

Analyzing the Geometry and Dynamics of Viral Structures: A Review of Computational Approaches Based on Alpha Shape Theory, Normal Mode Analysis, and Poisson–Boltzmann Theories

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Geometry of Viral Structures
2.1. Motivation and Background
- (i)
- (ii)
- Evaluate the hydrophobicity of a molecule. The most common use of molecular shape is the quantification of the hydrophobic effect. Eisenberg and McLachlan, for example [68], introduced the concept of a solvation free energy for large biomolecules, computed as a weighted sum of the accessible areas of all their atoms i. This solvation-free energy is a mean force potential that quantifies the energy that is required to solvate a molecule. Its nonpolar contribution is evaluated from geometric measures of the molecule, including surface area [68], volume [72], or even the curvature of the surface area in the so-called morphometric model [73].
- (iii)
- Identifying pockets and cavities in molecules: detecting and measuring internal cavities of biomolecules is often performed as a first step for drug design as those cavities map to putative binding sites.
2.2. Methodology
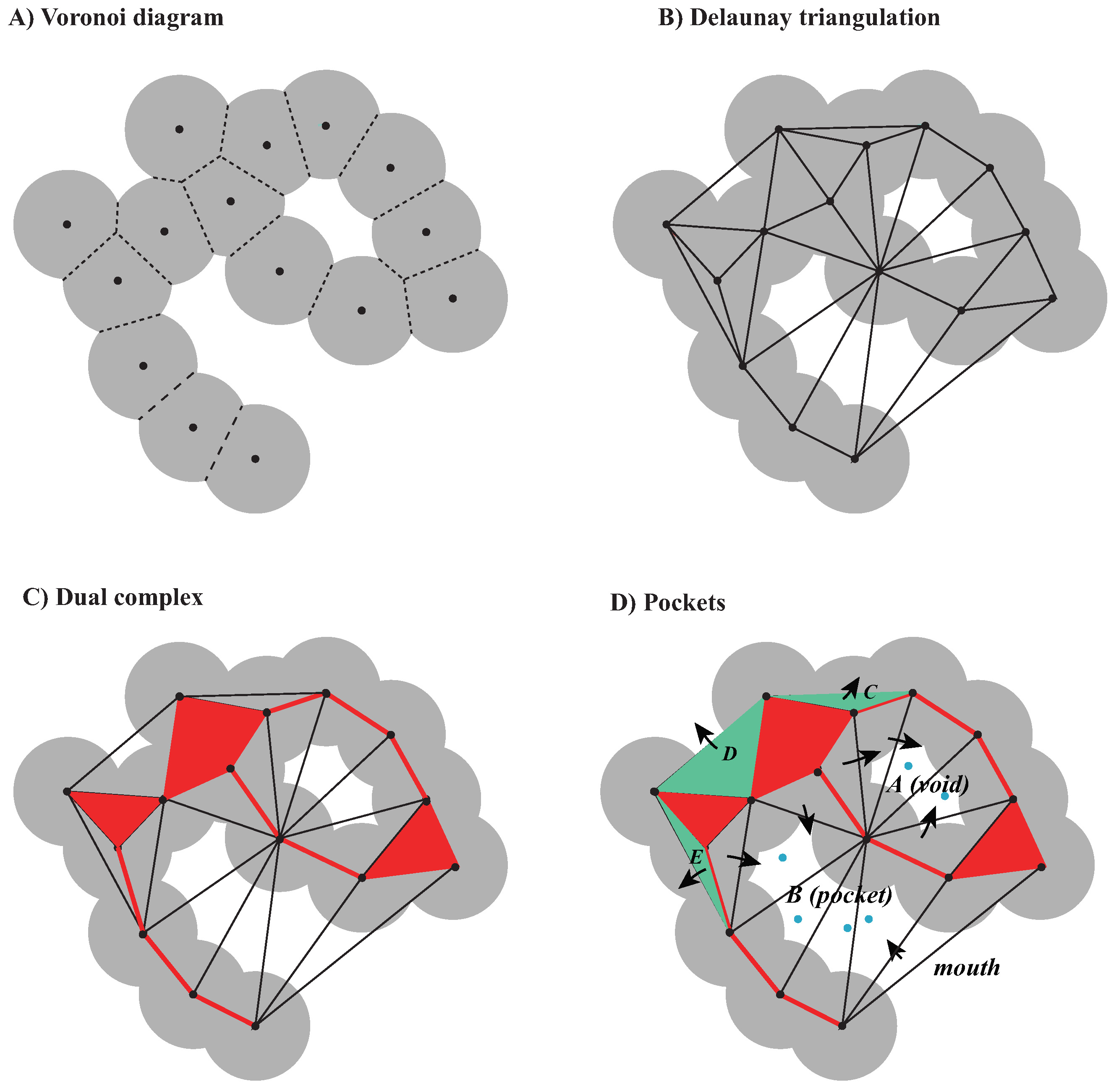
2.2.1. Voronoi Decompositions and Dual Complexes
2.2.2. Area and Volume Formulas
2.2.3. Voids and Pockets
- (i)
- and share a common triangle ;
- (ii)
- The interior of and the orthogonal center of lie on different sides of the plane defined by .
2.3. Examples
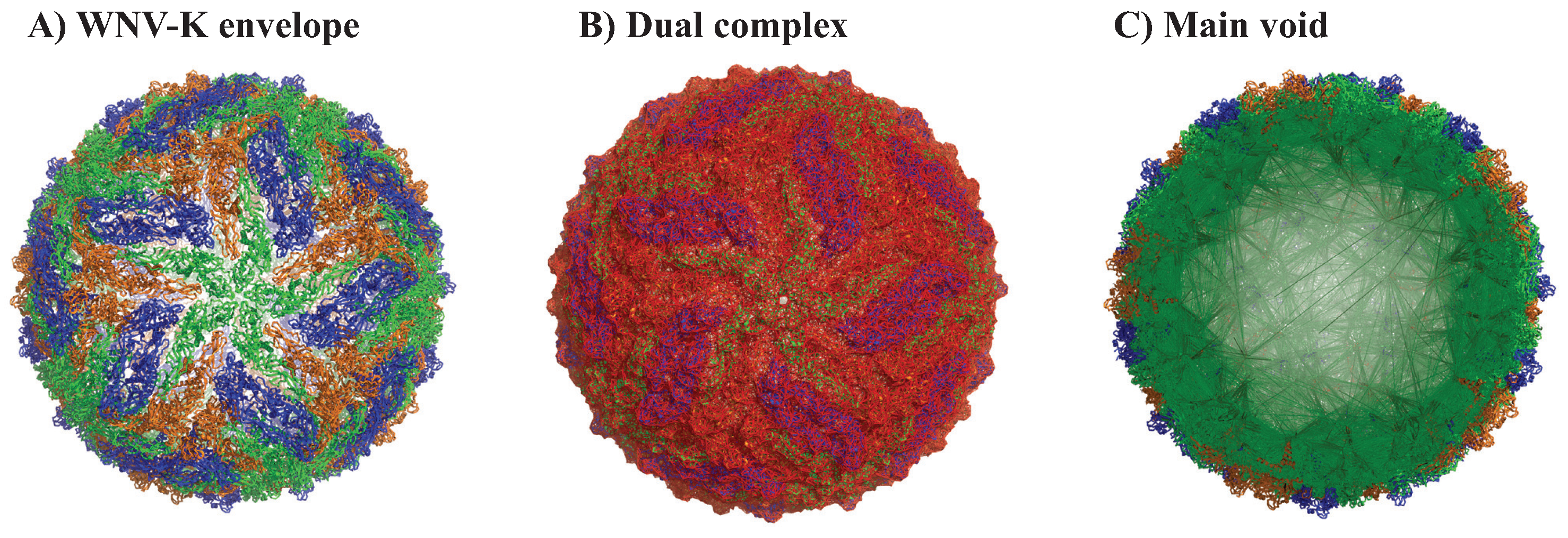
2.3.1. Full Viral Envelope
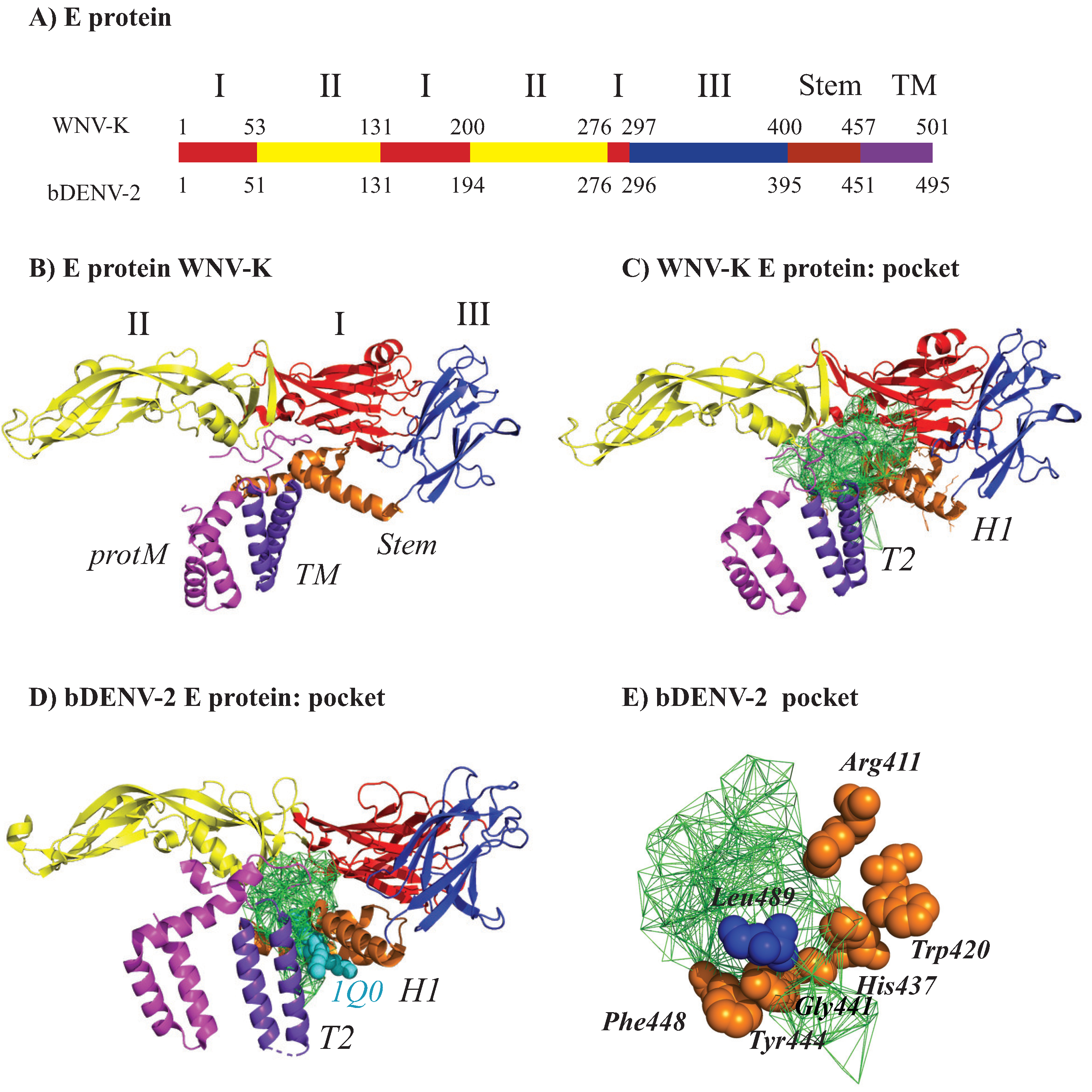
2.3.2. The E Protein–M Protein Complex
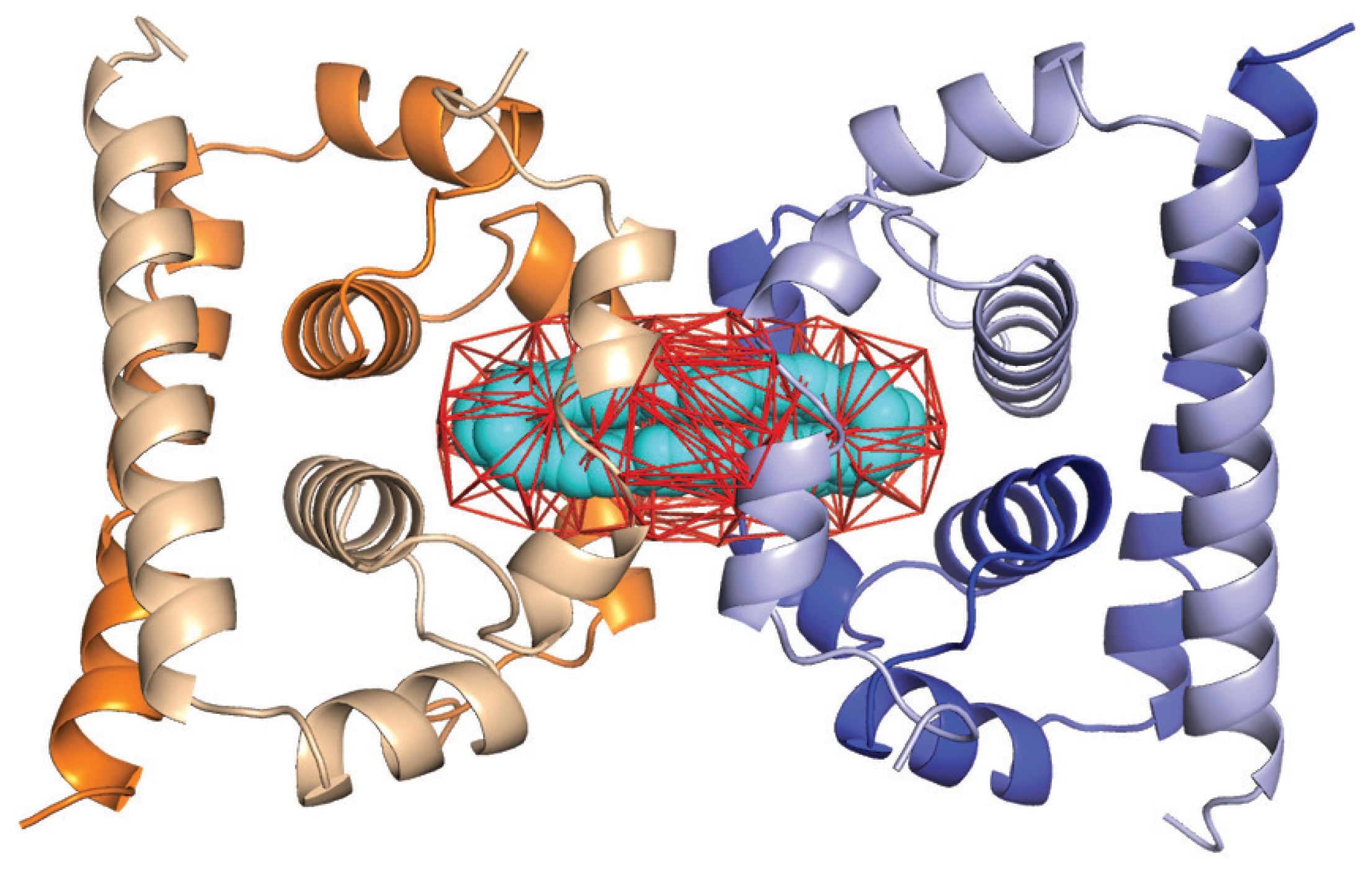
2.4. Application of UnionBall to Modeling: A Toy Problem on the Capsid Protein of Flaviviruses
3. Dynamics of Viral Structures
3.1. Motivation
3.2. Methodology
3.2.1. Coarse Grained Potentials for Normal Mode Analysis of Biomolecules
3.2.2. Diagonalizing the Hessian Matrix
- (i)
- The storage issue. As described above, the pairs of atoms that are included in the potential are filtered based on either a cutoff value or based on a geometric construction such as the Delaunay triangulation. As a consequence, the Hessian matrix is sparse, with the number of nonzero values only a fraction of the expected , and more of the order (see for example [151]). In addition, the forms of both the Tirion potential and the Go potential are such that their Hessian can be expressed as sums of tensor products, further reducing their storage needs [153].
- (ii)
- Computing eigenvalues and eigenvectors. In her original paper on coarse-grained normal mode analyses of proteins, Tirion showed that the lowest frequency normal modes based on a geometric potential capture most of the dynamics of the molecular system of interest [147]. She did not indicate, however, how many low frequency normal modes need to be considered, as this is most likely problem specific (see for example [154]). Still, only a fraction of the total eigenvalues and eigenvectors of the Hessian matrix need to be computed [131]. There are powerful iterative algorithms for computing a subset of the eigenpairs of a matrix. In Ref. [141], we compared four such methods, namely an implicitly restarted Arnoldi method as implemented in ARPACK [155], a simple modification of this method based on polynomial filtering [156,157], a variational method based on the minimization of an energy function [138,158], and a block Chebyshev–Davidson method [159,160]. We have shown that the latter provides the most efficient implementation when computing eigenpairs of extremely large Hessian matrices corresponding to large viral structures [141].
3.2.3. Correlated Motions within a Molecular System
3.3. Examples
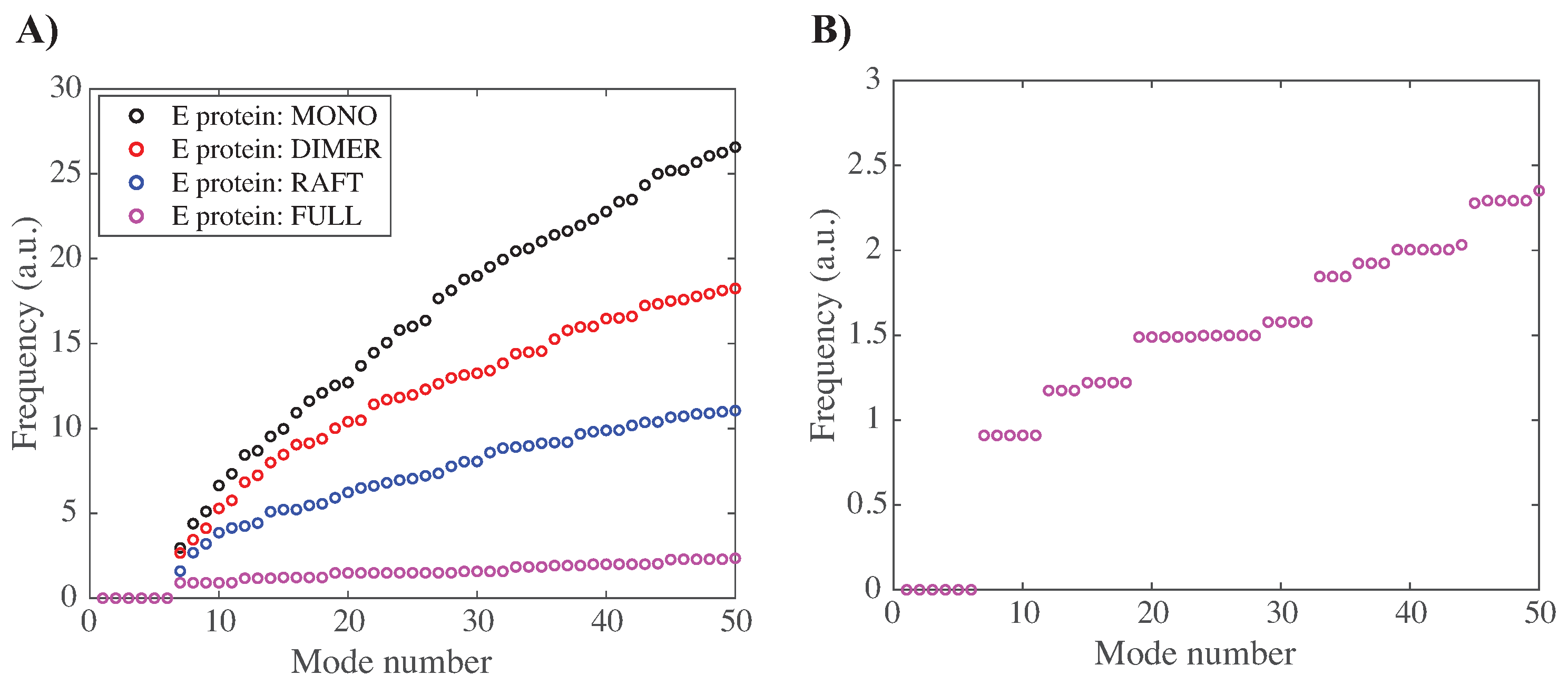
3.3.1. Characterizing the Low-Frequency Normal Modes of WNV-K
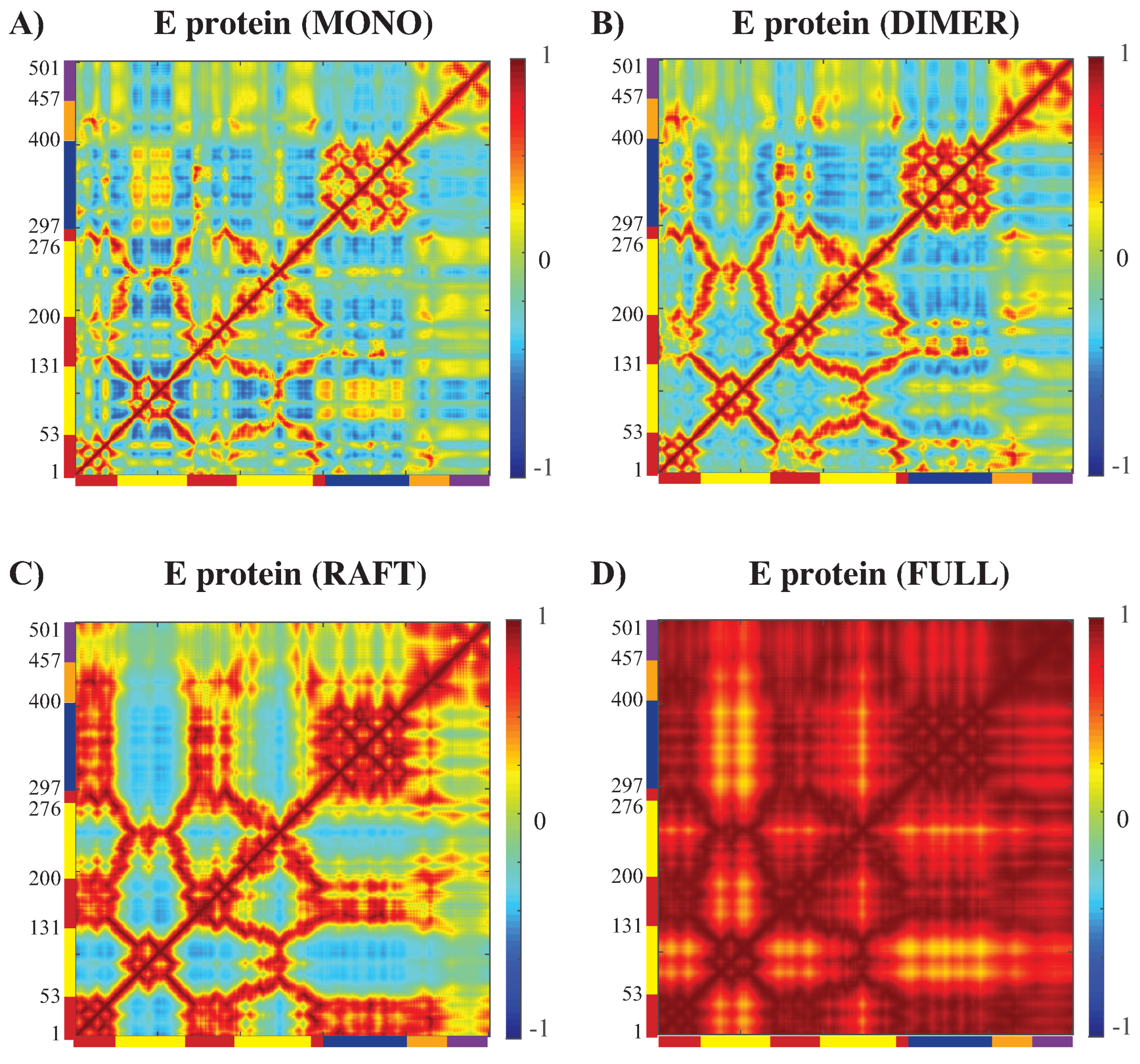
3.3.2. Concerted Motions of E Proteins in Different Environments
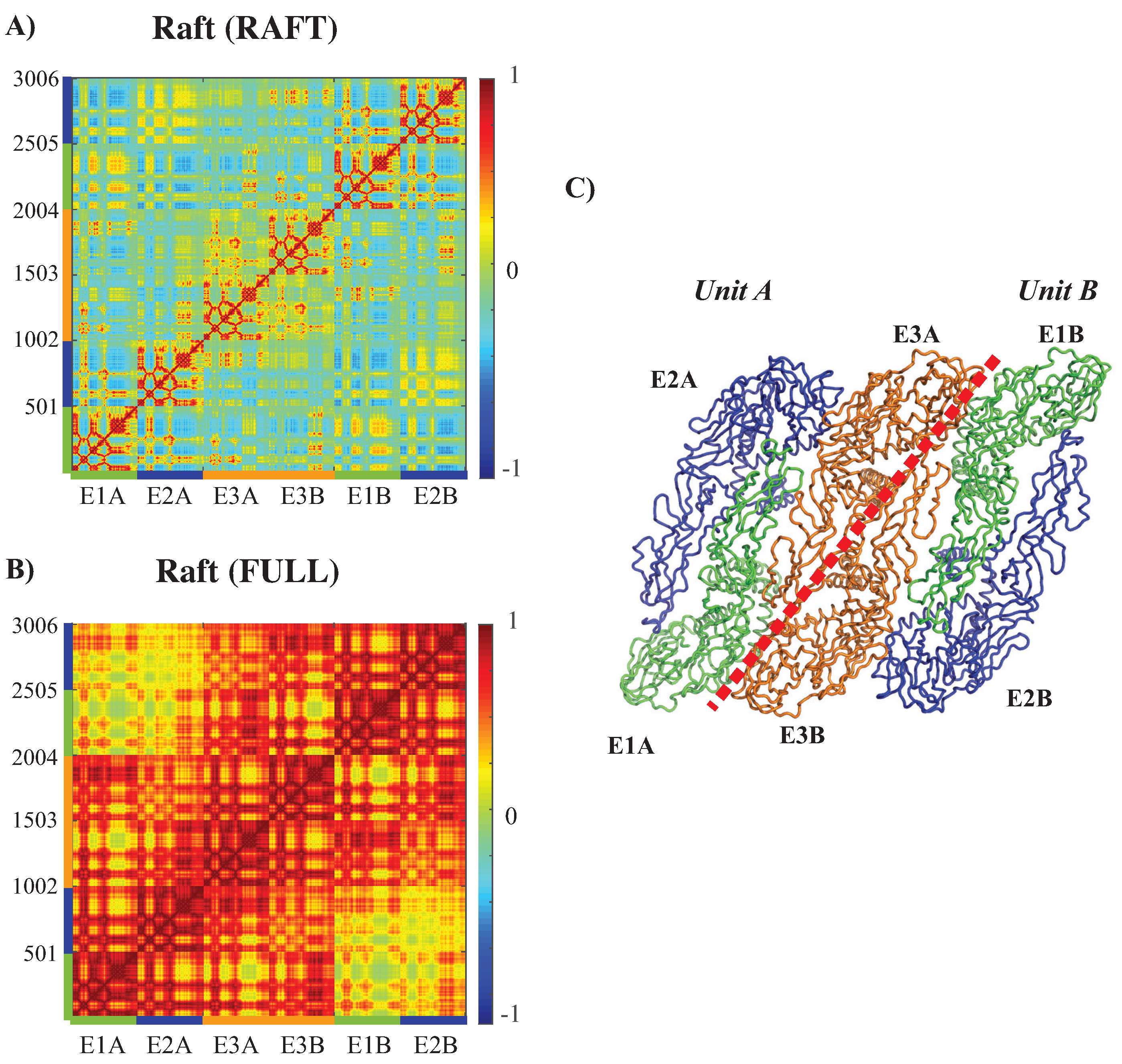
3.3.3. Concerted Motions of Rafts of E Proteins in Different Environments
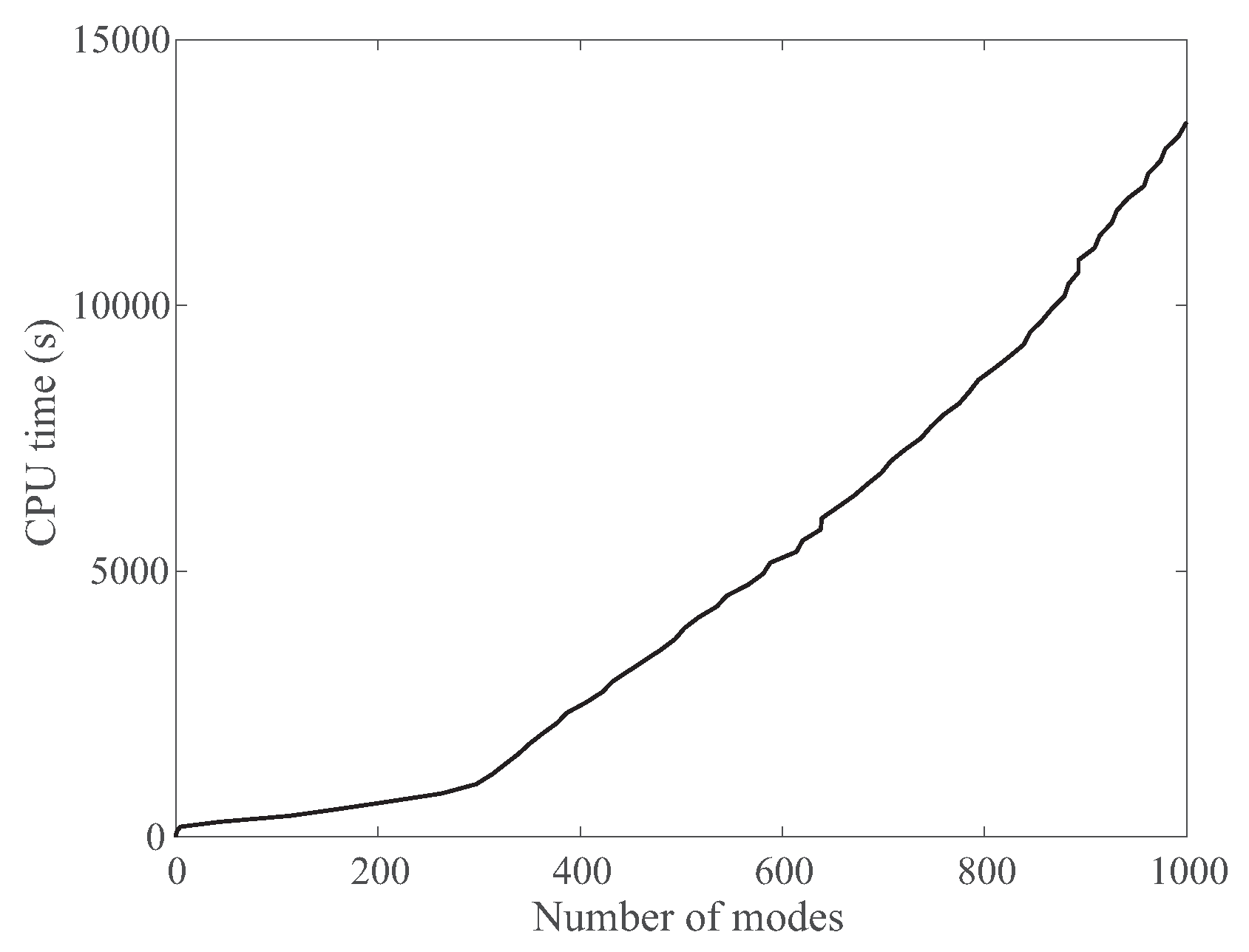
3.3.4. Computing Time for NormalModes
4. Energetics of Viral Structures
4.1. Motivation
4.2. Methodology
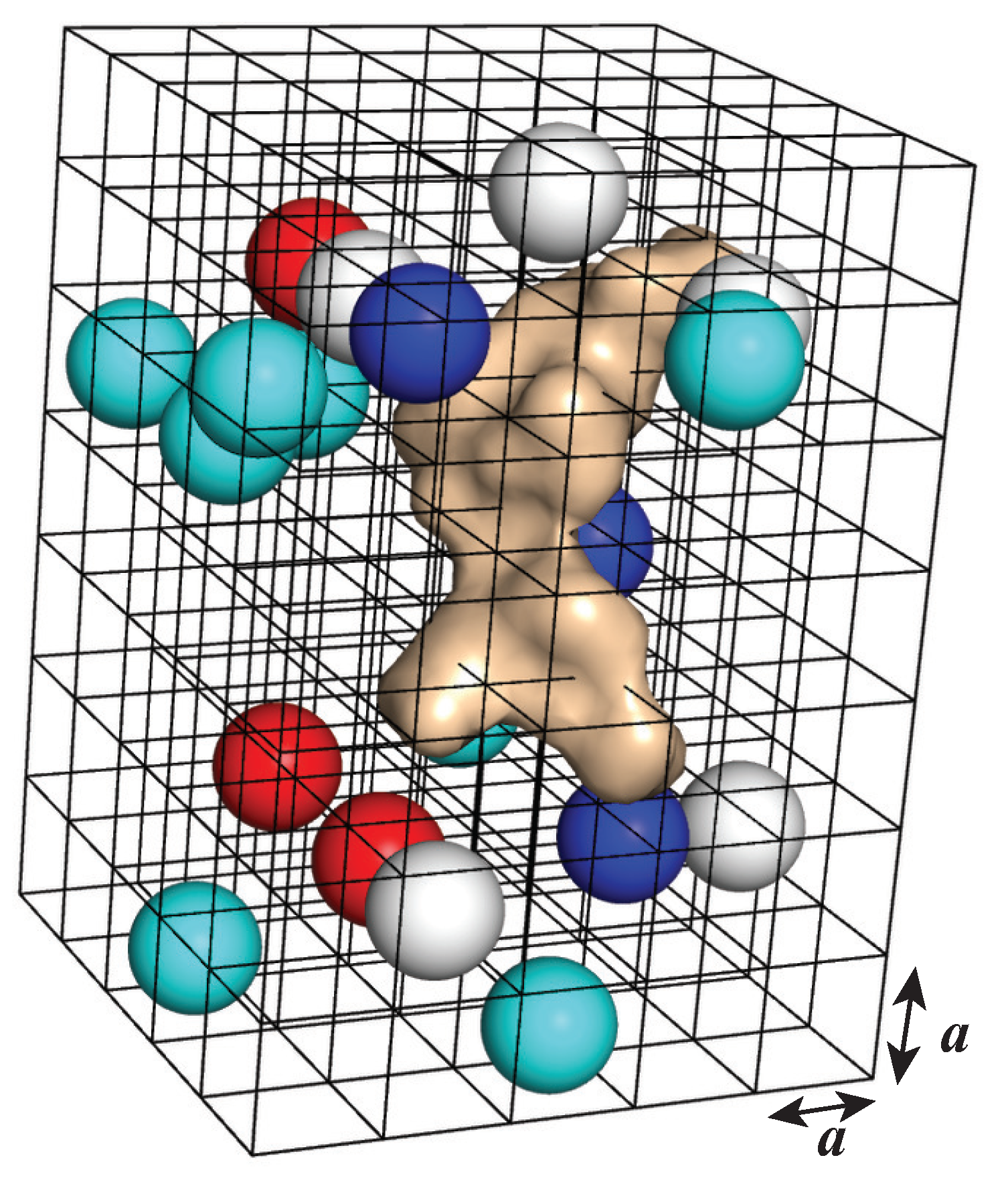
4.2.1. A Lattice Gas Model for the Environment of the Solute of Interest
4.2.2. A Free Energy Model for the Solute and Lattice Gas
- (i)
- Water molecule: the energy of one water dipole of constant magnitude at position is obtained as the Boltzmann-weighted average of the interaction over all orientations of , where is the local electric potential:where and is the fugacity of the water (see below).
- (ii)
- Ions: the energy of one ion with charge is simply . We assume that there are as many positive ions and negative ions in the environment, with charges and , respectively. Then:where is the fugacity of the ions.
- (iii)
