In Vitro Production and Immunogenicity of a Clostridium difficile Spore-Specific BclA3 Glycopeptide Conjugate Vaccine

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Production of Spores
2.3. Recombinant Expression and Purification of SgtA
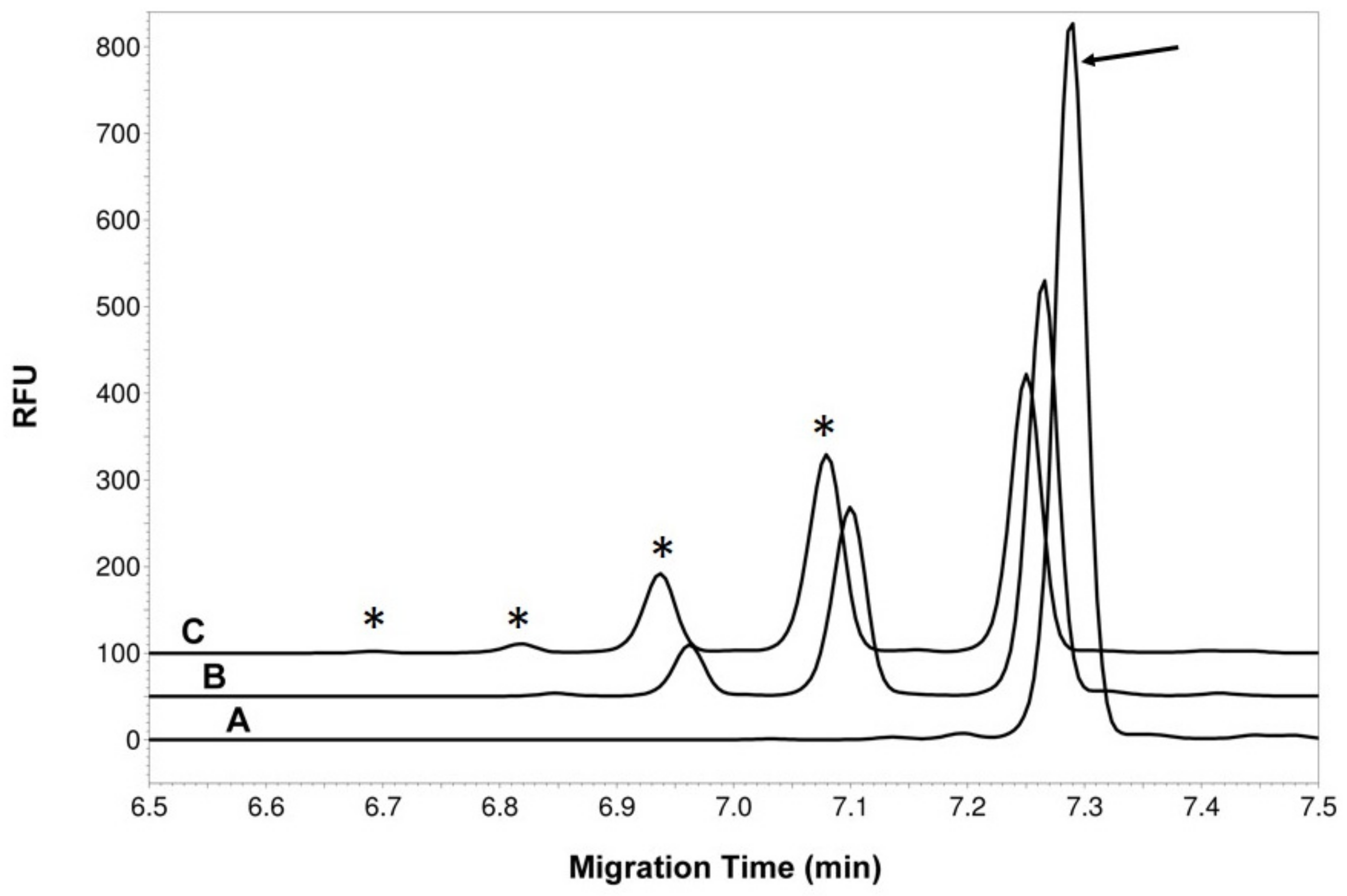
2.4. SgtA Glycosyltransferase Assay
2.5. BclA3 Peptide and Glycopeptide Conjugate Production
2.6. NMR Spectroscopy
2.7. KLH-Conjugate Production
2.8. Biotin-Peptide and Biotin-Glycopeptide Production
2.9. Animals
2.10. Immunization and Serum Preparation
2.11. SDS-PAGE Analysis and Western Blotting of Spore Extracts
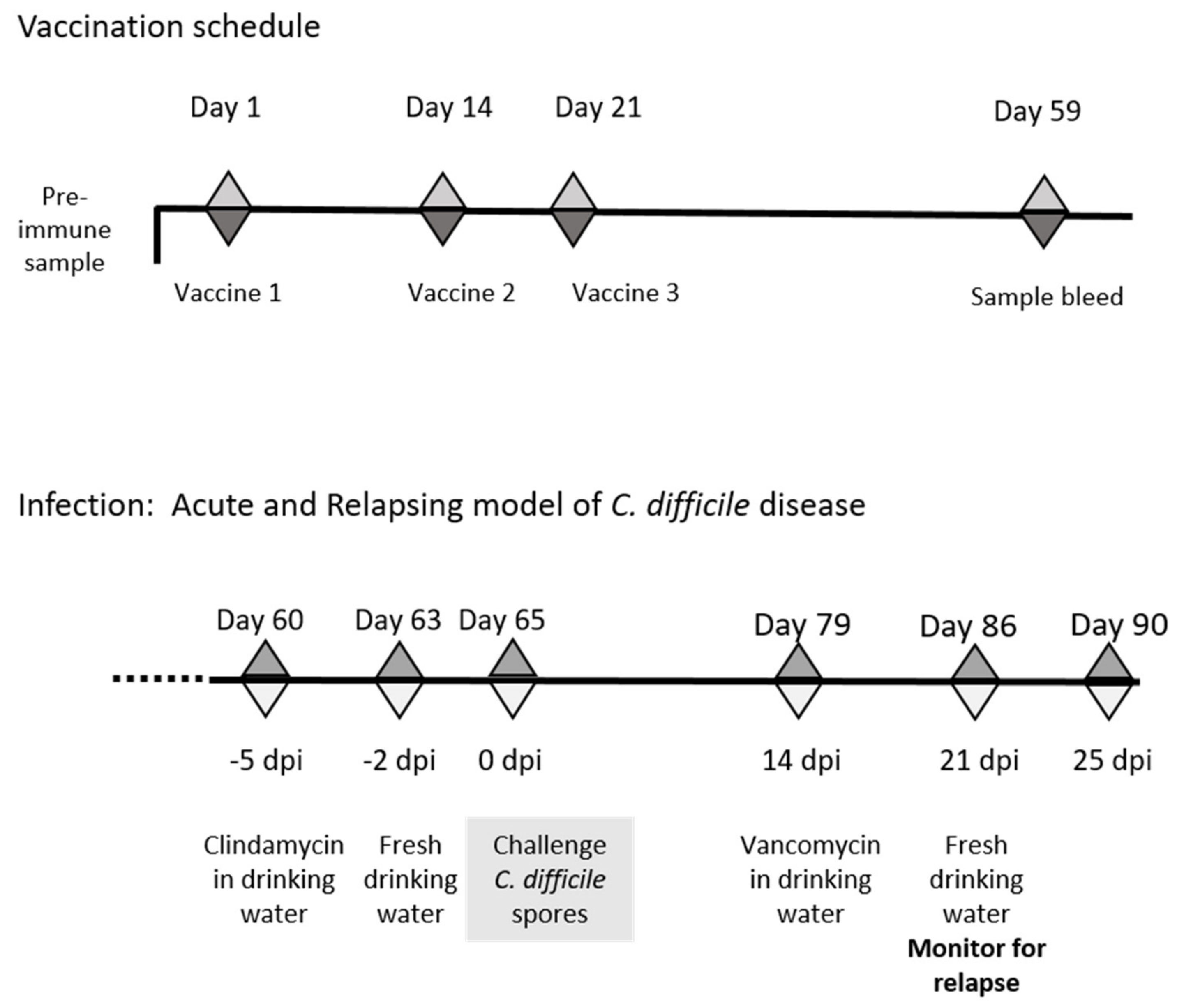
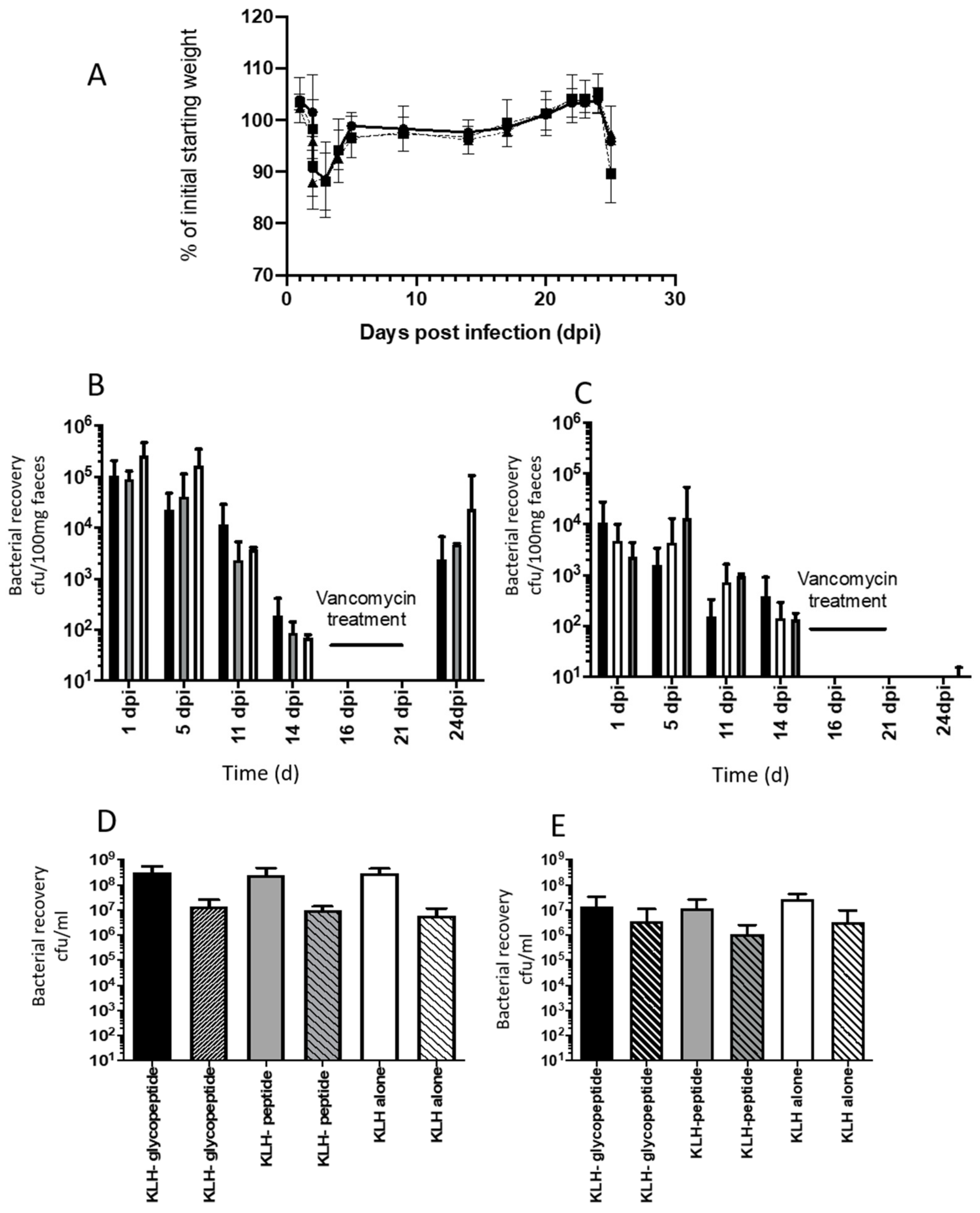
2.12. Evaluation of Vaccine Mediated Protection in the Recurrent Mouse Models of C. difficile Disease
2.13. ELISA Assays
3. Results
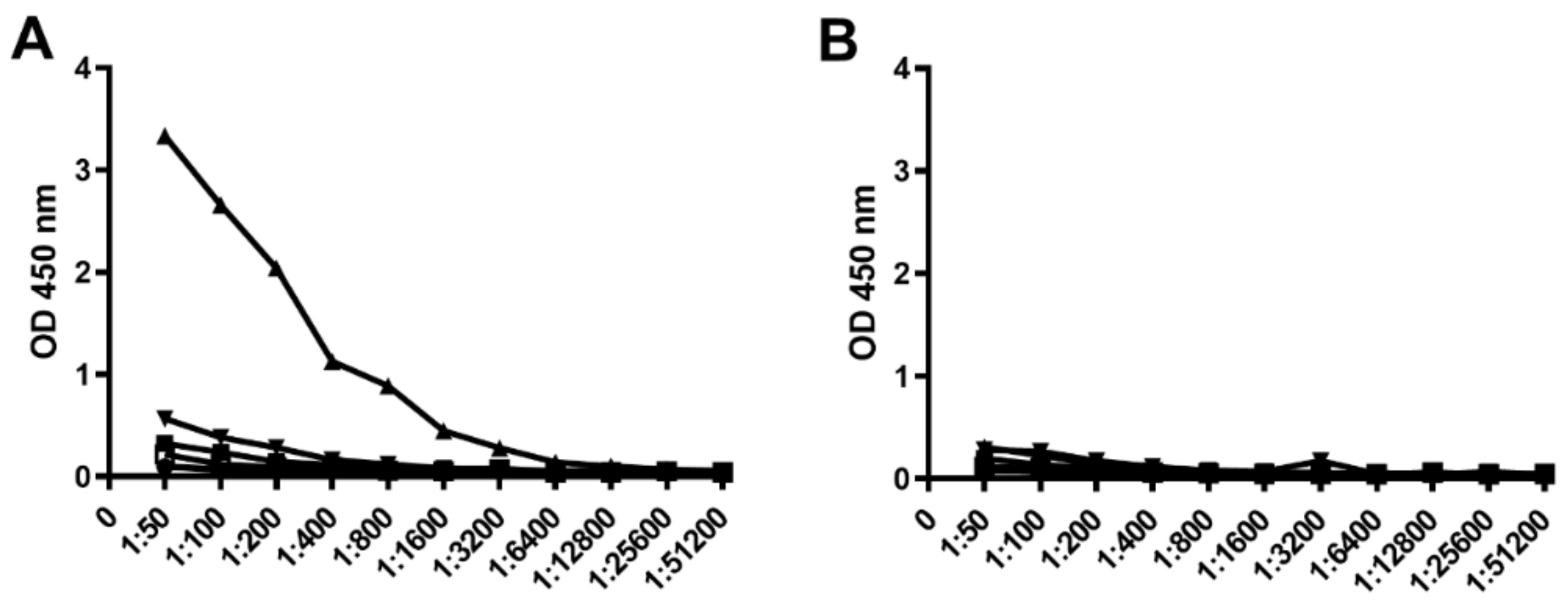
3.1. Immunogenicity of BclA3 Spore Glycoprotein
3.2. In Silico Analysis of BclA3 Glycoprotein and Selection of Synthetic Peptides
3.3. Recombinant Expression of SgtA Glycosyltransferase and in vitro Assay Optimization
3.4. Conjugation of BclA3 Peptide and BclA3 Glycopeptide to KLH
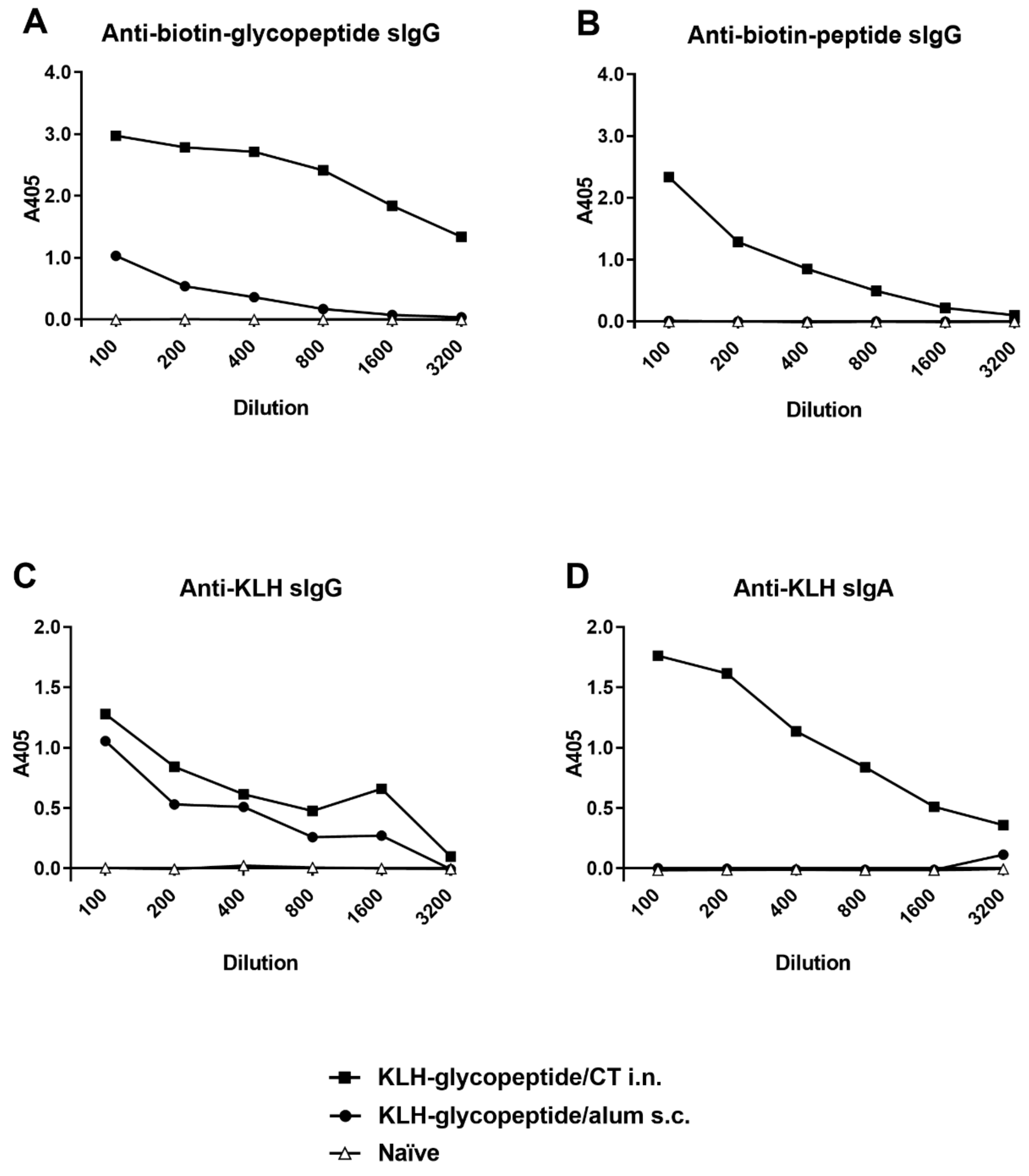
3.5. Evaluation of Peptide Antigenicity
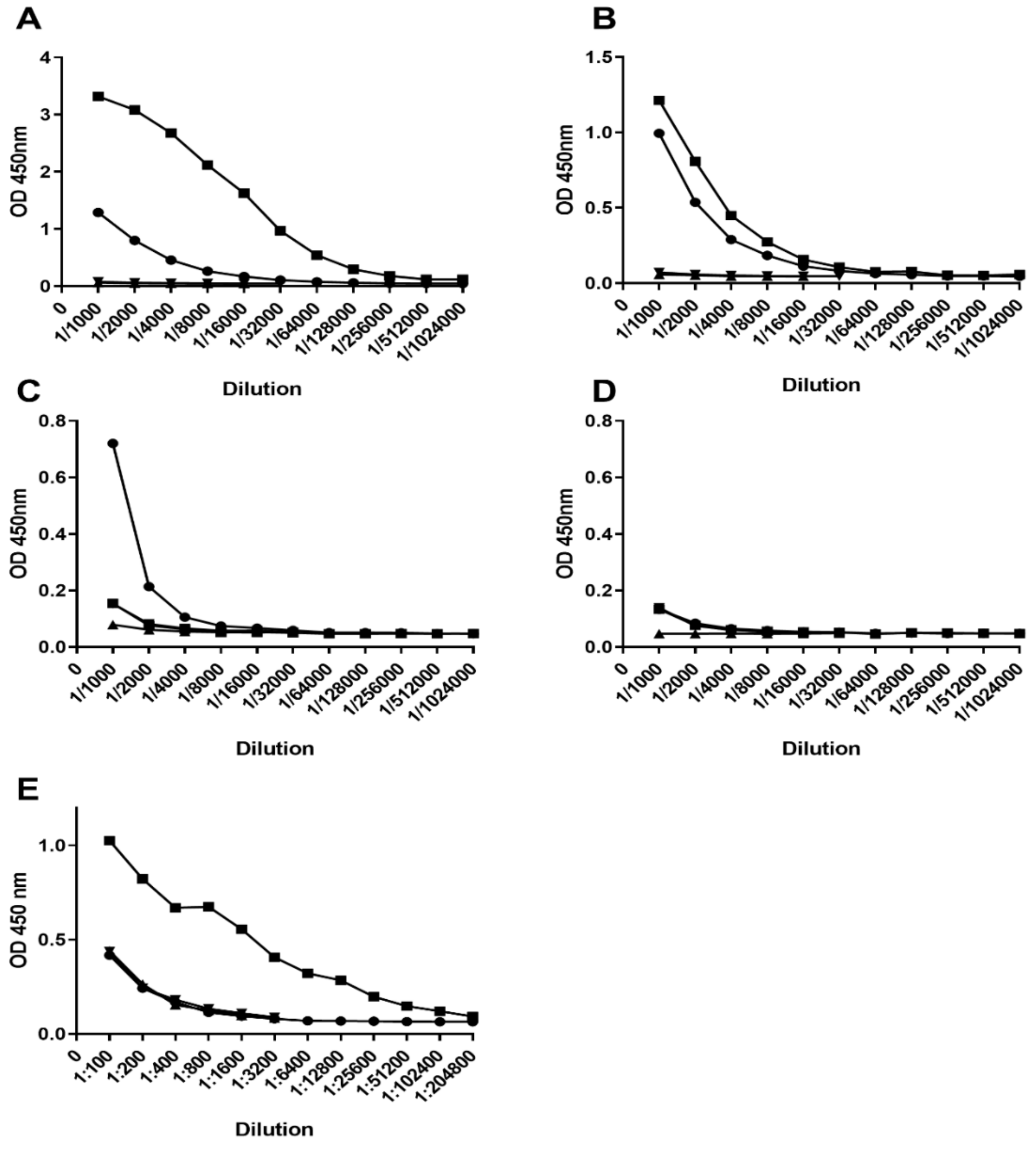
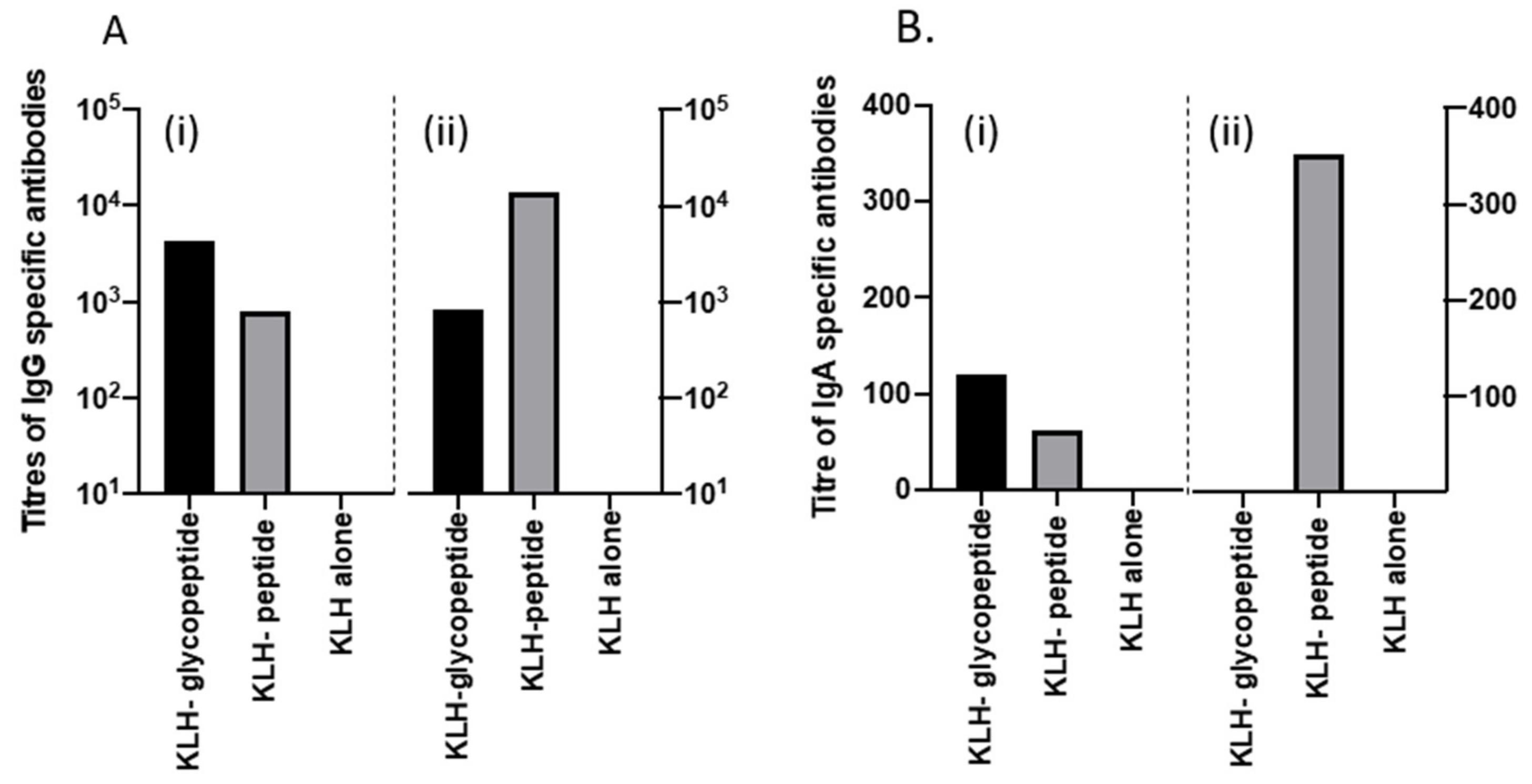
3.6. KLH-Conjugate Animal Immunization
3.7. Recurrent Mouse Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: new developments in epidiemiology and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Bouza, E. Consequences of Clostridium difficile infection: understanding the healthcare burden. Clin. Microbiol. Infect. 2012, 18 (Suppl. 6), 5–12. [Google Scholar]
