
Two Novel Yersinia pestis Bacteriophages with a Broad Host Range: Potential as Biocontrol Agents in Plague Natural Foci
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Identification of Phages
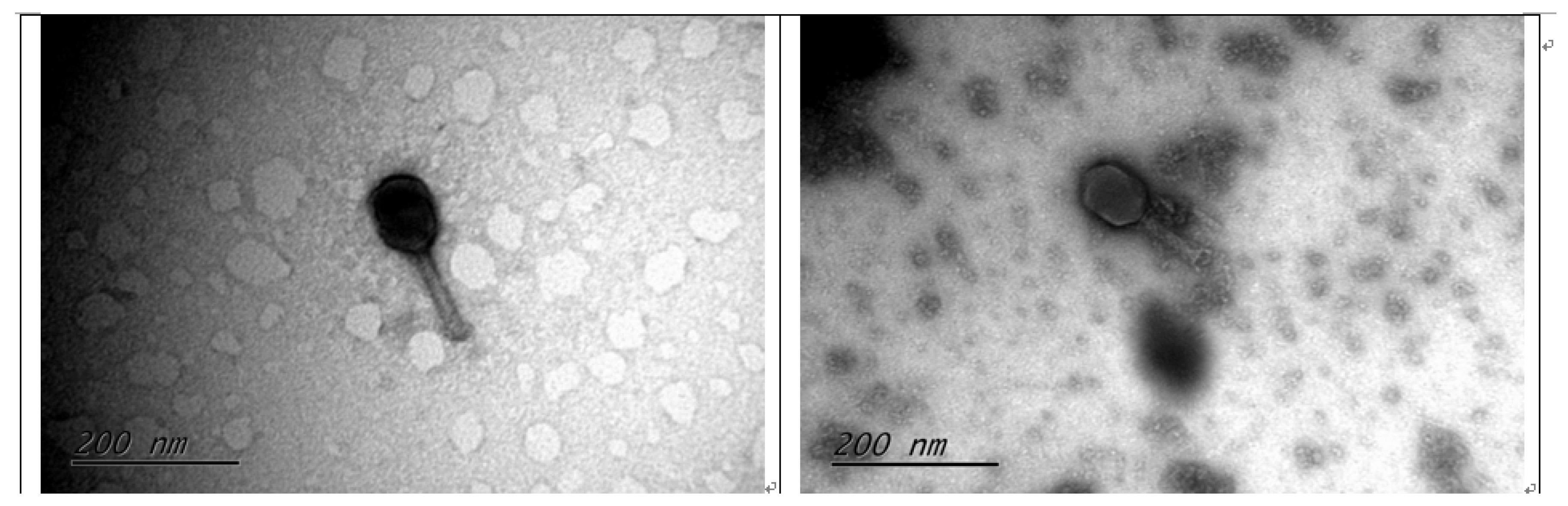
2.2. Transmission Electron Microscopy
2.3. Host Range Analysis
2.4. Growth Characteristics and Thermo, pH Stability Tests
2.5. Phage Genome Sequencing, Assembly, Annotation, and Comparison
2.6. Alignments of the Inferred Amino Acid Sequences of gp23
2.7. Accession Number
3. Results
3.1. Isolation and Morphology of the MHS112 and GMS130 Phages
3.2. Host Range Determination of MHS112 and GMS130 Phages
3.3. Growth Characteristics and Stability Assessment
3.4. Genomic Features of MHS112 and GMS130 Phages
3.5. Alignments of MHS112 and GMS130 with Phage SP18 and Phage vB_EcoM_VR20
3.6. Phylogenetic Relationship Analysis of Phages in the Genus Gaprivervirus and T4-Like Phages Infecting the Genus Yersinia
4. Discussion
4.1. Morphological Properties with T4-Like Phage Characteristics
4.2. Associated Ecological Function of Phages with Wider Host Ranges
4.3. The Ecological Barrier Function Endowed Phages with a Wider Host Range in Natural Plague Foci
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morelli, G.; Song, Y.; Mazzoni, C.J.; Eppinger, M.; Roumagnac, P.; Wagner, D.M.; Feldkamp, M.; Kusecek, B.; Vogler, A.J.; Li, Y.; et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 2010, 42, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Guiyoule, A.; Gerbaud, G.; Buchrieser, C.; Galimand, M.; Rahalison, L.; Chanteau, S.; Courvalin, P.; Carniel, E. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg. Infect. Dis. 2001, 7, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimand, M.; Guiyoule, A.; Gerbaud, G.; Rasoamanana, B.; Chanteau, S.; Carniel, E.; Courvalin, P. Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N. Engl. J. Med. 1997, 337, 677–680. [Google Scholar] [CrossRef]
- Dai, R.; He, J.; Zha, X.; Wang, Y.; Zhang, X.; Gao, H.; Yang, X.; Li, J.; Xin, Y.; Wang, Y.; et al. A novel mechanism of streptomycin resistance in Yersinia pestis: Mutation in the rpsL gene. PLoS Negl. Trop. Dis. 2021, 15, e0009324. [Google Scholar] [CrossRef] [PubMed]
- Andrianaivoarimanana, V.; Wagner, D.M.; Birdsell, D.N.; Nikolay, B.; Rakotoarimanana, F.; Randriantseheno, L.N.; Vogler, A.J.; Sahl, C.M.; Somprasong, N.; Cauchemez, S.; et al. Transmission of Antimicrobial Resistant Yersinia pestis during a Pneumonic Plague Outbreak. Clin. Infect. Dis. 2022, 74, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, H.; Li, M.; Qi, W.; Qi, Z.; Chen, W.; Dong, Y.; Xu, Z.; Zhang, W. Characterization and genome analysis of a broad lytic spectrum bacteriophage P479 against multidrug-resistant Escherichia coli. Virus Res. 2022, 308, 198628. [Google Scholar] [CrossRef]
- Zhao, X.; Skurnik, M. Bacteriophages of Yersinia pestis. Adv. Exp. Med. Biol. 2016, 918, 361–375. [Google Scholar] [CrossRef]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Huang, X.Z.; Gnade, B.T.; Mueller, A.J.; Fernandez-Prada, C.M.; Nikolich, M.P. Bacteriophage-resistant mutants in Yersinia pestis: Identification of phage receptors and attenuation for mice. PLoS ONE 2011, 6, e25486. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Cui, Y.; Yan, Y.; Du, Z.; Tan, Y.; Yang, H.; Bi, Y.; Zhang, P.; Zhou, L.; Zhou, D.; et al. Outer membrane proteins ail and OmpF of Yersinia pestis are involved in the adsorption of T7-related bacteriophage Yep-phi. J. Virol. 2013, 87, 12260–12269. [Google Scholar] [CrossRef] [Green Version]