- Hydrophobic particles: The hydrophobic interactions between the hydrophobic particles are defined by a Yukawa potential:where , defines the range of the hydrophobic interaction, and its strength. The negative sign denotes the attractive nature of the interaction. Setting to be the hydrophobic field associated with this potential gives the following:where is the fugacity of the hydrophobic particles.
- (iv)
- Possible empty sites: The system may be considered as incompressible, in which case all lattice sites are occupied by one particle, or compressible, in which case a lattice site may be empty. We model this behavior by introducing a pseudo-fugacity for vacancies such that if the system in incompressible, and otherwise. Note that this is set once at the beginning of the analysis.
4.2.3. Solving for the Electrostatic and Hydrophobic Fields
4.2.4. The Particle Fugacities
- (i)
- (ii)
- Incompressible system: There are no vacancies among the lattice sites and . The fugacities are then not independent. If we choose , the fugacities are defined as follows [189]:Finally, the bulk value is given by:
4.2.5. The Densities or Water Dipoles, Ions, and Hydrophobic Probes
- (i)
- Anions and cations:
- (ii)
- Water dipoles:
- (iii)
- Hydrophobic particles:
4.2.6. AquaVit
System Setup
4.3. Examples
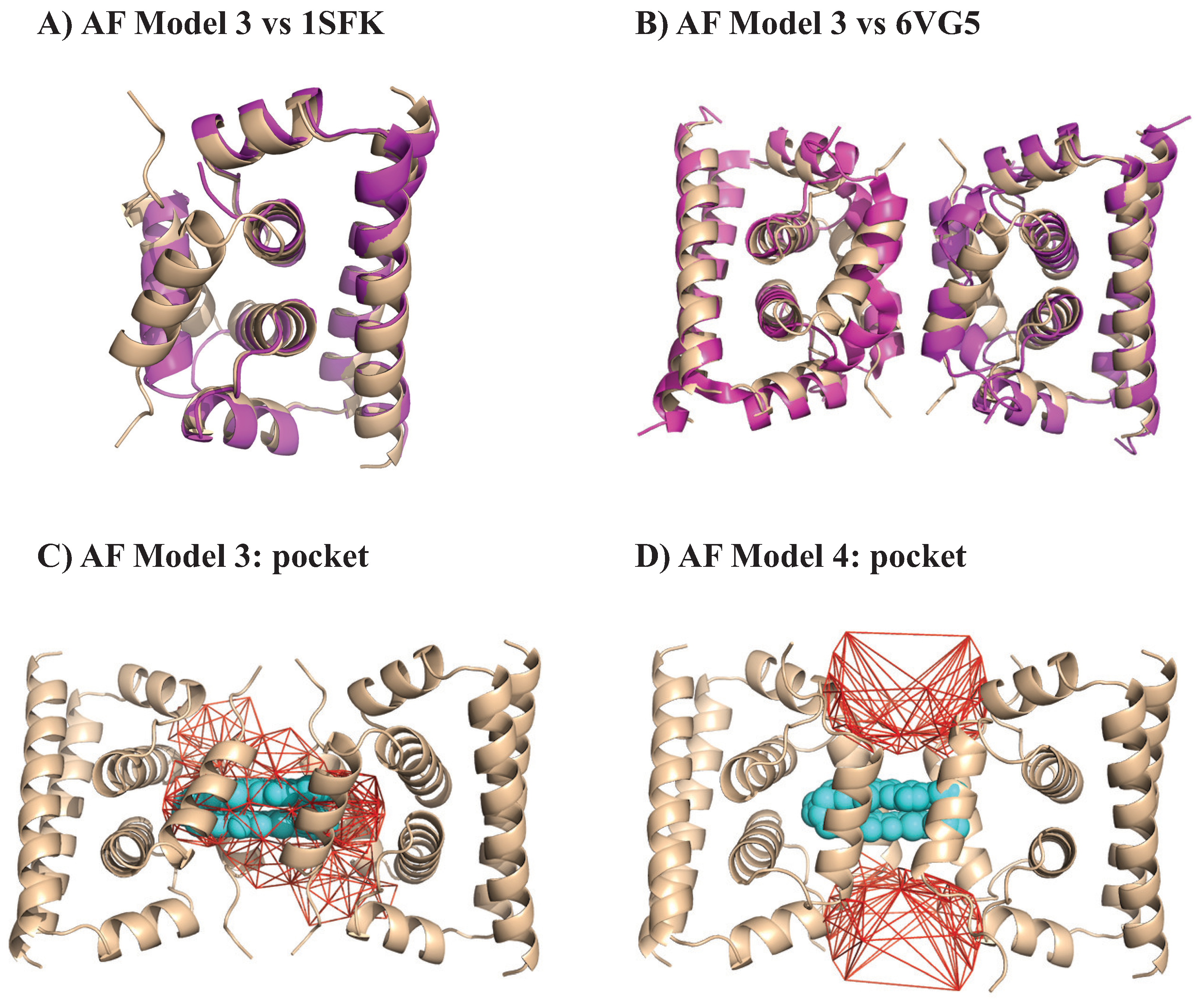
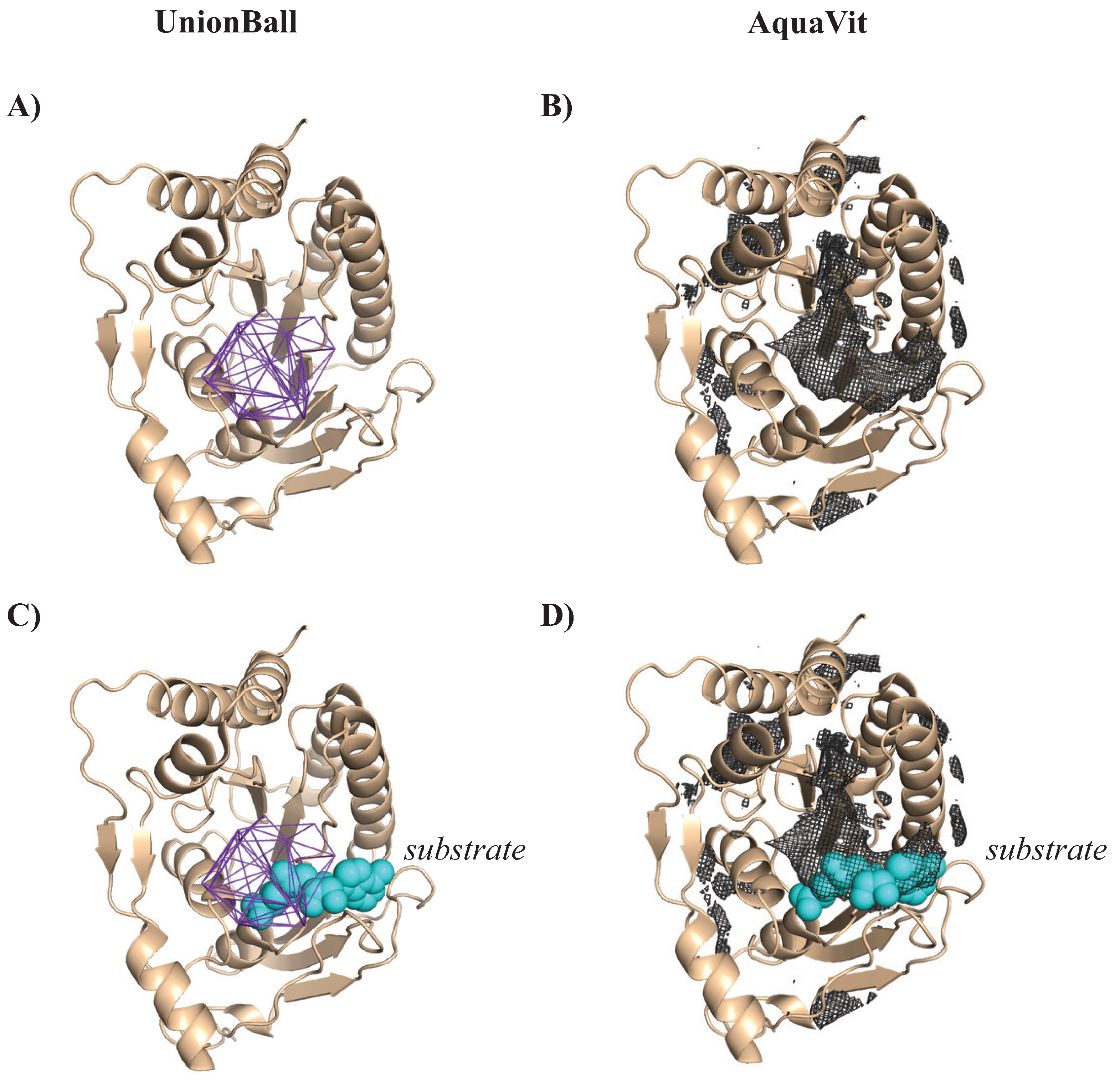
4.3.1. Identifying a Hydrophobic Pocket in a Methyltransferase
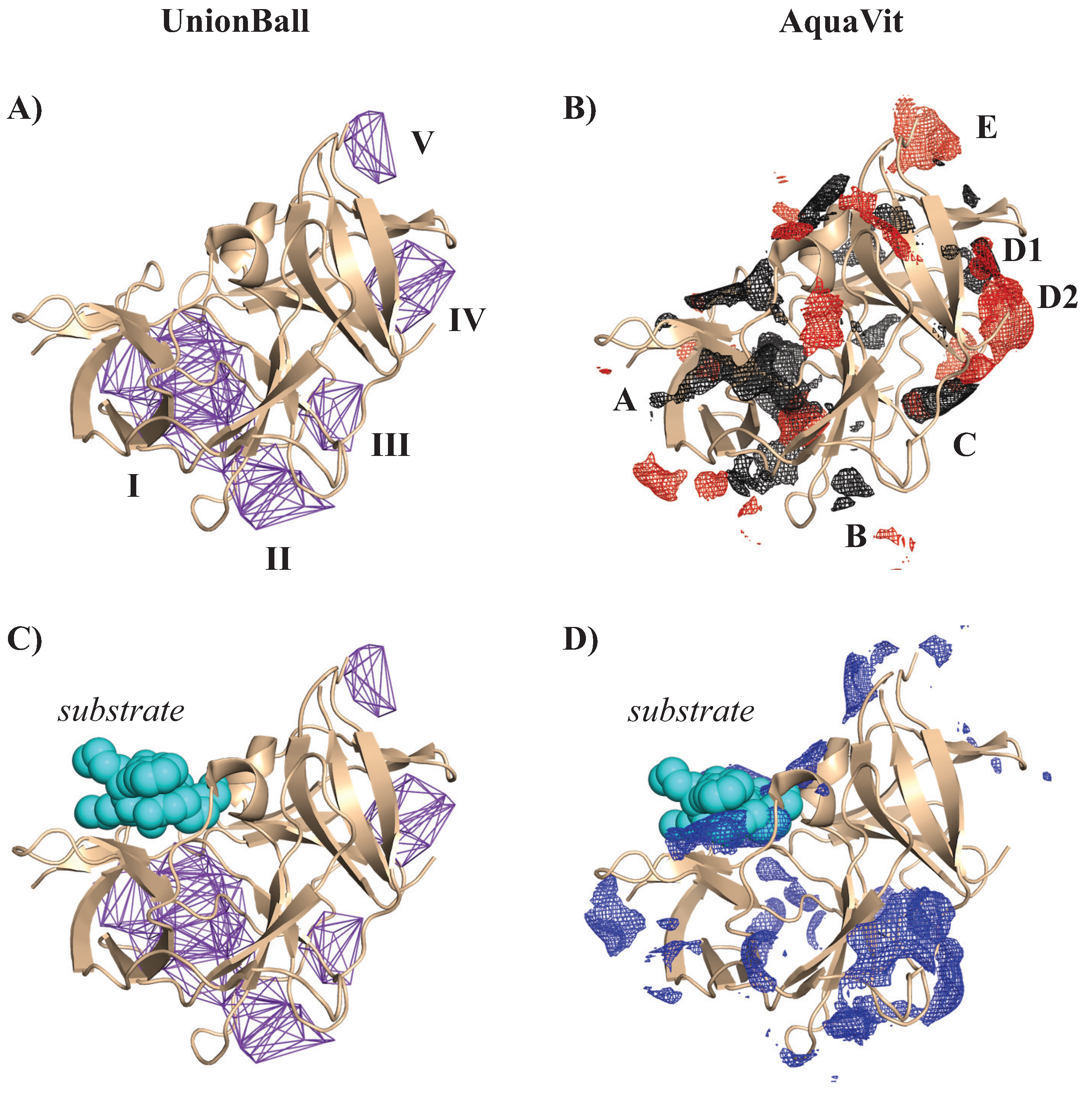
4.3.2. Identifying a Charged Pocket in a Protease
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Hamichi, S.; Gold, A.; Murray, T.G.; Graversen, V.K. Pandemics, climate change, and the eye. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 2597–2601. [Google Scholar] [CrossRef]
- Rothenberg, M.E. The climate change hypothesis for the allergy epidemic. J. Allergy Clin. Immunol. 2022, 149, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Rupasinghe, R.; Chomel, B.B.; Martínez-López, B. Climate change and zoonoses: A review of the current status, knowledge gaps, and future trends. Acta Trop. 2022, 226, 106225. [Google Scholar] [CrossRef] [PubMed]
- Leal Filho, W.; Ternova, L.; Parasnis, S.A.; Kovaleva, M.; Nagy, G.J. Climate change and zoonoses: A review of concepts, definitions, and bibliometrics. Int. J. Environ. Res. Public Health 2022, 19, 893. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.E.; Kuhn, M.; Prettner, K. Modern infectious diseases: Macroeconomic impacts and policy responses. J. Econ. Lit. 2022, 60, 85–131. [Google Scholar] [CrossRef]
- Callegari, B.; Feder, C. A literature review of pandemics and development: The long-term perspective. Econ. Disasters Clim. Chang. 2022, 6, 183–212. [Google Scholar] [CrossRef]
- Msemburi, W.; Karlinsky, A.; Knutson, V.; Aleshin-Guendel, S.; Chatterji, S.; Wakefield, J. The WHO estimates of excess mortality associated with the COVID-19 pandemic. Nature 2023, 613, 130–137. [Google Scholar] [CrossRef]
- Johnson, N.P.; Mueller, J. Updating the accounts: Global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull. Hist. Med. 2002, 76, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Puhach, O.; Meyer, B.; Eckerle, I. SARS-CoV-2 viral load and shedding kinetics. Nat. Rev. Microbiol. 2023, 21, 147–161. [Google Scholar] [CrossRef]
- Telenti, A.; Hodcroft, E.B.; Robertson, D.L. The evolution and biology of SARS-CoV-2 variants. Cold Spring Harb. Perspect. Med. 2022, 12, a041390. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 1–19. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Hardenbrook, N.J.; Zhang, P. A structural view of the SARS-CoV-2 virus and its assembly. Curr. Opin. Virol. 2022, 52, 123–134. [Google Scholar] [CrossRef]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of SARS-CoV-2 membrane protein essential for virus assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef]
- Li, S.; Zandi, R. Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding. Viruses 2022, 14, 2089. [Google Scholar] [CrossRef] [PubMed]
- Dolan, K.A.; Dutta, M.; Kern, D.M.; Kotecha, A.; Voth, G.A.; Brohawn, S.G. Structure of SARS-CoV-2 M protein in lipid nanodiscs. Elife 2022, 11, e81702. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Kaki, M.; Potluri, V.S.; Kahar, P.; Khanna, D. A comprehensive review of SARS-CoV-2 vaccines: Pfizer, moderna & Johnson & Johnson. Hum. Vaccines Immunother. 2022, 18, 2002083. [Google Scholar]
- Wagner, C.S.; Cai, X.; Zhang, Y.; Fry, C.V. One-year in: COVID-19 research at the international level in CORD-19 data. PLoS ONE 2022, 17, e0261624. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Huangfu, X.; Wang, H. Citation advantage of COVID-19-related publications. J. Inf. Sci. 2023. [Google Scholar] [CrossRef]
- Holly, E. How a torrent of COVID science changed research publishing. Nature 2020, 588, 553. [Google Scholar]
- Bernal, J.; Fankuchen, I.; Riley, D. Structure of the crystals of tomato bushy stunt virus preparations. Nature 1938, 142, 1075. [Google Scholar] [CrossRef]
- Winkler, F.; Schutt, C.; Harrison, S.; Bricogne, G. Tomato bushy stunt virus at 5.5-Å resolution. Nature 1977, 265, 509–513. [Google Scholar] [CrossRef]