- Deakin, L.J.; Clare, S.; Fagan, R.P.; Dawson, L.F.; Pickard, D.J.; West, M.R.; Wren, B.W.; Fairweather, N.F.; Dougan, G.; Lawley, T.D. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect. Immun. 2012, 80, 2704–2711. [Google Scholar]
- Ananthakrishnan, A.N. Clostridium difficile infection: epidiemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Redmond, C.; Baillie, L.W.; Hibbs, S.; Moir, A.J.; Moir, A. Identification of proteins in the exosporium of Bacillus anthracis. Microbiology 2004, 150, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylvestre, P.; Couture-Tosi, E.; Mock, M. A collagen-like glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol. Microbiol. 2002, 45, 169–178. [Google Scholar] [CrossRef]
- Paredes-Sabja, D.; Shen, A.; Sorg, J.A. Clostridium difficile spore biology: sporulation, germination and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Lawley, T.D.; Croucher, N.J.; Yu, L.; Clare, S.; Sebaihia, M.; Goulding, D.; Pickard, D.J.; Parkhill, J.; Choudhary, J.; Dougan, G. Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J. Bacteriol. 2009, 191, 5377–5386. [Google Scholar] [CrossRef] [Green Version]
- Permpoonpattana, P.; Phetcharaburanin, J.; Mikelsone, A.; Dembek, M.; Tan, S.; Brisson, M.C.; La Ragione, R.; Brisson, A.R.; Fairweather, N.; Hong, H.A.; et al. Functional characterization of Clostridium difficile spore coat proteins. J. Bacteriol. 2013, 195, 1492–1503. [Google Scholar] [CrossRef] [Green Version]
- Abhyankar, W.; Hossain, A.H.; Djajasaputra, A.; Permpoonpattana, P.; Ter Beek, A.; Dekker, H.L.; Cutting, S.M.; Brul, S.; de Koning, L.J.; de Koster, C.G. In pursuit of protein targets: proteomic characterisation of bacterial spore outer layers. J. Proteomic. Res. 2013, 12, 4507–4521. [Google Scholar] [CrossRef]
