

(+)-Usnic Acid and Its Derivatives as Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses
 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Procedure for the Synthesis of Compound 5b
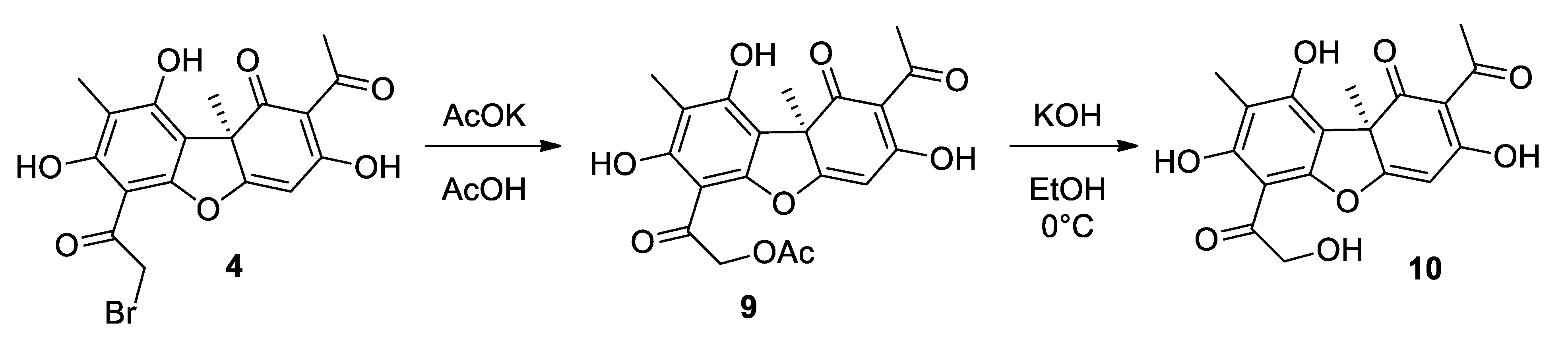
2.1.2. Procedure for Synthesis of Compound 10
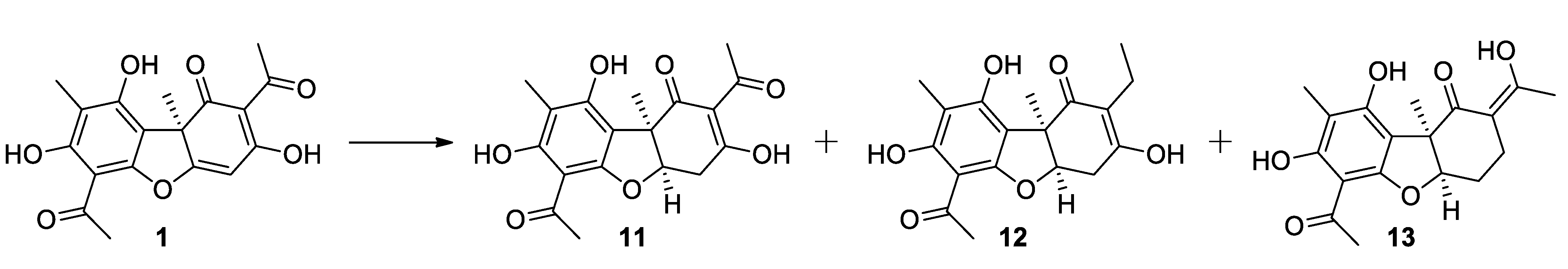
2.1.3. Procedure for Hydrogenation of Usnic Acid
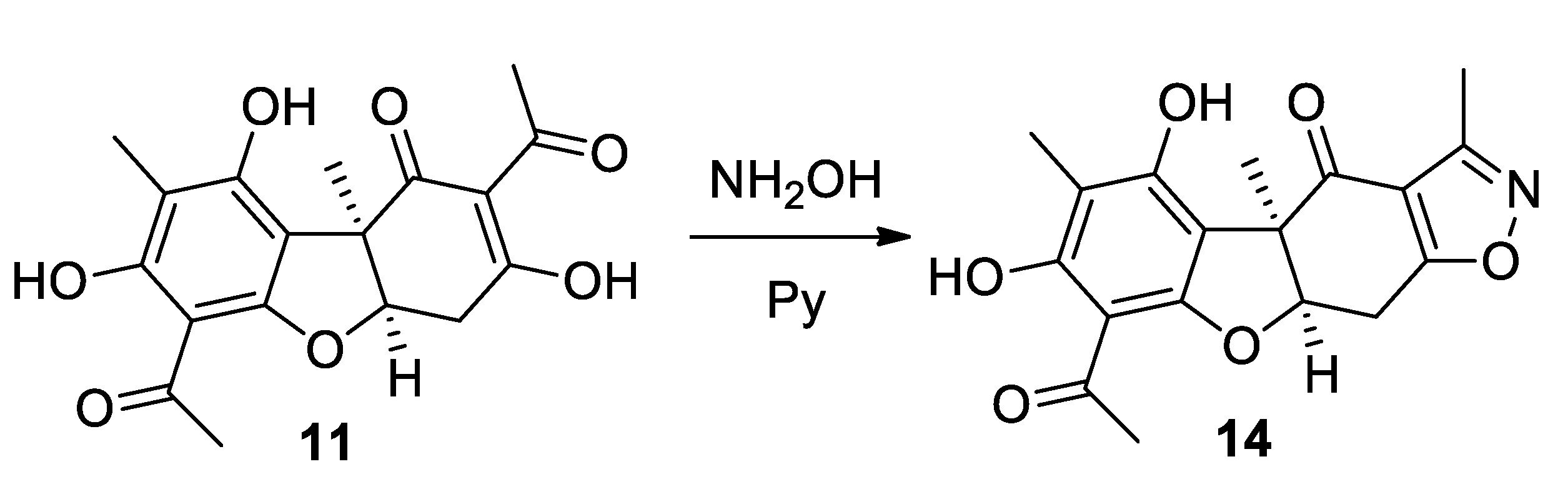
2.1.4. Procedure for the Synthesis of Compound 14
2.2. Biological Experiments
2.2.1. Evaluation of the Antiviral Activities against SARS-CoV-2 Viruses