- Bernstein, F.; Koetzle, T.; Williams, G.; Meyer, E.F., Jr.; Brice, M.; Rodgers, J.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Molec. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucl. Acids. Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C.L.; Dutta, S.; Westbrook, J.D.; Henrick, K.; Berman, H.M. Representation of viruses in the remediated PDB archive. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Olson, A.J. Icosahedral virus structures and the protein data bank. J. Biol. Chem. 2021, 296, 100554. [Google Scholar] [CrossRef]
- Reddy, V.S.; Natarajan, P.; Okerberg, B.; Li, K.; Damodaran, K.; Morton, R.T.; Brooks, C.L., III; Johnson, J.E. Virus Particle Explorer (VIPER), a website for virus capsid structures and their computational analyses. J. Virol. 2001, 75, 11943–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, C.M.; Borelli, I.A.; Lander, G.; Natarajan, P.; Siddavanahalli, V.; Bajaj, C.; Johnson, J.E.; Brooks, C.L., III; Reddy, V.S. VIPERdb: A relational database for structural virology. Nucleic Acids Res. 2006, 34, D386–D389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo-Tripp, M.; Shepherd, C.M.; Borelli, I.A.; Venkataraman, S.; Lander, G.; Natarajan, P.; Johnson, J.E.; Brooks, C.L., III; Reddy, V.S. VIPERdb2: An enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 2009, 37, D436–D442. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.T.; Montiel-Garcia, D.J.; Wong, J.J.; Carrillo-Tripp, M.; Brooks, C.L., III; Johnson, J.E.; Reddy, V.S. VIPERdb: A tool for virus research. Annu. Rev. Virol. 2018, 5, 477–488. [Google Scholar] [CrossRef]
- Montiel-Garcia, D.; Santoyo-Rivera, N.; Ho, P.; Carrillo-Tripp, M.; Iii, C.L.B.; Johnson, J.E.; Reddy, V.S. VIPERdb v3.0: A structure-based data analytics platform for viral capsids. Nucleic Acids Res. 2021, 49, D809–D816. [Google Scholar] [CrossRef]
- Chapman, M.S.; Liljas, L. Structural folds of viral proteins. Adv. Protein Chem. 2003, 64, 125–196. [Google Scholar]
- Cheng, S.; Brooks, C.L., III. Viral capsid proteins are segregated in structural fold space. PLoS Comput. Biol. 2013, 9, e1002905. [Google Scholar] [CrossRef] [PubMed]
- Sevvana, M.; Klose, T.; Rossmann, M.G. Principles of virus structure. Encycl. Virol. 2021, 1, 257–277. [Google Scholar]
- Senior, A.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Callaway, E. “It will change everything”: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Thornton, J. The impact of AlphaFold2 one year on. Nat. Methods 2022, 19, 15–20. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, Artificial Intelligence, and Allostery. J. Phys. Chem. B 2022, 126, 6372–6383. [Google Scholar] [CrossRef]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stärk, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, allosteric, and orthosteric drug discovery: Ways forward. Drug Discov. Today 2023, 28, 103551. [Google Scholar] [CrossRef] [PubMed]
- Twarock, R. Mathematical virology: A novel approach to the structure and assembly of viruses. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2006, 364, 3357–3373. [Google Scholar] [CrossRef] [PubMed]
- Cieplak, M.; Roos, W.H. Special Issue on the Physics of Viral Capsids. J. Phys. Condens. Matter 2018, 30, 290201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, R.; Dragnea, B.; Travesset, A.; Podgornik, R. On virus growth and form. Phys. Rep. 2020, 847, 1–102. [Google Scholar] [CrossRef]
- Twarock, R.; Bingham, R.J.; Dykeman, E.C.; Stockley, P.G. A modelling paradigm for RNA virus assembly. Curr. Opin. Virol. 2018, 31, 74–81. [Google Scholar] [CrossRef]
- Twarock, R.; Luque, A. Structural puzzles in virology solved with an overarching icosahedral design principle. Nat. Commun. 2019, 10, 4414. [Google Scholar] [CrossRef] [Green Version]
- Martín-Bravo, M.; Llorente, J.M.G.; Hernández-Rojas, J.; Wales, D.J. Minimal design principles for icosahedral virus capsids. ACS Nano 2021, 15, 14873–14884. [Google Scholar] [CrossRef]
- Indelicato, G.; Cermelli, P.; Twarock, R. Local rules for the self-assembly of a non-quasi-equivalent viral capsid. Phys. Rev. E 2022, 105, 064403. [Google Scholar] [CrossRef]
- Tetter, S.; Terasaka, N.; Steinauer, A.; Bingham, R.J.; Clark, S.; Scott, A.J.; Patel, N.; Leibundgut, M.; Wroblewski, E.; Ban, N.; et al. Evolution of a virus-like architecture and packaging mechanism in a repurposed bacterial protein. Science 2021, 372, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.E.; Šulc, P.; Sciortino, F.; Russo, J. Design strategies for the self-assembly of polyhedral shells. Proc. Natl. Acad. Sci. USA 2023, 120, e2219458120. [Google Scholar] [CrossRef]
- Li, S.; Orland, H.; Zandi, R. Self consistent field theory of virus assembly. J. Phys. Condens. Matter 2018, 30, 144002. [Google Scholar] [CrossRef] [PubMed]
- van der Holst, B.; Kegel, W.K.; Zandi, R.; van der Schoot, P. The different faces of mass action in virus assembly. J. Biol. Phys. 2018, 44, 163–179. [Google Scholar] [CrossRef] [Green Version]