- Permpoonpattana, P.; Tolls, E.H.; Nadem, R.; Tan, S.; Brisson, A.; Cutting, S.M. Surface layers of Clostridium difficile endospores. J. Bacteriol. 2011, 193, 6461–6470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.N.; Tamayo, R.; McBride, S.M. A novel regulator controls Clostridium difficile sporulation, motility and toxin production. Mol. Micro. 2016, 100, 954–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.N.; Anjuwon-Foster, B.R.; McBride, S.M. RstA is a major regulator of Clostridioides difficile toxin production and motility. Mbio. 2019, 10, e01991-18. [Google Scholar] [CrossRef] [PubMed]
- Strong, P.C.; Fulton, K.M.; Aubry, A.; Foote, S.; Twine, S.M.; Logan, S.M. Identification and characterization of glycoproteins on the spore surface of Clostridium difficile. J. Bacteriol. 2014, 196, 2627–2637. [Google Scholar] [CrossRef] [Green Version]
- Pizarro-Guajardo, M.; Calderón-Romero, P.; Castro-Córdova, P.; Mora-Uribe, P.; Paredes-Sabja, D. Ultrastructural variability of the exosporium layer of Clostridium difficile spores. Appl. Environ. Microbiol. 2016, 82, 2202–2209. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, M.H.; Gerding, D.N.; Poxton, I.R.; Kelly, C.; Nathan, R.; Birch, T.; Cornely, O.A.; Rahav, G.; Bouza, E.; Lee, C.; et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. New Eng. J. Med. 2017, 376, 305–317. [Google Scholar] [CrossRef]
- Simeon, R.; Jiang, M.; Chamoun-Emanuelli, A.M.; Yu, H.; Zhang, Y.; Meng, R.; Peng, Z.; Jakana, J.; Zhang, J.; Feng, H.; et al. Selection and characterisation of ultrahigh potency designed ankyrin repeat protein inhibitors of C. difficile toxin B. PLoS Biol. 2019, 17, e3000311. [Google Scholar]
