
Adenovirus E1A/E1B Transformed Amniotic Fluid Cells Support Human Cytomegalovirus Replication

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Viruses
2.3. Analysis of Viral Protein Expression in Suspension CAP Cells
2.4. Indirect Immunofluorescence (IF)
2.5. Flow Cytometry of Viral Protein Expression
2.6. Quantitative PCR Analysis of HCMV Genome Replication
2.7. Replication Kinetics and Release of Viral Genomes into the Cell Culture Supernatant of Infected CAP Cells
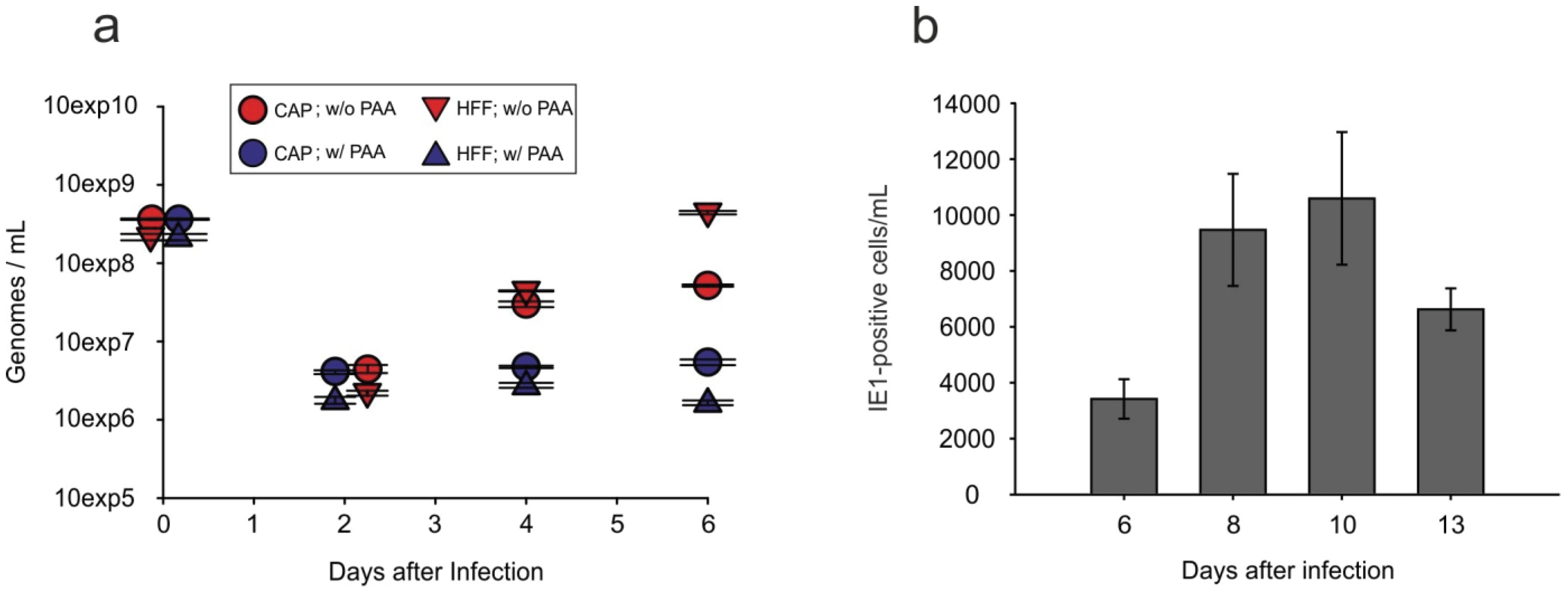
2.8. Release of Infectious HCMV into the Culture Supernatant of Infected CAP Cells, Measured by IE1-Staining
2.9. Electron Microscopy
2.10. Analysis of Dense Bodies (DBs) Production from Infected CAP Cells
3. Results
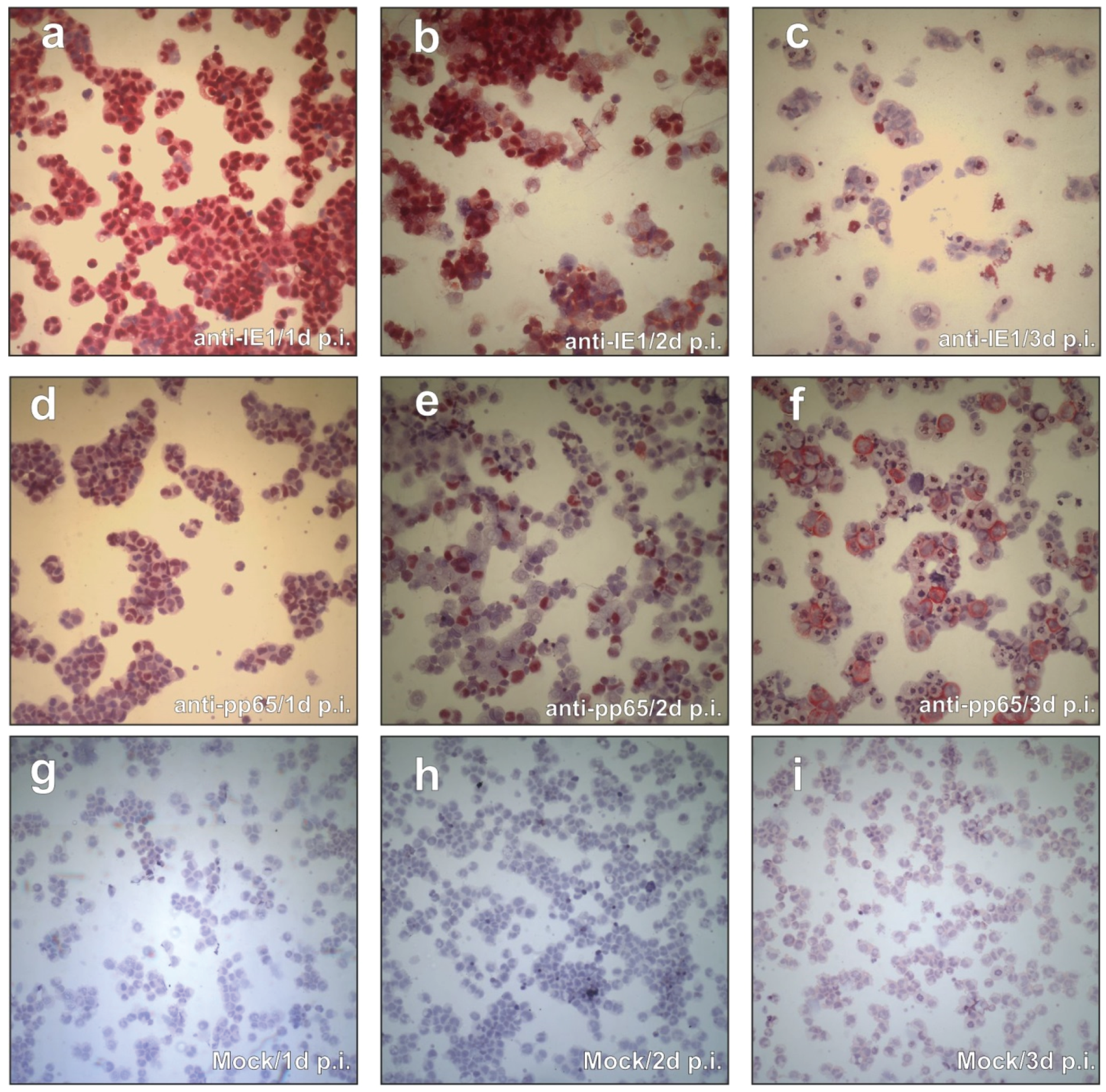
3.1. CAP Cells Support IE- and pp65-Gene Expression


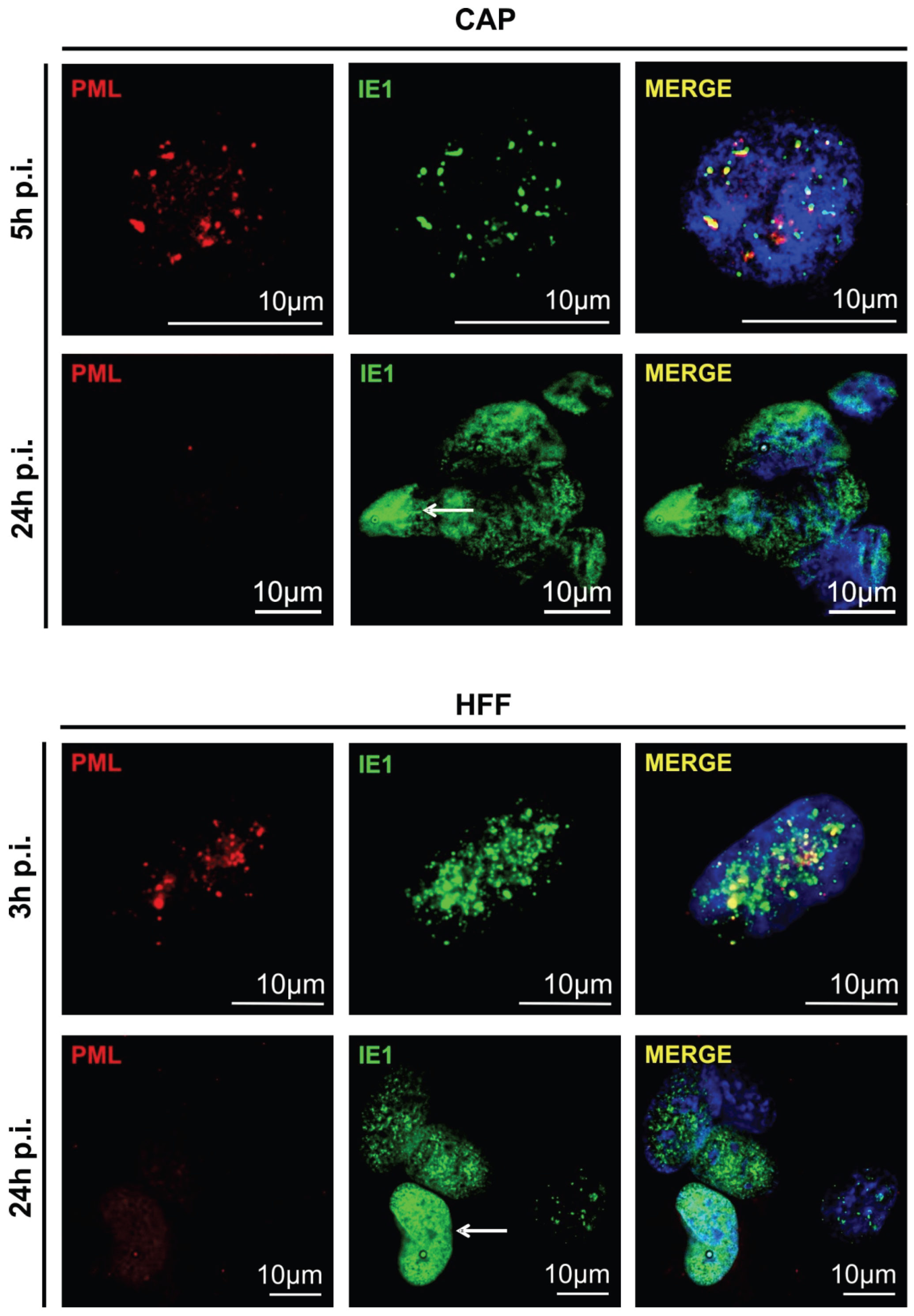
3.2. Cellular PML-Bodies Are Dissolved in HCMV-Infected CAP Cells

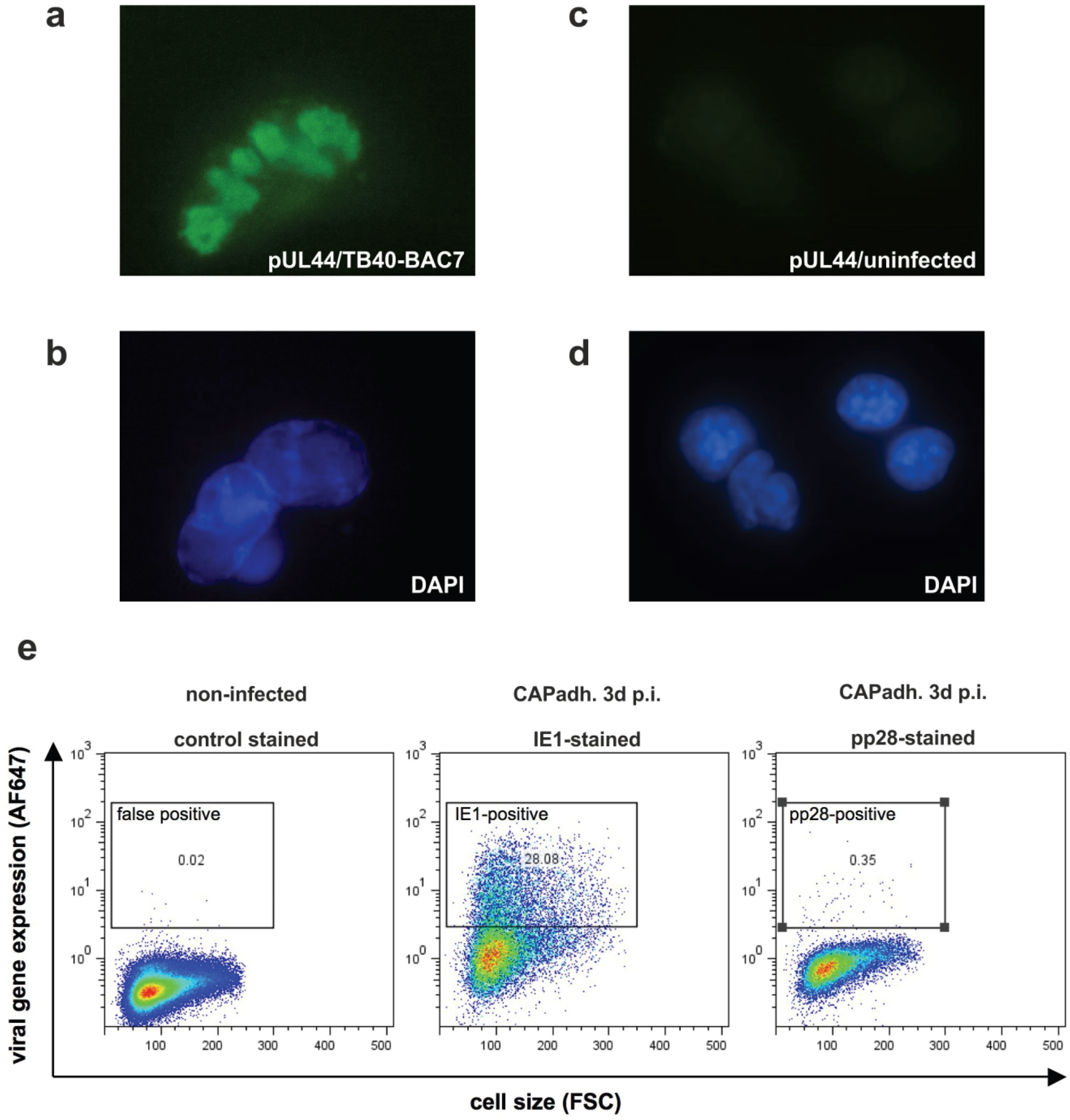
3.3. Detection of Nuclear Replication Compartments and Late Protein pp28 in HCMV Infected CAP Cells

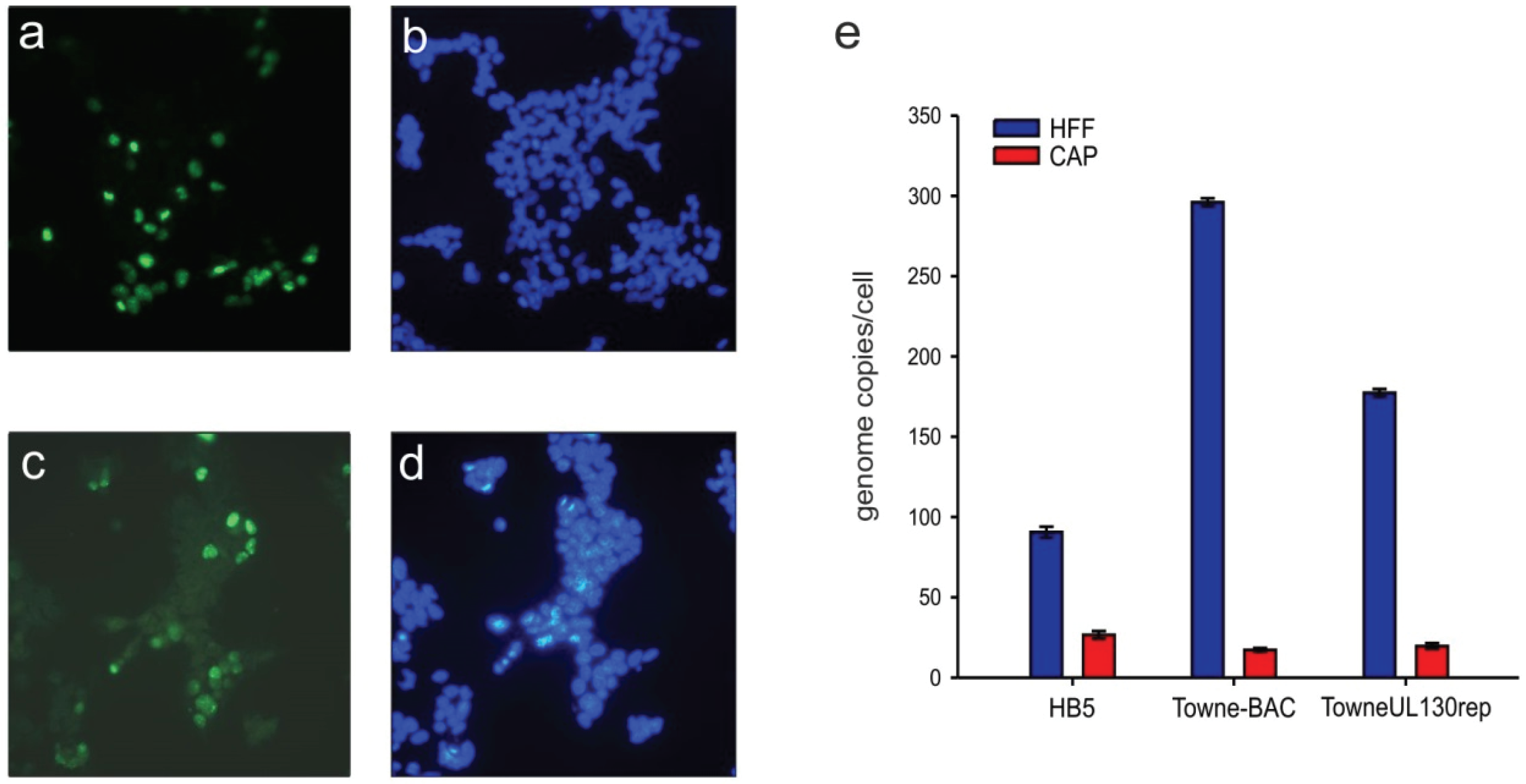
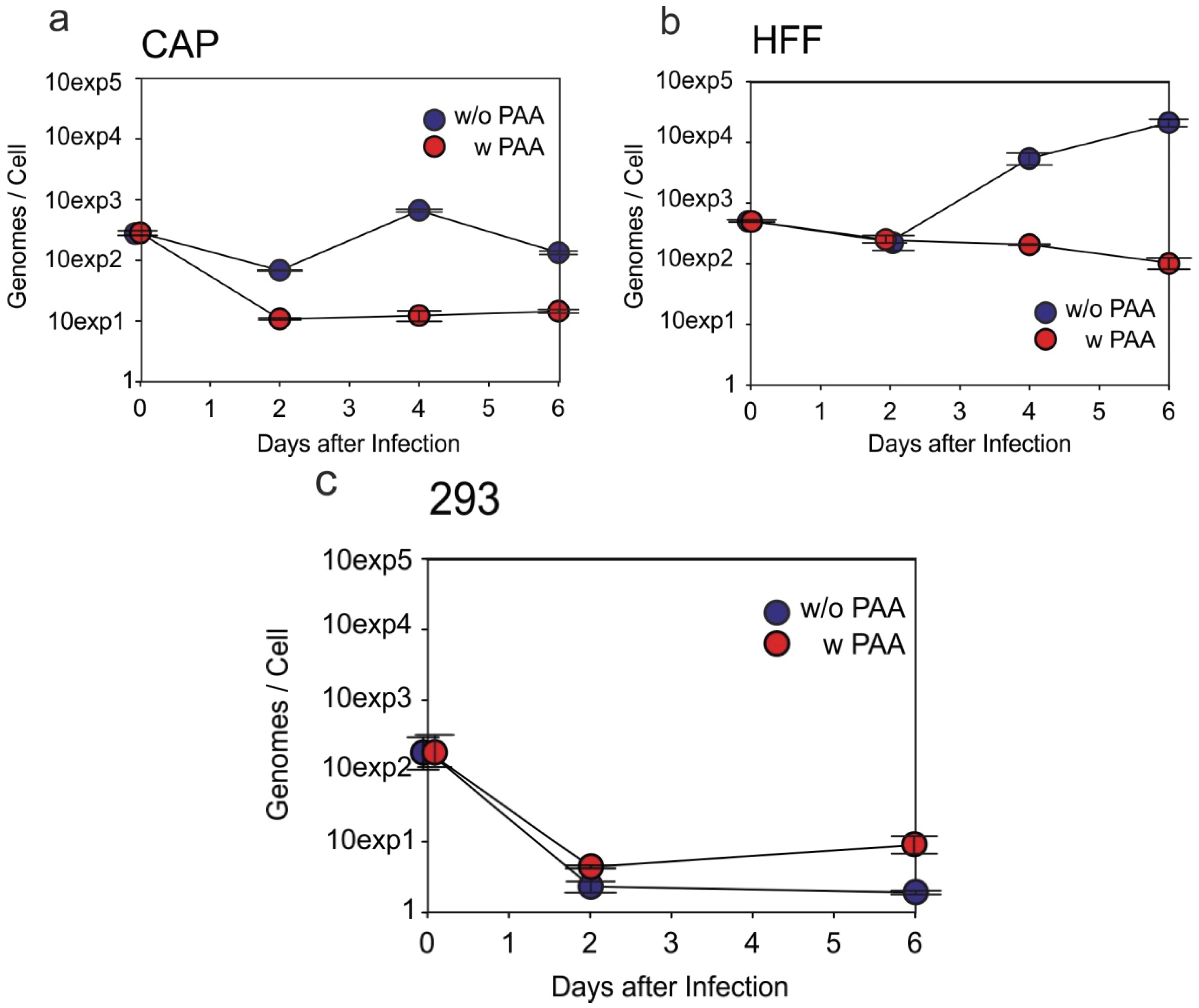
3.4. HCMV-DNA Is Replicated in CAP Cells

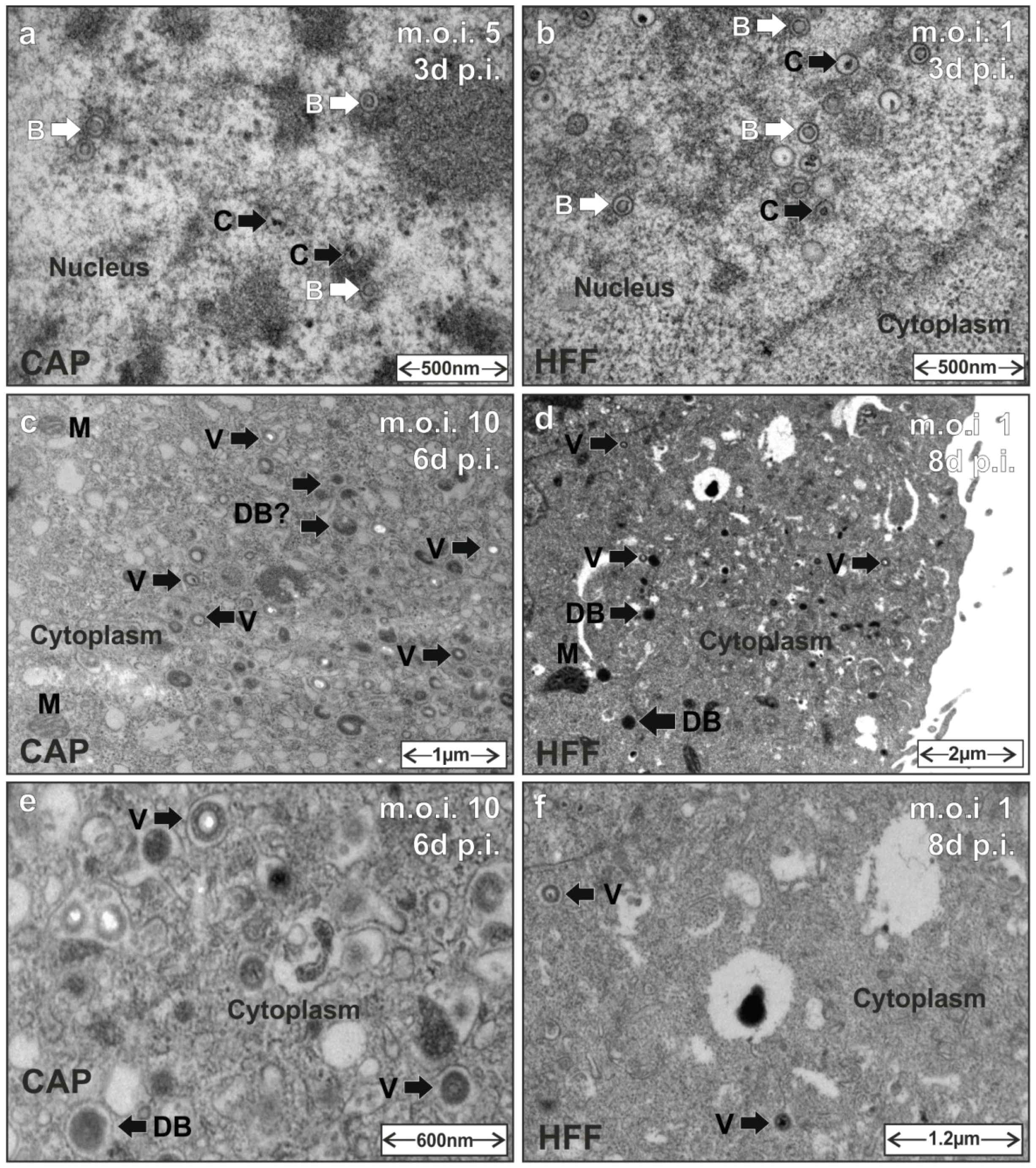
3.5. HCMV Capsid and Virion Morphogenesis Is Supported by CAP Cells

3.6. Viral Progeny Is Released from Infected CAP Cells

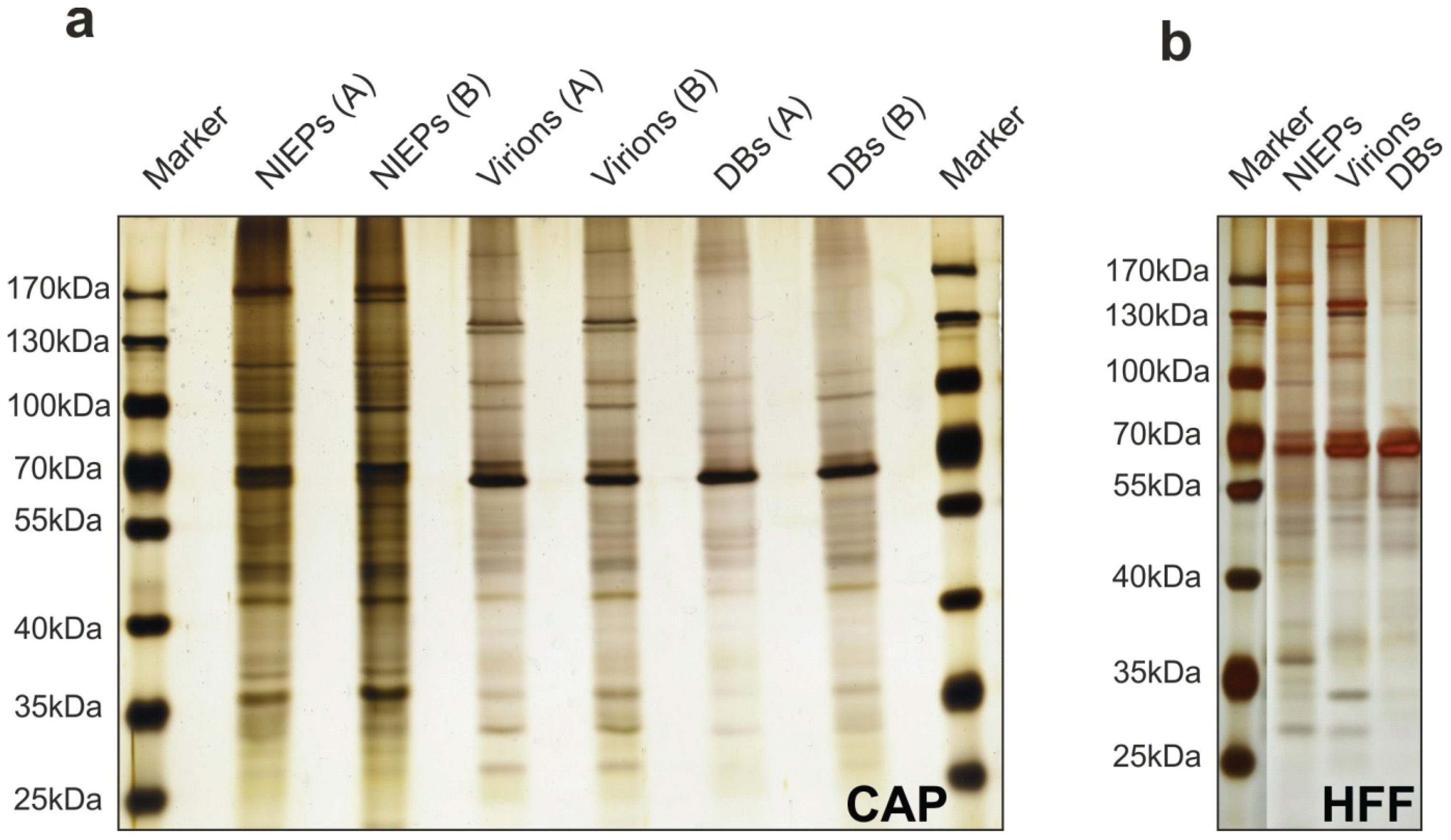
3.7. Purification of Viral Particles from Infected CAP Cells

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boppana, S.B.; Britt, W.J. Synopsis of clinical aspects of human cytomegalovirus disease. In Cytomegaloviruses from Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 1–25. [Google Scholar]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Mussi-Pinhata, M.M.; Yamamoto, A.Y.; Moura Brito, R.M.; de Lima Isaac, M.; de Carvalho e Oliveira, P.F.; Boppana, S.; Britt, W.J. Birth prevalence and natural history of congenital cytomegalovirus