- Panahandeh, S.; Li, S.; Dragnea, B.; Zandi, R. Virus assembly pathways inside a host cell. ACS Nano 2022, 16, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Pak, A.J.; Voth, G.A. Critical mechanistic features of HIV-1 viral capsid assembly. Sci. Adv. 2023, 9, eadd7434. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.B.; Ramezani, A.; Montalvo, T.; Nguyen, M.; van der Schoot, P.; van Hest, J.C.; Zandi, R. The dynamics of viruslike capsid assembly and disassembly. J. Am. Chem. Soc. 2022, 144, 12608–12612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.; Yufenyuy, E.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzinek, J.K.; Huber, R.G.; Bond, P.J. Multiscale modelling and simulation of viruses. Curr. Opin. Struct. Biol. 2020, 61, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.G.; Marzinek, J.K.; Boon, P.L.; Yue, W.; Bond, P.J. Computational modelling of flavivirus dynamics: The ins and outs. Methods 2021, 185, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.E.; Pérez-Segura, C.; Bryer, A.J.; Perilla, J.R.; Hadden-Perilla, J.A. Molecular dynamics of the viral life cycle: Progress and prospects. Curr. Opin. Virol. 2021, 50, 128–138. [Google Scholar] [CrossRef]
- Lynch, D.L.; Pavlova, A.; Fan, Z.; Gumbart, J.C. Understanding Virus Structure and Dynamics through Molecular Simulations. J. Chem. Theory Comput. 2023. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Corey, R.B.; Pauling, L. Molecular models of amino acids, peptides, and proteins. Rev. Sci. Instruments 1953, 24, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Koltun, W.L. Precision space-filling atomic models. Biopolym. Orig. Res. Biomol. 1965, 3, 665–679. [Google Scholar] [CrossRef]
- Eisenberg, D.; McLachlan, A.D. Solvation energy in protein folding and binding. Nature 1986, 319, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Ooi, T.; Oobatake, M.; Nemethy, G.; Scheraga, H.A. Accessible surface-areas as a measure of the thermodynamic parameters of hydration of peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3086–3090. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Edelsbrunner, H.; Fu, P.; Sudhakar, P.V.; Subramaniam, S. Analytical shape computation of macromolecules. I. Molecular area and volume through Alpha Shape. Proteins Struct. Func. Genet. 1998, 33, 1–17. [Google Scholar] [CrossRef]
- Liang, J.; Edelsbrunner, H.; Fu, P.; Sudhakar, P.V.; Subramaniam, S. Analytical shape computation of macromolecules. II. Inaccessible cavities in proteins. Proteins Struct. Func. Genet. 1998, 33, 18–29. [Google Scholar] [CrossRef]
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at small and large length scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar] [CrossRef]
- König, P.M.; Roth, R.; Mecke, K. Morphological thermodynamics of fluids: Shape dependence of free energies. Phys. Rev. Lett. 2004, 93, 160601. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Richards, F.M. Interpretation of protein structures: Estimation of static accessibility. J. Molec. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Molec. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Legrand, S.M.; Merz, K.M. Rapid approximation to molecular-surface area via the use of boolean logic and look-up tables. J. Comp. Chem. 1993, 14, 349–352. [Google Scholar] [CrossRef]
- Wang, H.; Levinthal, C. A vectorized algorithm for calculating the accessible surface area of macromolecules. J. Comp. Chem. 1991, 12, 868–871. [Google Scholar] [CrossRef]
- Futamura, N.; Alura, S.; Ranjan, D.; Hariharan, B. Efficient parallel algorithms for solvent accessible surface area of proteins. IEEE Trans. Parallel Dist. Syst. 2002, 13, 544–555. [Google Scholar] [CrossRef]
- Till, M.; Ullmann, G.M. McVol—A program for calculating protein volumes and identifying cavities by a Monte Carlo algorithm. J. Mol. Model. 2010, 16, 419–429. [Google Scholar] [CrossRef]
- Wodak, S.J.; Janin, J. Analytical approximation to the accessible surface-area of proteins. Proc. Natl. Acad. Sci. USA 1980, 77, 1736–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasel, W.; Hendrikson, T.F.; Still, W.C. A rapid approximation to the solvent accessible surface areas of atoms. Tetrahed. Comp. Method. 1988, 1, 103–106. [Google Scholar] [CrossRef]
- Petitjean, M. On the analytical calculation of van-der-Waals surfaces and volumes: Some numerical aspects. J. Comp. Chem. 1994, 15, 507–523. [Google Scholar] [CrossRef]
- Street, A.G.; Mayo, S.L. Pairwise calculation of protein solvent-accessible surface areas. Fold. Des. 1998, 3, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo, L.; Kleinjung, J.; Fraternali, F. POPS: A fast algorithm for solvent accessible surface areas at atomic and residue level. Nucl. Acids. Res. 2003, 31, 3364–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dynerman, D.; Butzlaff, E.; Mitchell, J. CUSA and CUDE: GPU-accelerated methods for estimating solvent accessible surface area and desolvation. J. Comput. Biol. 2009, 16, 523–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richmond, T.J. Solvent accessible surface-area and excluded volume in proteins. Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Molec. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Connolly, M.L. Computation of molecular volume. J. Am. Chem. Soc. 1985, 107, 1118–1124. [Google Scholar] [CrossRef]