- Hussack, G.; Ryan, S.; van Faassen, H.; Rossotti, M.; MacKenzie, C.R.; Tanha, J. Neutralization of Clostridium difficile toxin B with VHH-Fc fusions targeting the delivery and CROPs domains. PLoS Biol. 2018, 13, e0208978. [Google Scholar] [CrossRef]
- Hong, H.A.; Ferreira, W.T.; Hosseini, S.; Anwar, S.; Hitri, K.; Wilkinson, A.J.; Vahjen, W.; Zentek, J.; Soloviev, M.; Cutting, S.M. The spore coat protein cotE facilitates host colonization by Clostridium difficile. J. Infect Dis. 2017, 216, 1452–1459. [Google Scholar] [CrossRef]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Carter, G.P.; Minton, N.P. The ClosTron: a universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods. 2007, 70, 452–464. [Google Scholar] [CrossRef]
- Heap, J.T.; Cartman, S.T.; Kuehne, S.A.; Cooksley, C.; Minton, N.P. ClosTron-targeted mutagenesis. Methods Mol. Biol. 2010, 646, 165–182. [Google Scholar] [PubMed]
- Bernatchez, S.; Gilbert, M.; Blanchard, M.C.; Karwaski, M.F.; Li, J.; Defrees, S.; Wakarchuk, W.W. Variants of the beta 1,3-galactosyltransferase CgtB from the bacterium Campylobacter jejuni have distinct acceptor specificities. Glycobiology 2007, 17, 1333–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Katchar, K.; Goldsmith, J.D.; Nanthakumar, N.; Cheknis, A.; Gerding, D.N.; Kelly, C.P. A mouse model of Clostridium difficile-associated disease. Gastroenterology 2008, 135, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Fleites, C.; Macauley, M.S.; He, Y.; Shen, D.L.; Vocadlo, D.J.; Davies, G.J. Structure of an O-GlcNAc transferase homolog provides insight into intracellular glycosylation. Nat. Struct. Mol. Biol. 2008, 15, 764–765. [Google Scholar] [CrossRef]