infection in a highly seroimmune population. Clin. Infec. Dis. 2009, 49, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Controversies in the natural history of congenital human cytomegalovirus infection: The paradox of infection and disease in offspring of women with immunity prior to pregnancy. Med. Microbiol. Immunol. 2015, 204, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Fowler, K.B.; Boppana, S.B. Congenital cytomegalovirus (CMV) infection and hearing deficit. J. Clin. Virol. 2006, 35, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Ross, S.A.; Fowler, K.B. Congenital cytomegalovirus infection: Clinical outcome. Clin. Infect. Dis. 2013, 57, S178–S181. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Petitt, M.; Fong, A.; Tsuge, M.; Tabata, T.; Fang-Hoover, J.; Maidji, E.; Zydek, M.; Zhou, Y.; Inoue, N.; et al. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J. Infect. Dis. 2014, 209, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Tabata, T.; Petitt, M.; Hoover, J.F. Cytomegalovirus replication in the developing human placenta. In Cytomegaloviruses from Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 74–87. [Google Scholar]
- Emery, V.C.; Milne, R.S.B.; Griffiths, P.D. Clinical cytomegalovirus research: Liver and kidney transplantation. In Cytomegaloviruses from Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 301–311. [Google Scholar]
- Avery, R.K. Clinical cytomegalovirus research: Thoracic organ transplantation. In Cytomegaloviruses from Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 286–300. [Google Scholar]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Haematopoietic cell transplantation. In Cytomegaloviruses from Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 337–353. [Google Scholar]
- Michel, D.; Chevillotte, M.; Mertens, T. Antiviral therapy, drug resistance and computed resistance profiling. In Cytomegaloviruses from Molecular Pathogenesis to Intervention, 2th ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 402–423. [Google Scholar]