- Gibson, K.D.; Scheraga, H.A. Exact calculation of the volume and surface-area of fused hard-sphere molecules with unequal atomic radii. Mol. Phys. 1987, 62, 1247–1265. [Google Scholar] [CrossRef]
- Edelsbrunner, H. The union of balls and its dual shape. Discret. Comput. Geom. 1995, 13, 415–440. [Google Scholar] [CrossRef] [Green Version]
- Levitt, D.; Banaszak, L. POCKET: A computer graphics method for identifying and displaying protein cavities and their surrounding amino acids. J. Mol. Graph. 1992, 10, 229–234. [Google Scholar] [CrossRef]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, C.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Weisel, M.; Proschak, E.; Schneider, G. PocketPicker: Analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph. 1995, 13, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Stouten, P. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Go, N. Detection of pockets on protein surfaces using small and large probe spheres to find putative ligand binding sites. Proteins: Struct. Func. Genet. 2007, 68, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelsbrunner, H.; Koehl, P. The weighted-volume derivative of a space-filling diagram. Proc. Natl. Acad. Sci. USA 2003, 100, 2203–2208. [Google Scholar] [CrossRef] [Green Version]
- Bryant, R.; Edelsbrunner, H.; Koehl, P.; Levitt, M. The area derivative of a space-filling diagram. Discret. Comput. Geom. 2004, 32, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Edelsbrunner, H.; Koehl, P. The geometry of biomolecular solvation. Discret. Comput. Geom. 2005, 52, 243–275. [Google Scholar]
- Yaffe, E.; Fishelovitch, D.; Wolfson, H.; Halperin, D.; Nussinov, R. MolAxis: A server for identification of channels in macromolecules. Nucl. Acids. Res. 2008, 36, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Albou, L.P.; Schwarz, B.; Poch, O.; Wurtz, J.M.; Moras, D. Defining and characterizing protein surface using alpha shapes. Proteins Struct. Funct. Bioinform. 2009, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mach, P.; Koehl, P. Geometric measures of large biomolecules: Surface, volume, and pockets. J. Comp. Chem. 2011, 32, 3023–3038. [Google Scholar] [CrossRef] [Green Version]
- Akopyan, A.; Edelsbrunner, H. The Weighted Mean Curvature Derivative of a Space-Filling Diagram. Comput. Math. Biophys. 2020, 8, 51–67. [Google Scholar] [CrossRef]
- Akopyan, A.; Edelsbrunner, H. The Weighted Gaussian Curvature Derivative of a Space-Filling Diagram. Comput. Math. Biophys. 2020, 8, 74–88. [Google Scholar] [CrossRef]
- Koehl, P.; Akopyan, A.; Edelsbrunner, H. Computing the Volume, Surface Area, Mean, and Gaussian Curvatures of Molecules and Their Derivatives. J. Chem. Inf. Model. 2023, 63, 973–985. [Google Scholar] [CrossRef]
- Li, J.; Mach, P.; Koehl, P. Measuring the shapes of macromolecules and why it matters. Comp. Struct. Biotech. J. 2013, 8, e201309001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelsbrunner, H.; Facello, M.A.; Liang, J. On the definition and construction of pockets in macromolecules. Discret. Appl. Math. 1998, 88, 83–102. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Verma, C. Straightforward incorporation of multiple ligand types into molecular dynamics simulations for efficient binding site detection and characterization. J. Chem. Theory Comput. 2020, 16, 6633–6644. [Google Scholar] [CrossRef]
- Hardy, J.M.; Newton, N.D.; Modhiran, N.; Scott, C.A.; Venugopal, H.; Vet, L.J.; Young, P.R.; Hall, R.A.; Hobson-Peters, J.; Coulibaly, F.; et al. A unified route for flavivirus structures uncovers essential pocket factors conserved across pathogenic viruses. Nat. Commun. 2021, 12, 3266. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.4; Schrödinger, LLC: New York, NY, USA, 2020.
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S. A ligand-binding pocket in the Dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef] [Green Version]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S. Structure of the Dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Dokland, T.; Walsh, M.; Mackenzie, J.M.; Khromykh, A.A.; Ee, K.H.; Wang, S. West Nile virus core protein: Tetramer structure and ribbon formation. Structure 2004, 12, 1157–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Z.; Song, H.; Shi, Y.; Qi, J.; Gao, G.F. Crystal structure of the capsid protein from Zika virus. J. Molec. Biol. 2018, 430, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Xie, X.; Zou, J.; Noble, C.G.; Russell, W.K.; Holthauzen, L.M.F.; Choi, K.H.; White, M.A.; Shi, P.Y. A cocrystal structure of dengue capsid protein in complex of inhibitor. Proc. Natl. Acad. Sci. USA 2020, 117, 17992–18001. [Google Scholar] [CrossRef]