- Lubas, W.A.; Hanover, J.A. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J. Biol. Chem. 2000, 275, 10983–10988. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.P.; Hart, G.W. Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J. Biol. Chem. 2003, 278, 24608–24616. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Tropea, J.E.; Waugh, D.S. Enhancing the solubility of recombinant proteins in Escherichia coli by using hexahistidine-tagged maltose-binding protein as a fusion partner. Methods. Mol. Biol. 2011, 705, 259–274. [Google Scholar]
- Matsuoka, O.; Patel, D.M.; Sasaki, S.; Oka, H.; Sasaki, T.; Pietrobon, P.J.; Laot, T.; Bouckenooghe, A.; Menezes, J.; de Bruyn, G. Safety and immunogenicity of Clostridium difficile toxoid vaccine in Japanese adults. Hum. Vaccin. Immunother. 2018, 14, 322–328. [Google Scholar] [CrossRef]
- De Bruyn, G.; Saleh, J.; Workman, D.; Pollak, R.; Elinoff, V.; Fraser, N.J.; Lefebvre, G.; Martens, M.; Mills, R.E.; Nathan, R.; et al. Defining the optimal formulation and schedule of a candidate toxoid vaccine against Clostridium difficile infection: A randomized phase 2 clinical trial. Vaccine 2016, 34, 2170–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bézay, N.; Ayad, A.; Dubischar, K.; Firbas, C.; Hochreiter, R.; Kiermayr, S.; Kiss, I.; Pinl, F.; Jilma, B.; Westritschnig, K. Safety, immunogenicity and dose response of VLA84, a new vaccine candidate against Clostridium difficile, in healthy volunteers. Vaccine 2016, 34, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.B.; Austrian, R.; Lee, C.J.; Rastogi, S.C.; Schiffman, G.; Henrichsen, J.; Mäkelä, P.H.; Broome, C.V.; Facklam, R.R.; Tiesjema, R.H.; et al. Considerations for formulating the second-generation pneumococcal capsular polysaccharide vaccine with emphasis on the cross-reactive types within groups. J. Infect. Dis. 1983, 148, 1136–1159. [Google Scholar] [CrossRef]