- Plotkin, S.A.; Plachter, B. Cytomegalovirus vaccine: On the way to the future? In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 2th ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 424–449. [Google Scholar]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.M.; An, Z.; Wang, D. Progress on pursuit of human cytomegalovirus vaccines for prevention of congenital infection and disease. Vaccine 2014, 32, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, T.M. Progress on human cytomegalovirus vaccines for prevention of congenital infection and disease. Curr. Opin. Virol. 2014, 6, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Sauer, C.; Klobuch, S.; Herr, W.; Thomas, S.; Plachter, B. Subviral dense bodies of human cytomegalovirus stimulate maturation and activation of monocyte-derived immature dendritic cells. J. Virol. 2013, 87, 11287–11291. [Google Scholar] [CrossRef] [PubMed]
- Pepperl, S.; Münster, J.; Mach, M.; Harris, J.R.; Plachter, B. Dense bodies of human cytomegalovirus induce both humoral and cellular immune responses in the absence of viral gene expression. J. Virol. 2000, 74, 6132–6146. [Google Scholar] [CrossRef] [PubMed]
- Becke, S.; Aue, S.; Thomas, D.; Schader, S.; Podlech, J.; Bopp, T.; Sedmak, T.; Wolfrum, U.; Plachter, B.; Reyda, S. Optimized recombinant dense bodies of human cytomegalovirus efficiently prime virus specific lymphocytes and neutralizing antibodies without the addition of adjuvant. Vaccine 2010, 28, 6191–6198. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Müntefering, H.; Löning, T.; Stöss, H.; Plachter, B.; Jahn, G. Cell types infected in human cytomegalovirus placentitis identified by immunohistochemical double staining. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Plachter, B.; Sinzger, C.; Jahn, G. Cell types involved in replication and distribution of human cytomegalovirus. Adv. Virus Res. 1996, 46, 195–261. [Google Scholar] [PubMed]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [PubMed]