2.2.2. Evaluation of Inhibitory Activity against the Main Viral Protease
2.2.3. Evaluation of Antiviral Activity Using the Pseudovirus System
Cell Cultures
Plasmids
Determination of Cytotoxicity of Compounds on HEK293T Cells
Pseudoviruses Neutralizing Assay
2.3. Molecular Modeling
2.3.1. Protein and Ligand Preparation
2.3.2. Analysis of a Potential Binding Site
2.3.3. Molecular Docking Procedure
3. Results and Discussions
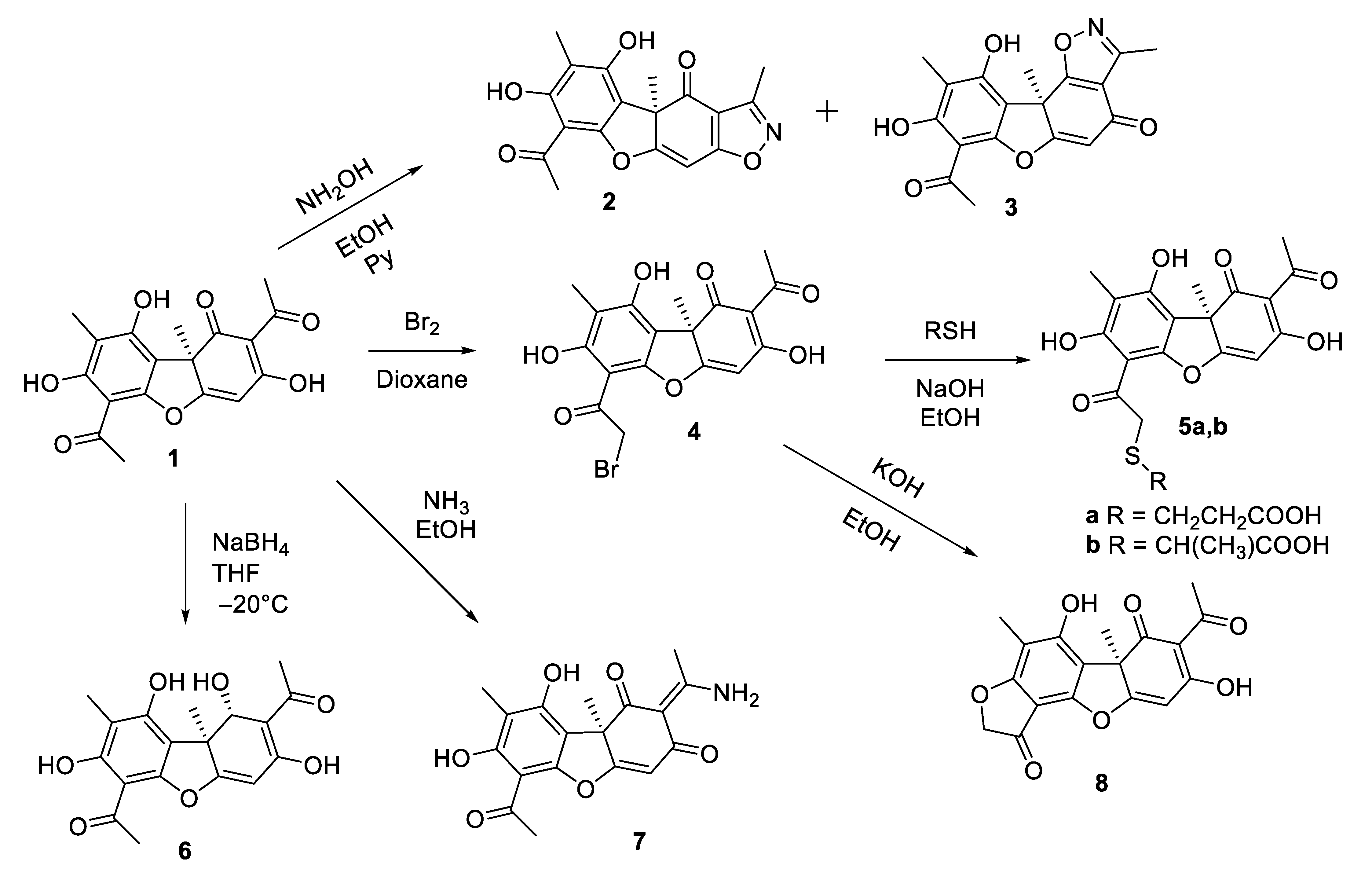
3.1. Synthesis of Usnic Acid Derivatives
3.2. Study of Antiviral Activity
Biological Testing for an Infectious Virus