- Tan, T.Y.; Fibriansah, G.; Kostyuchenko, V.A.; Ng, T.S.; Lim, X.X.; Zhang, S.; Lim, X.N.; Wang, J.; Shi, J.; Morais, M.C.; et al. Capsid protein structure in Zika virus reveals the flavivirus assembly process. Nat. Commun. 2020, 11, 895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, C.M.; Dai, D.; Grosenbach, D.W.; Berhanu, A.; Jones, K.F.; Cardwell, K.B.; Schneider, C.; Wineinger, K.A.; Page, J.M.; Harver, C.; et al. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob. Agents Chemother. 2013, 57, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Shaw, D.; Deneroff, M.; Dror, R.; Kuskin, J.; Larson, R.; Salmon, J.; Young, C.; Batson, B.; Bowers, K.; Chao, J.; et al. Anton, a Special-purpose Machine for Molecular Dynamics Simulation. Commun. ACM 2008, 51, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.; Hardy, D.; Ufimtsev, I.; Schulten, K. GPU-accelerated molecular modeling coming of age. J. Molec. Graph. Model. 2010, 29, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Harrison, C.; Schulten, K.; McCammon, J. Implementation of accelerated molecular dynamics in NAMD. Comput. Sci. Discov. 2011, 4, 015002. [Google Scholar] [CrossRef]
- Pierce, L.; Salomon-Ferrer, R.; Augusto, F.; de Oliveira, C.; McCammon, J.A.; Walker, R.C. Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theor. Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Sweet, J.; Nowling, R.; Cickovski, T.; Sweet, C.; Pande, V.; Izaguirre, J. Long Timestep Molecular Dynamics on the Graphical Processing Unit. J. Chem. Theor. Comput. 2013, 13, 3267–3281. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC14: International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Eastman, P.; Pande, V. OpenMM: A Hardware Independent Framework for Molecular Simulations. Comput. Sci. Eng. 2015, 12, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Sener, M.; Strumpfer, J.; Singharoy, A.; Hunter, C.; Schulten, K. Overall energy conversion efficiency of a photosynthetic vesicle. Elife 2016, 5, e09541. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.; Hardy, D.; Maia, J.; Stone, J.; Ribeiro, J.; Bernardi, R.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kobayashi, C.; Kasahara, K.; Tan, C.; Kuroda, A.; Minami, K.; Ishiduki, S.; Nishiki, T.; Inoue, H.; Ishikawa, Y.; et al. New parallel computing algorithm of molecular dynamics for extremely huge scale biological systems. J. Comp. Chem. 2021, 42, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.; Sarkar, D.; Tieleman, D.; Singharoy, A. The ugly, bad, and good stories of large-scale biomolecular simulations. Curr. Opin. Struct. Biol. 2022, 73, 102338. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Sanejouand, Y.H. On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys. 2015, 567, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.; Voth, G. Coarse-graining Methods for Computational Biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Lopez-Blanco, J.; Chacon, P. New generation of elastic network models. Curr. Opin. Struct. Biol. 2016, 37, 46–53. [Google Scholar] [CrossRef]
- Noguti, T.; Go, N. Collective variable description of small-amplitude conformational fluctuations in a globular protein. Nature 1982, 296, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.; Bruccoleri, R.; Olafson, B. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Levitt, M.; Sander, C.; Stern, P. Protein normal-mode dynamics: Trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Molec. Biol. 1985, 181, 423–447. [Google Scholar] [CrossRef]
- Tama, F.; Brooks, C., III. Symmetry, form, and shape: Guiding principles for robustness in macromolecular machines. Ann. Rev. Biophys. Biomol. Struct. 2006, 35, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Dykeman, E.; Sankey, O. Normal mode analysis and applications in biological physics. J. Phys. Condens. Matter 2010, 22, 423202. [Google Scholar] [CrossRef] [PubMed]
- Lezon, T.; Shrivastava, I.; Yan, Z.; Bahar, I. Elastic Network Models For Biomolecular Dynamics: Theory and Application to Membrane Proteins and Viruses. In Handbook on Biological Networks; Boccaletti, S., Latora, V., Moreno, Y., Eds.; World Scientific Publishing Co: Singapore, 2010; pp. 129–158. [Google Scholar]
- Hsieh, Y.C.; Poitevin, F.; Delarue, M.; Koehl, P. Comparative normal mode analysis of the dynamics of DENV and ZIKV capsids. Front. Bio. Sci. 2016, 3, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehl, P. Large eigenvalue problems in coarse-grained dynamic analyses of supramolecular systems. J. Chem. Theory Comput. 2018, 14, 3903–3919. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L. Modern protein force fields behave comparably in molecular dynamics simulations. J. Comp. Chem. 2002, 23, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirion, M. Large amplitude elastic motions in proteins from a single parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Taketomi, H.; Gō, N. Studies on protein folding, unfolding and fluctuations by computer simulation. I. The effects of specific amino acid sequence represented by specific inter-unit interactions. Int. J. Pept. Res. 1975, 7, 445–459. [Google Scholar]