- Filippov, A.A.; Sergueev, K.V.; He, Y.; Nikolich, M.P. Bacteriophages capable of lysing Yersinia pestis and Yersinia pseudotuberculosis: Efficiency of plating tests and identification of receptors in escherichia coli K-12. Adv. Exp. Med. Biol. 2012, 954, 123–134. [Google Scholar] [CrossRef]
- Rashid, M.H.; Revazishvili, T.; Dean, T.; Butani, A.; Verratti, K.; Bishop-Lilly, K.A.; Sozhamannan, S.; Sulakvelidze, A.; Rajanna, C. A Yersinia pestis-specific, lytic phage preparation significantly reduces viable Y. pestis on various hard surfaces experimentally contaminated with the bacterium. Bacteriophage 2012, 2, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Kiljunen, S.; Datta, N.; Dentovskaya, S.V.; Anisimov, A.P.; Knirel, Y.A.; Bengoechea, J.A.; Holst, O.; Skurnik, M. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage phiA1122. J. Bacteriol. 2011, 193, 4963–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wu, W.; Qi, Z.; Cui, Y.; Yan, Y.; Guo, Z.; Wang, Z.; Wang, H.; Deng, H.; Xue, Y.; et al. The complete genome sequence and proteomics of Yersinia pestis phage Yep-phi. J. Gen. Virol. 2011, 92, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Chouikha, I.; Charrier, L.; Filali, S.; Derbise, A.; Carniel, E. Insights into the infective properties of YpfPhi, the Yersinia pestis filamentous phage. Virology 2010, 407, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, E.; Chain, P.; Elliott, J.M.; Bobrov, A.G.; Motin, V.L.; Kirillina, O.; Lao, V.; Calendar, R.; Filippov, A.A. Molecular characterization of L-413C, a P2-related plague diagnostic bacteriophage. Virology 2008, 372, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Meng, B.; Wei, X.; Li, X.; Peng, H.; Li, Y.; Feng, Q.; Huang, Y.; Zhang, Q.; Xu, X.; et al. Identification and characterization of P2-like bacteriophages of Yersinia pestis. Virus Res. 2022, 322, 198934. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Pajunen, M.I.; Jun, J.W.; Skurnik, M. T4-like Bacteriophages Isolated from Pig Stools Infect Yersinia pseudotuberculosis and Yersinia pestis Using LPS and OmpF as Receptors. Viruses 2021, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M.; Jaakkola, S.; Mattinen, L.; von Ossowski, L.; Nawaz, A.; Pajunen, M.I.; Happonen, L.J. Bacteriophages fEV-1 and fD1 Infect Yersinia pestis. Viruses 2021, 13, 384. [Google Scholar] [CrossRef]
- Yuan, Y.; Xi, H.; Dai, J.; Zhong, Y.; Lu, S.; Wang, T.; Yang, L.; Guan, Y.; Wang, P. The characteristics and genome analysis of the novel Y. pestis phage JC221. Virus Res. 2020, 283, 197982. [Google Scholar] [CrossRef]
- Anand, T.; Vaid, R.K.; Bera, B.; Barua, S.; Riyesh, T.; Virmani, N.; Yadav, N.; Malik, P. Isolation and characterization of a bacteriophage with broad host range, displaying potential in preventing bovine diarrhoea. Virus Genes 2015, 51, 315–521. [Google Scholar] [CrossRef]
- Liao, Y.T.; Zhang, Y.; Salvador, A.; Harden, L.A.; Wu, V.C.H. Characterization of a T4-like Bacteriophage vB_EcoM-Sa45lw as a Potential Biocontrol Agent for Shiga Toxin-Producing Escherichia coli O45 Contaminated on Mung Bean Seeds. Microbiol. Spectrum 2022, 10, e0222021. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetart, F.; Desplats, C.; Kutateladze, M.; Monod, C.; Ackermann, H.W.; Krisch, H.M. Phylogeny of the major head and tail genes of the wide-ranging T4-type bacteriophages. J Bacteriol. 2001, 183, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Chang, H.W.; Nam, Y.D.; Roh, S.W.; Bae, J.W. Phenotypic characterization and genomic analysis of the Shigella sonnei bacteriophage SP18. J. Microbiol. 2010, 48, 213–222. [Google Scholar] [CrossRef]