- Riegler, S.; Hebart, H.; Einsele, H.; Brossart, P.; Jahn, G.; Sinzger, C. Monocyte-derived dendritic cells are permissive to the complete replicative cycle of human cytomegalovirus. J. Gen. Virol. 2000, 81, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Jochemsen, H.; Hertoghs, J.J.; Lupker, J.H.; Davis, A.; van der Eb, A.J. In vitro synthesis of adenovirus type 5 T antigens. II. Translation of virus-specific RNA from cells transformed by fragments of adenovirus type 5 DNA. J. Virol. 1981, 37, 530–534. [Google Scholar] [PubMed]
- Genzel, Y. Designing cell lines for viral vaccine production: Where do we stand? Biotechnol. J. 2015, 10, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, K.; Smith, M.S.; Malouli, D.; Mansouri, M.; Nelson, J.A.; Früh, K. BST2/Tetherin enhances entry of human cytomegalovirus. PLoS Pathog. 2011, 7, e1002332. [Google Scholar] [CrossRef] [PubMed]
- Ellsmore, V.; Reid, G.G.; Stow, N.D. Detection of human cytomegalovirus DNA replication in non-permissive Vero and 293 cells. J. Gen. Virol. 2003, 84, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; et al. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Pau, M.G.; Ophorst, C.; Koldijk, M.H.; Schouten, G.; Mehtali, M.; Uytdehaag, F. The human cell line PER.C6 provides a new manufacturing system for the production of influenza vaccines. Vaccine 2001, 19, 2716–2721. [Google Scholar] [CrossRef]
- Schiedner, G.; Hertel, S.; Kochanek, S. Efficient transformation of primary human amniocytes by E1 functions of Ad5: Generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 2000, 11, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Genzel, Y.; Behrendt, I.; Rodig, J.; Rapp, E.; Kueppers, C.; Kochanek, S.; Schiedner, G.; Reichl, U. CAP, a new human suspension cell line for influenza virus production. Appl. Microbiol. Biotechnol. 