- Peltola, H.; Kayhty, H.; Sivonen, A. Haemophilus influenzae type b capsular polysaccharide vaccine in children: A double—blind field trial of 100,000 vaccinees 3 months to 5 years of age in Finland. Pediatrics. 1977, 60, 730–737. [Google Scholar] [PubMed]
- Ramsay, M.E.; Andrews, N.; Kaczmarski, E.B.; Miller, E. Efficacy of meningococcal serogroup C conjugate vaccine in teenagers and toddlers in England. Lancet. 2001, 357, 195–196. [Google Scholar] [CrossRef]
- Cox, A.D.; St Michael, F.; Aubry, A.; Cairns, C.M.; Strong, P.C.; Hayes, A.C.; Logan, S.M. Investigating the candidacy of a lipoteichoic acid-based glycoconjugate as a vaccine to combat Clostridium difficile infection. Glycoconj. J. 2013, 30, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Mora-Uribe, P.; Miranda-Cárdenas, C.; Castro-Córdova, P.; Gil, F.; Calderón, I.; Fuentes, J.A.; Rodas, P.I.; Banawas, S.; Sarker, M.R.; Paredes-Sabja, D. Characterization of the adherence of Clostridium difficile spores: the integrity of the outermost layer affects adherence properties of spores of the epidemic strain R20291 to components of the intestinal mucosa. Front Cell Infect Microbial. 2016, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Daubenspeck, J.M.; Zeng, H.; Chen, P.; Dong, S.; Steichen, C.T.; Krishna, N.R.; Pritchard, D.G.; Turnbough, C.L., Jr. Novel oligosaccharide side chains of the collagen-like region of BclA, the major glycoprotein of the Bacillus anthracis exosporium. J. Biol. Chem. 2004, 279, 30945–30953. [Google Scholar] [CrossRef] [Green Version]