- Kaliniene, L.; Klausa, V.; Truncaite, L. Low-temperature T4-like coliphages vB_EcoM-VR5, vB_EcoM-VR7 and vB_EcoM-VR20. Arch. Virol. 2010, 155, 871–880. [Google Scholar] [CrossRef]
- Kazibwe, G.; Ndekezi, C.; Alinaitwe, R.; Alafi, S.; Nanteza, A.; Kimuda, M.P.; Nakavuma, J.L. Genome Sequences of Bacteriophages UPEC01, UPEC03, UPEC06, and UPEC07 Infecting Avian Pathogenic Escherichia coli. Microbiol. Resour. Announc. 2022, 11, e0081121. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, H.W.; Krisch, H.M. A catalogue of T4-type bacteriophages. Arch. Virol. 1997, 142, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Chibani-Chennoufi, S.; Canchaya, C.; Bruttin, A.; Brussow, H. Comparative genomics of the T4-Like Escherichia coli phage JS98: Implications for the evolution of T4 phages. J. Bacteriol. 2004, 186, 8276–8286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignacio-Espinoza, J.C.; Sullivan, M.B. Phylogenomics of T4 cyanophages: Lateral gene transfer in the 'core' and origins of host genes. Environ. Microbiol. 2012, 14, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Desplats, C.; Krisch, H.M. The diversity and evolution of the T4-type bacteriophages. Res. Microbiol. 2003, 154, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Denou, E.; Bruttin, A.; Barretto, C.; Ngom-Bru, C.; Brussow, H.; Zuber, S. T4 phages against Escherichia coli diarrhea: Potential and problems. Virology 2009, 388, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Bertrand, C.; Letarov, A.; Tetart, F.; Krisch, H.M. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virology 2007, 362, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.S.; Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Durkin, A.S.; Ciecko, A.; Feldblyum, T.V.; White, Q.; Paulsen, L.T.; Nierman, W.C.; et al. Complete genome sequence of the broad-host-range vibriophage KVP40: Comparative genomics of a T4-related bacteriophage. J. Bacteriol. 2003, 185, 5220–5233. [Google Scholar] [CrossRef] [Green Version]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Hamdi, S.; Rousseau, G.M.; Labrie, S.J.; Tremblay, D.M.; Kourda, R.S.; Ben Slama, K.; Moineau, S. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 2017, 7, 40349. [Google Scholar] [CrossRef]
- Lee, H.; Ku, H.J.; Lee, D.H.; Kim, Y.T.; Shin, H.; Ryu, S.; Lee, J.-H. Characterization and Genomic Study of the Novel Bacteriophage HY01 Infecting Both Escherichia coli O157:H7 and Shigella flexneri: Potential as a Biocontrol Agent in Food. PLoS ONE 2016, 11, e0168985. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Zhong, Y.; Wang, Y.; Zhang, C.; Guo, J.; Shen, X.; Li, C.; Huang, Y.; Xiong, H.; Wang, P.; et al. Two Novel Yersinia pestis Bacteriophages with a Broad Host Range: Potential as Biocontrol Agents in Plague Natural Foci. Viruses 2022, 14, 2740. https://doi.org/10.3390/v14122740
Jin H, Zhong Y, Wang Y, Zhang C, Guo J, Shen X, Li C, Huang Y, Xiong H, Wang P, et al. Two Novel Yersinia pestis Bacteriophages with a Broad Host Range: Potential as Biocontrol Agents in Plague Natural Foci. Viruses. 2022; 14(12):2740. https://doi.org/10.3390/v14122740
Chicago/Turabian StyleJin, Haixiao, Youhong Zhong, Yiting Wang, Chuanyu Zhang, Jin Guo, Xiaona Shen, Cunxiang Li, Ying Huang, Haoming Xiong, Peng Wang, and et al. 2022. "Two Novel Yersinia pestis Bacteriophages with a Broad Host Range: Potential as Biocontrol Agents in Plague Natural Foci" Viruses 14, no. 12: 2740. https://doi.org/10.3390/v14122740