- Lin, T.L.; Song, G. Generalized spring tensor models for protein fluctuation dynamics and conformation changes. BMC Struct. Biol. 2010, 10, S3. [Google Scholar] [CrossRef] [Green Version]
- Na, H.; Lin, T.L.; Song, G. Generalized spring tensor models for protein fluctuation dynamics and conformation changes. Adv. Exp. Med. Biol. 2014, 805, 107–135. [Google Scholar]
- Xia, F.; Tong, D.; Yang, L.; Wang, D.; Doi, S.; Koehl, P.; Lu, L. Identifying essential pairwise interactions in elastic network model using the alpha shape theory. J. Comp. Chem. 2014, 35, 1111–1121. [Google Scholar] [CrossRef]
- Anderson, E.; Bai, Z.; Bischof, C.; Demmel, J.; Dongarra, J.; Ducroz, J.; Greenbaum, A.; Hammarling, S.; Mckenney, A.; Sorensen, D. LAPACK: A Portable Linear Algebra Library for High-Performance Computers; Technical Report CS-90-105; Computer Science Department University of Tennessee: Knoxville, TN, USA, 1990. [Google Scholar]
- Koehl, P.; Delarue, M. Coarse-grained dynamics of supramolecules: Conformational changes in outer shells of Dengue viruses. Prog. Biophys. Mol. Biol. 2019, 143, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Petrone, P.; Pande, V. Can conformational change be described by only a few normal modes? Biophys. J. 2006, 90, 1583–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehoucq, R.; Sorensen, D.; Yang, C. ARPACK Users’ Guide: Solution of Large-Scale Eigenvalue Problems with Implicitly Restarted Arnoldi Methods; SIAM: Philadelphia, PA, USA, 1998. [Google Scholar]
- Rutishauser, H. Computational aspects of F.L. Bauer’s simultaneous iteration method. Numer. Math. 1969, 13, 4–13. [Google Scholar] [CrossRef]
- Saad, Y. Chebishev acceleration techniques for solving nonsymmetric eigenvalue problems. Math. Comput. 1984, 42, 567–588. [Google Scholar] [CrossRef]
- Dykeman, E.; Sankey, O. Low frequency mechanical modes of viral capsids: An atomistic approach. Phys. Rev. Lett. 2008, 100, 028101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Saad, Y. A Chebishev-Davidson algorithm for large symmetric eigenvalue problems. SIAM J. Matrix Anal. Appl. 2006, 29, 341–359. [Google Scholar]
- Zhou, Y. A block Chebishev-Davidson method with inner-outer restart for large eigenvalue problems. J. Comput. Phys. 2010, 229, 9188–9200. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins: Struct. Func. Genet. 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Brooks, C., III. The mechanism and pathway of pH induced swelling in cowpea chlorotic mottle virus. J. Molec. Biol. 2002, 318, 733–747. [Google Scholar] [CrossRef]
- Tama, F.; Brooks, C., III. Diversity and identity of mechanical properties of icosahedral viral capsids studied with elastic network normal mode analysis. J. Molec. Biol. 2005, 345, 299–314. [Google Scholar] [CrossRef]
- Rader, A.; Vlad, D.; Bahar, I. Maturation dynamics of bacteriophage HK97 capsid. Structure 2005, 13, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Chennubotla, C.; Rader, A.; Yang, L.; Bahar, I. Elastic network models for understanding biomolecular machinery: From enzymes to supramolecular assemblies. Phys. Biol. 2005, 2, S173–S180. [Google Scholar] [CrossRef]
- Kim, M.; Jernigan, R.; Chirikjian, G. An elastic network model of HK97 capsid maturation. J. Struct. Biol. 2003, 143, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Polles, G.; Indelicato, G.; Potestio, R.; Cermelli, P.; Twarock, R.; Micheletti, C. Mechanical and assembly units of viral capsids identified via quasi-rigid domain decomposition. PLoS Comput. Biol. 2013, 9, e1003331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ssentongo, P.; Ssentongo, A.E.; Voleti, N.; Groff, D.; Sun, A.; Ba, D.M.; Nunez, J.; Parent, L.J.; Chinchilli, V.M.; Paules, C.I. SARS-CoV-2 vaccine effectiveness against infection, symptomatic and severe COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2022, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Henrich, S.; Salo-Ahen, O.; Huang, B.; Rippmann, F.; Cruciani, G.; Wade, R. Computational approaches to identifying and characterizing protein binding sites for ligand design. J. Molec. Recognit. 2010, 23, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janežič, D. Binding site comparison for function prediction and pharmaceutical discovery. Curr. Opin. Struct. Biol. 2014, 25, 34–39. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, Y.; Zhang, L. Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J. 2020, 18, 417–426. [Google Scholar] [CrossRef]
- Laurie, A.; Jackson, R. Q-SiteFinder: An energy-based method for theprediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef]