3.3. Investigating the Mechanism of Action
3.3.1. Main Viral Protease 3CLPro as a Target of Activity
3.3.2. Study of Early Viral Replication
Testing with the Use of Pseudo-Viral Systems
Competitive Inhibition of the RBD/ACE2 Interaction Based on ELISA
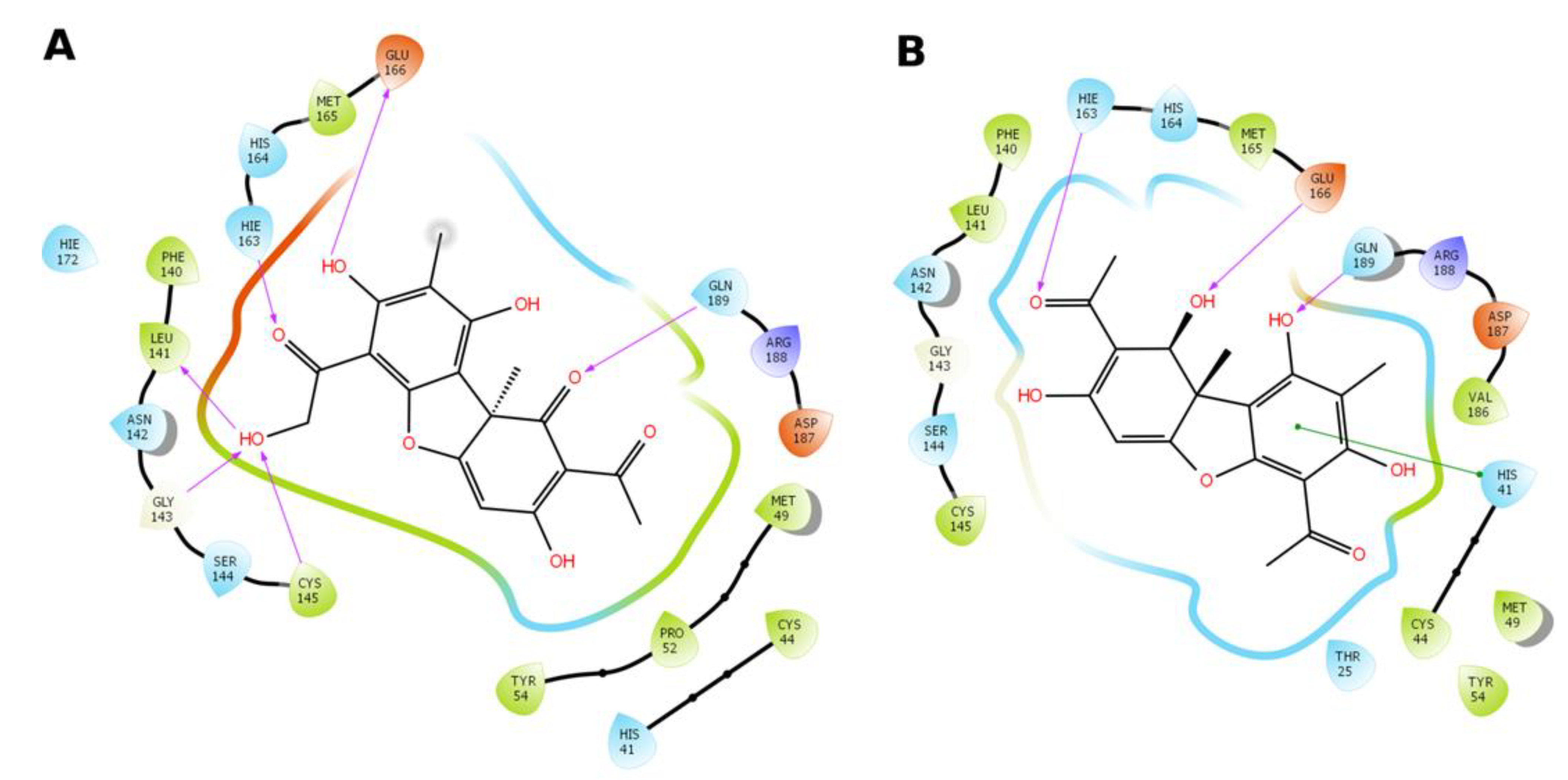
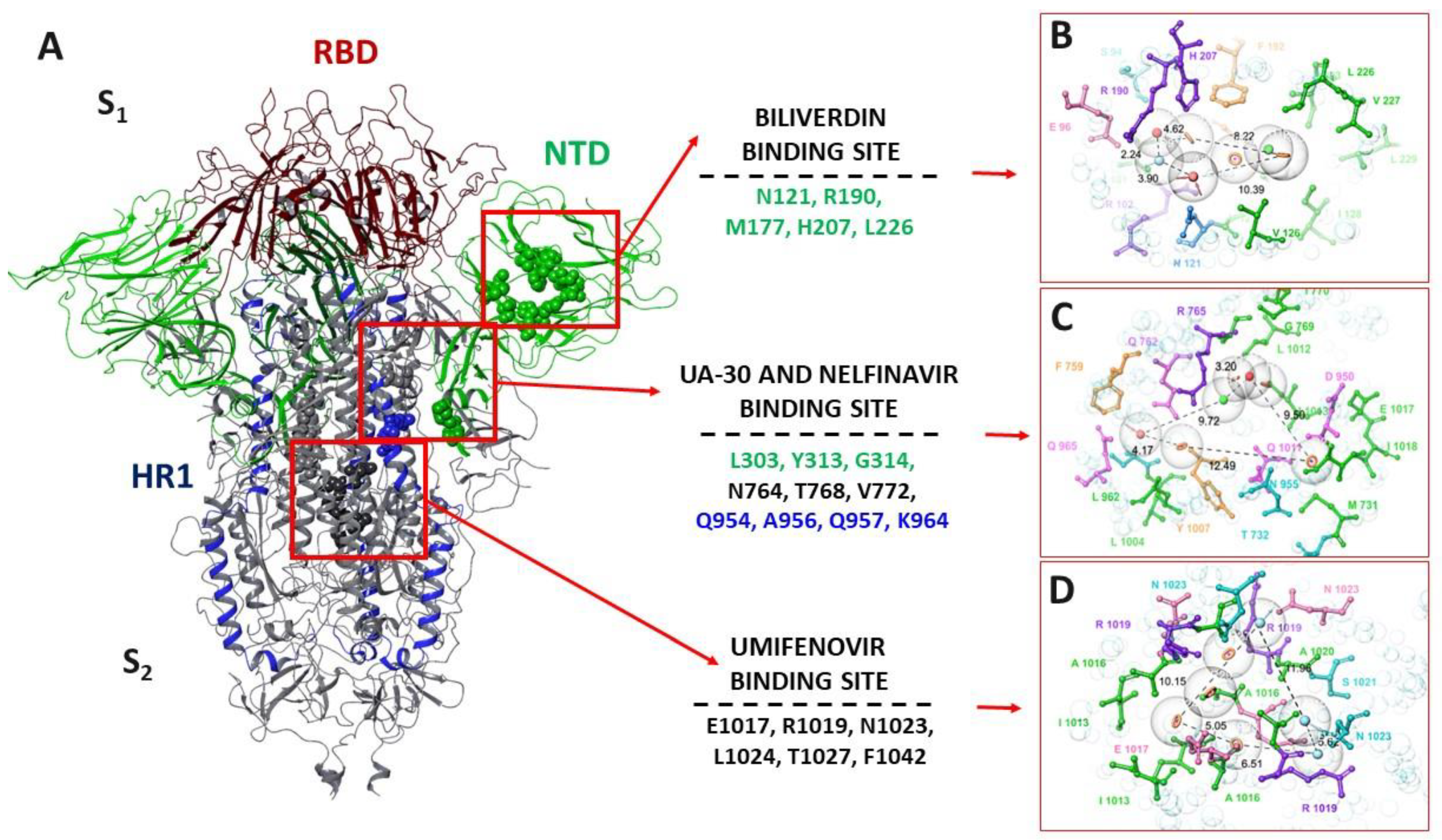
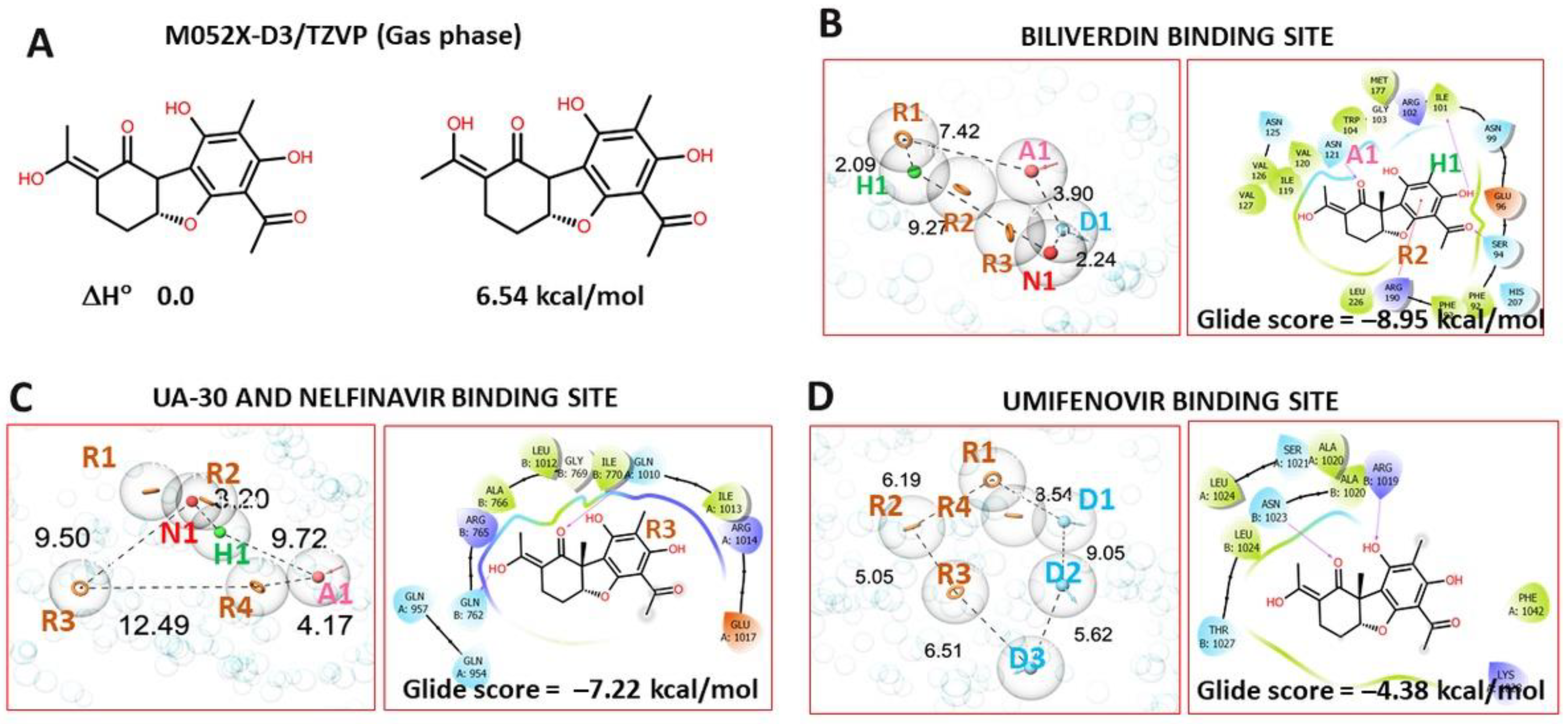
3.4. Molecular Modeling Study
3.4.1. Pharmacophore Profile of Entry Inhibitor Binding Sites
3.4.2. Molecular Docking Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://www.arcgis.com/apps/dashboards/bda7594740fd40299423467b48e9ecf6 (accessed on 18 August 2022).