2013, 97, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Genzel, Y.; Vogel, T.; Buck, J.; Behrendt, I.; Ramirez, D.V.; Schiedner, G.; Jordan, I.; Reichl, U. High cell density cultivations by alternating tangential flow (ATF) perfusion for influenza A virus production using suspension cells. Vaccine 2014, 32, 2770–2781. [Google Scholar] [CrossRef] [PubMed]
- Vlecken, D.H.; Pelgrim, R.P.; Ruminski, S.; Bakker, W.A.; van der Pol, L.A. Comparison of initial feasibility of host cell lines for viral vaccine production. J. Virol. Methods 2013, 193, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Essers, R.; Kewes, H.; Schiedner, G. Improving volumetric productivity of a stable human CAP cell line by bioprocess optimization. BMC Proc. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Borst, E.M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: A new approach for construction of HCMV mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [PubMed]
- Marchini, A.; Liu, H.; Zhu, H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed]
- Digel, M.; Sampaio, K.L.; Jahn, G.; Sinzger, C. Evidence for direct transfer of cytoplasmic material from infected to uninfected cells during cell-associated spread of human cytomegalovirus. J. Clin. Virol. 2006, 37, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, K.L.; Jahn, G.; Sinzger, C. Applications for a dual fluorescent human cytomegalovirus in the analysis of viral entry. Methods Mol. Biol. 2013, 1064, 201–209. [Google Scholar] [PubMed]
- Scrivano, L.; Sinzger, C.; Nitschko, H.; Koszinowski, U.H.; Adler, B. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 2011, 7, e1001256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreoni, M.; Faircloth, M.; Vugler, L.; Britt, W.J. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 1989, 23, 157–167. [Google Scholar] [CrossRef]
- Wiebusch, L.; Hagemeier, C. Use of 5-ethynyl-2’-deoxyuridine labelling and flow cytometry to study cell cycle-dependent regulation of human cytomegalovirus gene expression. Methods Mol. Biol. 2014, 1119, 123–132. [Google Scholar] [PubMed]