- Murray, C.; Rees, D. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug delivery: What does it hold? Expert Opin. Drug Discov. 2019, 14, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Goodford, P. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Miranker, A.; Karplus, M. Functionality maps of binding-sites—A multiple copy simultaneous search method. Proteins 1991, 11, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.Y.; Begloc, D.; Hall, D.; Landon, M.; Mattos, C.; Vajda, S. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Grove, L.; Hall, D.; Bohnuud, T.; Mottarella, S.; Luo, L.; Xia, B.; Begloz, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Ivetac, A.; McCammon, J. A molecular dynamics ensemble-based approach for the mapping of druggable binding sites. Methods Mol. Biol. 2018, 819, 3–12. [Google Scholar]
- Feng, T.; Barakat, K. Molecular dynamics simulation and prediction of druggable binding sites. Methods Mol. Biol. 2018, 1762, 87–103. [Google Scholar]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, M.; Sturlese, M.; Moro, S. The rise of molecular simulations in fragment-based drug design (FBDD): An overview. Drug Discov. Today 2020, 25, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Basse, N.; Kaar, J.; Settanni, G.; Joerger, A.; Rutherford, T.; Fersht, A. Toward the rational design of p53-stabilizing drugs: Probing the surface of the oncogenic Y220C mutant. Chem. Biol. 2010, 17, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Sledz, P.; Lang, S.; Stubbs, C.; Spring, D.; Abell, C.; Best, R. Using ligand-mapping simulations to design a ligand selectively targeting a cryptic surface pocket of Polo-Like kinase 1. Angew. Chem. Int. Ed. 2012, 51, 10078–10081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Spring, D.; Verma, C. The use of chlorobenzene as a probe molecule in molecular dynamics simulations. J. Chem. Inf. Model. 2014, 54, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Kalenkiewicz, A.; Grant, B.; Yang, C.Y. Enrichment of druggable conformations from apo protein structures using cosolvent-accelerated molecular dynamics. Biology 2015, 4, 344–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Hu, H.; Ruvinsky, A.; Sherman, W.; Favia, A. Deciphering cryptic binding sites on proteins by mixed-solvent molecular dynamics. J. Chem. Inf. Model. 2017, 57, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Boehm, M.; McClendon, C.; Torella, R.; Gohlke, H. Cosolvent-enhanced sampling and unbiased identification of cryptic pockets suitable for structure-based drug design. J. Chem. Theory Comput. 2019, 15, 3331–3343. [Google Scholar] [CrossRef]
- Koehl, P.; Delarue, M.; Orland, H. Simultaneous identification of multiple binding sites in proteins: A statistical mechanics approach. J. Phys. Chem. B 2021, 125, 5052–5067. [Google Scholar] [CrossRef]
- Azuara, C.; Lindahl, E.; Koehl, P.; Orland, H.; Delarue, M. PDB_Hydro. Incorporating dipolar solvents with variable density in the Poisson–Boltzmann treatment of macromolecule electrostatics. Nucl. Acids. Res. 2006, 34, W38–W42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuara, A.; Orland, H.; Bon, M.; Koehl, P.; Delarue, M. Incorporating dipolar solvents with variable density in Poisson-Boltzmann electrostatics. Biophys. J. 2008, 95, 5587–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehl, P.; Delarue, M. AQUASOL: An efficient solver for the dipolar Poisson-Boltzmann-Langevin equation. J. Chem. Phys. 2010, 132, 064101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, M. Multilevel Methods for the Poisson–Boltzmann Equation. Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 1993. [Google Scholar]
- Dolinsky, T.; Nielsen, J.; Cammon, J.M.; Baker, N. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucl. Acids. Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, Y.; Shatkin, A.J. Viral and cellular mRNA capping: Past and prospects. Adv. Virus Res. 2000, 55, 135–184. [Google Scholar]
- Muthukrishnan, S.; Both, G.; Furuichi, Y.; Shatkin, A. 5’-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 1975, 255, 33–37. [Google Scholar] [CrossRef]
- Dong, H.; Liu, L.; Zou, G.; Zhao, Y.; Li, Z.; Lim, S.P.; Shi, P.Y.; Li, H. Structural and functional analyses of a conserved hydrophobic pocket of flavivirus methyltransferase. J. Biol. Chem. 2010, 285, 32586–32595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, Y.-C.; Delarue, M.; Orland, H.; Koehl, P. Analyzing the Geometry and Dynamics of Viral Structures: A Review of Computational Approaches Based on Alpha Shape Theory, Normal Mode Analysis, and Poisson–Boltzmann Theories. Viruses 2023, 15, 1366. https://doi.org/10.3390/v15061366
Hsieh Y-C, Delarue M, Orland H, Koehl P. Analyzing the Geometry and Dynamics of Viral Structures: A Review of Computational Approaches Based on Alpha Shape Theory, Normal Mode Analysis, and Poisson–Boltzmann Theories. Viruses. 2023; 15(6):1366. https://doi.org/10.3390/v15061366
Chicago/Turabian StyleHsieh, Yin-Chen, Marc Delarue, Henri Orland, and Patrice Koehl. 2023. "Analyzing the Geometry and Dynamics of Viral Structures: A Review of Computational Approaches Based on Alpha Shape Theory, Normal Mode Analysis, and Poisson–Boltzmann Theories" Viruses 15, no. 6: 1366. https://doi.org/10.3390/v15061366