- Díaz-González, F.; Milano, M.; Olguin-Araneda, V.; Pizarro-Cerda, J.; Castro-Córdova, P.; Tzeng, S.C.; Maier, C.S.; Sarker, M.R.; Paredes-Sabja, D. Protein composition of the outermost exosporium-like layer. J. Proteomics. 2015, 123, 1–13. [Google Scholar] [CrossRef]
- Ghose, C.; Eugenis, I.; Edwards, A.N.; Sun, X.; McBride, S.M.; Ho, D.D. Immunogenicity and protective efficacy of Clostridium difficile spore proteins. Anaerobe 2016, 37, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Broecker, F.; Wegner, E.; Seco, B.M.S.; Kaplonek, P.; Bräutigam, M.; Ensser, A.; Pfister, F.; Daniel, C.; Martin, C.E.; Mattner, J.; et al. Synthetic oligosaccharide-based vaccines protect mice from Clostridioides difficile infections. ACS Chem. Biol. 2019, 14, 2720–2728. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, M.A. The design of a Clostridium difficile carbohydrate-based vaccine. Methods. Mol. Biol. 2016, 1403, 397–408. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Sequence (R20291 BclA3) | Tag/Conjugate |
|---|---|---|
| (BclA3 aa Sequence) | ||
| SML1 (308–319) 12 aa | AGLIGPTGATGV | FITC-Ahx |
| SML2 (391–404) 15 aa | VGPTGATGATGADGV | FITC-Ahx |
| SML3 (278–292) 15 aa | TGATGLIGPTGATGA | FITC-Ahx |
| SML4 (391–404) 15 aa | VGPTGATGATGADGV | KLH and BSA conjugate |
| SML9 (271–295) 25 aa | GATGIGITGATGLIGPTGATGATGA | FITC-Ahx |
| SML10 (278–292) 16 aa | (C)TGATGLIGPTGATGA | N terminal cysteine/KLH and BSA conjugate |
| SML11 single site | DGAVGLIGPTGAGADGA | FITC-Ahx |
| SML12 T to S, 4 sites | SGASGLIGPSGAGASGA | FITC-Ahx |
| SML13 single T site | DGATGLIGPDGAGADGA | FITC-Ahx |
| SML14 single S site | DGASGLIGPDGAGADGA | FITC-Ahx |
| SML7 (1–15) 15aa | (C)MSRNKYFGPFDDNDYN | N terminal cysteine/BSA conjugate |