- Mersseman, V.; Besold, K.; Reddehase, M.J.; Wolfrum, U.; Strand, D.; Plachter, B.; Reyda, S. Exogenous introduction of an immunodominant peptide from the non-structural IE1 protein of human cytomegalovirus into the MHC class I presentation pathway by recombinant dense bodies. J. Gen. Virol. 2008, 89, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.; Orenstein, J.M. Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research. Nat. Protoc. 2007, 2, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [PubMed]
- Lafemina, R.L.; Pizzorno, M.C.; Mosca, J.D.; Hayward, G.S. Expression of the acidic nuclear immediate-early protein (IE1) of human cytomegalovirus in stable cell lines and its preferential association with metaphase chromosomes. Virology 1989, 172, 584–600. [Google Scholar] [CrossRef]
- Krömmelbein, N. Analyse der Cytomegalovirus-Infektion in Adenovirus E1A/E1B Transformierten Zellen zur Entwicklung eines Zellsubstrats zur Impfstoffproduktion. PhD Thesis, Johannes Gutenberg-Universität Mainz, Mainz, Germany, February 2016. [Google Scholar]
- Tavalai, N.; Stamminger, T. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses 2009, 1, 1240–1264. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Kim, D.J.; Lee, J.M.; Choi, C.Y.; Ahn, B.Y.; Hayward, G.S.; Ahn, J.H. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J. Virol. 2004, 78, 6527–6542. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Wiebusch, L.; Neuwirth, A.; Grabenhenrich, L.; Voigt, S.; Hagemeier, C. Cell cycle-independent expression of immediate-early gene 3 results in G1 and G2 arrest in murine cytomegalovirus-infected cells. J. Virol. 2008, 82, 10188–10198. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.; Bogner, E. Morphogenesis of the cytomegalovirus virion and subviral particles. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 2th ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 230–246. [Google Scholar]
