499) | Sorrento Therapeutics | S protein | Phase 1 | NCT04454398 (Recruiting) | 2021 | |
| TY027 | Tychan Pte Ltd. | S protein | Phase 1 | NCT04429529 (Active, not recruiting) | 2020 | |
| Phase 3 | NCT04649515 (Not yet recruiting) | 2021 | ||||
| BGB-DXP593 | BeiGene | S protein | Phase 1 | NCT04532294 (Recruiting) | 2021 | |
| Phase 2 | NCT04551898 (Not yet recruiting) | 2021 | ||||
| VIR-7831 | Vir Biotechnology—GlaxoSmithKline | S protein | Phase 2/3 | NCT04545060 (Recruiting) | 2021 | |
| MW33 | Mabwell Bioscience | S protein | Phase 1 | NCT04533048 (Active, not recruiting) | 2020 | |
| Phase 2 | NCT04627584 (Not yet recruiting) | 2021 | ||||
| 47D11 | AbbVie | S protein | Phase 1 | NCT04644120 (Not yet recruiting) | 2021 | |
| AZD7442 (AZD8895 + AZD1061) | AstraZeneca | S protein | Phase 1 | NCT04507256 (Active, not recruiting) | 2021 | |
| Phase 3 | NCT04625725 (Recruiting), NCT04625972 (Recruiting) | 2022 | ||||
| LY3832479 | Junshi/Eli Lilly | Phase 2/3 | NCT04441931 (Completed) | 2020 | ||
| HFB30132A | Hifibio | S protein | Phase1 | NCT04590430 (Recruiting) | 2021 | |
| BI767551 DZIF-10c | Boehringer Ingelheim | Phase 1/2 | NCT04631705 (Not yet recruiting) NCT04631666 (Not yet recruiting) | 2021 | ||
| HLX71 fusion protein | Hengenix biotech/Henlius | S protein | Phase1 | NCT04583228 (Not yet recruiting) | 2021 | |
| HLX70 | Hengenix biotech/Henlius | S protein | Phase 1/2 | NCT04561076 (Not yet recruiting) | ||
| AMD03820 | Olygo Bioservice | Phase 1/2 | NCT04592549 (Recruiting) | 2021 | ||
| CT-P59 | Celltrion | S protein | Phase 1/2 | NCT04525079 (Recruiting) | 2020 | |
| Phase 2/3 | NCT04602000 (Recruiting) | 2021 |
| Indication | Generic Name | Sponsoring Company | Target | Development Stage | NCT | Completion Date | Status |
|---|---|---|---|---|---|---|---|
| Staphylococcus aureus | MEDI4893 | AstraZeneca | α-toxin | Phase 2 | NCT02296320 | 2018 | Active on AZ pipeline |
| AR-301 | Aridis Pharmaceuticals | α-toxin | Phase 3 | NCT03816956 | 2020 | Active on Aridis pipeline | |
| Bacillus anthracis | Obiltoxaximab (anthim) | Elusys Therapeutics | PA | Phase 4 | NCT03088111 | 2020 | |
| Pseudomonas aeruginosa | AR-101 (aerumab) | Aridis Pharmaceuticals | LPS | Phase 2 | NCT00851435 | 2009 | Active on Aridis pipeline |
| AR-105 (aerucin) | Aridis Pharmaceuticals | Alginate | Phase2 | NCT03027609 | 2019 | Active on Aridis pipeline |
| Indication | Generic Name | Sponsoring Company | Target | Development Stage | NCT | Completion Date | Status |
|---|---|---|---|---|---|---|---|
| Aspergillus fumigatus | Benralizumab | MedImmune | IL-5Rα | Phase 4 | NCT04108962 | 2022 | Recruiting |
| Dupilumab | Regeneron Pharmaceuticals | IL-4/IL-13R | Phase 3 | NCT04442269 | 2023 | Active, not yet recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayor, A.; Chesnay, A.; Desoubeaux, G.; Ternant, D.; Heuzé-Vourc’h, N.; Sécher, T. Therapeutic Antibodies for the Treatment of Respiratory Tract Infections—Current Overview and Perspectives. Vaccines 2021, 9, 151. https://doi.org/10.3390/vaccines9020151
Mayor A, Chesnay A, Desoubeaux G, Ternant D, Heuzé-Vourc’h N, Sécher T. Therapeutic Antibodies for the Treatment of Respiratory Tract Infections—Current Overview and Perspectives. Vaccines. 2021; 9(2):151. https://doi.org/10.3390/vaccines9020151
Chicago/Turabian StyleMayor, Alexie, Adélaïde Chesnay, Guillaume Desoubeaux, David Ternant, Nathalie Heuzé-Vourc’h, and Thomas Sécher. 2021. "Therapeutic Antibodies for the Treatment of Respiratory Tract Infections—Current Overview and Perspectives" Vaccines 9, no. 2: 151. https://doi.org/10.3390/vaccines9020151