| SML8 (46–60) 15aa | (C)VGPTGPMGPRGRTGP | N terminal cysteine/ BSA conjugate |
| Peptide (T/S, # of Potential Sites) | % Conversion (1 h) | % Conversion (24 h) |
|---|---|---|
| SML1 (T,2) | 13.0 | 17.0 |
| SML2 (T,3) | 21.5 | 45.5 |
| SML3 (T,4) | 38.9 | 52.6 |
| SML11 (T,1) | 17.1 | 28.1 |
| SML12 (S,4) | 10.0 | 46.6 |
| SML13 (T,1) | 24.1 | 39.0 |
| SML14 (S,1) | 40.6 | 70.6 |
| H/C-1 | H/C-2 | H/C-3 | H/C-4 | H/C-5 | H/C-6 | |
|---|---|---|---|---|---|---|
| β-GlcNAc A | 4.56 | 3.69 | 3.55 | 3.45 | 3.44 | 3.76; 3.90 |
| 101.0 | 56.6 | 74.8 | 71.0 | 77.0 | 61.9 | |
| Thr x | 4.52 | 4.31 | 1.18 | |||
| 58.7 | 75.9 | 17.1 | ||||
| Thr y | 4.54 | 4.34 | 1.18 | |||
| 58.7 | 75.9 | 17.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubry, A.; Zou, W.; Vinogradov, E.; Williams, D.; Chen, W.; Harris, G.; Zhou, H.; Schur, M.J.; Gilbert, M.; Douce, G.R.; et al. In Vitro Production and Immunogenicity of a Clostridium difficile Spore-Specific BclA3 Glycopeptide Conjugate Vaccine. Vaccines 2020, 8, 73. https://doi.org/10.3390/vaccines8010073
Aubry A, Zou W, Vinogradov E, Williams D, Chen W, Harris G, Zhou H, Schur MJ, Gilbert M, Douce GR, et al. In Vitro Production and Immunogenicity of a Clostridium difficile Spore-Specific BclA3 Glycopeptide Conjugate Vaccine. Vaccines. 2020; 8(1):73. https://doi.org/10.3390/vaccines8010073
Chicago/Turabian StyleAubry, Annie, Wei Zou, Evguenii Vinogradov, Dean Williams, Wangxue Chen, Greg Harris, Hongyan Zhou, Melissa J. Schur, Michel Gilbert, Gillian R. Douce, and et al. 2020. "In Vitro Production and Immunogenicity of a Clostridium difficile Spore-Specific BclA3 Glycopeptide Conjugate Vaccine" Vaccines 8, no. 1: 73. https://doi.org/10.3390/vaccines8010073