- Roby, C.; Gibson, W. Characterization of phosphoproteins and protein kinase activity of virions, noninfectious enveloped particles, and dense bodies of human cytomegalovirus. J. Virol. 1986, 59, 714–727. [Google Scholar] [PubMed]
- Nowlin, D.M.; Cooper, N.R.; Compton, T. Expression of a human cytomegalovirus receptor correlates with infectibility of cells. J. Virol. 1991, 65, 3114–3121. [Google Scholar] [PubMed]
- De Coppi, P.; Bartsch, G., Jr.; Siddiqui, M.M.; Xu, T.; Santos, C.C.; Perin, L.; Mostoslavsky, G.; Serre, A.C.; Snyder, E.Y.; Yoo, J.J.; et al. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 2007, 25, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Lee, K.B.; Kim, M.K. The potential of mesenchymal stem cells derived from amniotic membrane and amniotic fluid for neuronal regenerative therapy. BMB Rep. 2014, 47, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Toda, A.; Okabe, M.; Yoshida, T.; Nikaido, T. The potential of amniotic membrane/amnion-derived cells for regeneration of various tissues. J. Pharmacol. Sci. 2007, 105, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Priest, R.E.; Marimuthu, K.M.; Priest, J.H. Origin of cells in human amniotic fluid cultures: Ultrastructural features. Lab. Invest. 1978, 39, 106–109. [Google Scholar] [PubMed]
- Weisblum, Y.; Panet, A.; Haimov-Kochman, R.; Wolf, D.G. Models of vertical cytomegalovirus (CMV) transmission and pathogenesis. Semin. Immunopathol. 2014, 36, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Panet, A.; Zakay-Rones, Z.; Haimov-Kochman, R.; Goldman-Wohl, D.; Ariel, I.; Falk, H.; Natanson-Yaron, S.; Goldberg, M.D.; Gilad, R.; et al. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J. Virol. 2011, 85, 13204–13213. [Google Scholar] [CrossRef] [PubMed]
- Morere, L.; Andouard, D.; Labrousse, F.; Saade, F.; Calliste, C.A.; Cotin, S.; Aubard, Y.; Rawlinson, W.D.; Esclaire, F.; Hantz, S.; et al. Ex vivo model of congenital cytomegalovirus infection and new combination therapies. Placenta 2015, 36, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Geder, L.; Rapp, F. Infection of human amnion cells with cytomegalovirus. J. Med. Virol. 1978, 2, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.; Sinzger, C. Cytomegalovirus interstrain variance in cell type tropism. In Cytomegaloviruses, 2th ed.; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 297–329. [Google Scholar]
- Zhou, M.; Lanchy, J.M.; Ryckman, B.J. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.H. Human cytomegalovirus riding the cell cycle. Med. Microbiol. Immunol. 2015, 204, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Pari, G.S.; Anders, D.G. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt- dependent DNA replication. J. Virol. 1993, 67, 6979–6988. [Google Scholar] [PubMed]
- Sanchez, V.; Greis, K.D.; Sztul, E.; Britt, W.J. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: Characterization of a potential site of virus assembly. J. Virol. 2000, 74, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krömmelbein, N.; Wiebusch, L.; Schiedner, G.; Büscher, N.; Sauer, C.; Florin, L.; Sehn, E.; Wolfrum, U.; Plachter, B. Adenovirus E1A/E1B Transformed Amniotic Fluid Cells Support Human Cytomegalovirus Replication. Viruses 2016, 8, 37. https://doi.org/10.3390/v8020037
Krömmelbein N, Wiebusch L, Schiedner G, Büscher N, Sauer C, Florin L, Sehn E, Wolfrum U, Plachter B. Adenovirus E1A/E1B Transformed Amniotic Fluid Cells Support Human Cytomegalovirus Replication. Viruses. 2016; 8(2):37. https://doi.org/10.3390/v8020037
Chicago/Turabian StyleKrömmelbein, Natascha, Lüder Wiebusch, Gudrun Schiedner, Nicole Büscher, Caroline Sauer, Luise Florin, Elisabeth Sehn, Uwe Wolfrum, and Bodo Plachter. 2016. "Adenovirus E1A/E1B Transformed Amniotic Fluid Cells Support Human Cytomegalovirus Replication" Viruses 8, no. 2: 37. https://doi.org/10.3390/v8020037