- Wang, H.; Paulson, K.R.; Pease, S.A.; Watson, S.; Comfort, H.; Zheng, P.; Aravkin, A.Y.; Bisignano, C.; Barber, R.M.; Alam, T.; et al. Estimating excess mortality due to the COVID-19 pandemic: A systematic analysis of COVID-19-related mortality, 2020–21. Lancet 2022, 399, 1513–1536. [Google Scholar] [CrossRef]
- Novikov, F.N.; Stroylov, V.S.; Svitanko, I.V.; Nebolsin, V.E. Molecular basis of COVID-19 pathogenesis. Russ. Chem. Rev. 2020, 89, 858–878. [Google Scholar] [CrossRef]
- Cho, N.J.; Glenn, J.S. Materials science approaches in the development of broad-spectrum antiviral therapies. Nat. Mater. 2020, 19, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, X.-E. Construction and applications of SARS-CoV-2 pseudoviruses: A mini review. Int. J. Biol. Sci. 2021, 17, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, Q.; Huang, W.; Li, X.; Wang, Y. Current status on the development of pseudoviruses for enveloped viruses. Rev. Med. Virol. 2018, 28, e1963. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakova, N.S.; Zaykovskaya, A.V.; Baev, D.S.; Tolstikova, T.G.; Shcherbakov, D.N.; Pyankov, O.V.; et al. Monoterpenoid-based inhibitors of filoviruses targeting the glycoprotein-mediated entry process. Eur. J. Med. Chem. 2020, 207, 112726. [Google Scholar] [CrossRef]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 Proteases for COVID-19 Antiviral Development. Front. Chem. 2022, 9, 1–31. [Google Scholar] [CrossRef]
- Chen, C.; Yu, X.; Kuo, C.; Min, J.; Chen, S.; Ma, L.; Liu, K.; Guo, R. Overview of antiviral drug candidates targeting coronaviral 3C-like main proteases. FEBS J. 2021, 288, 5089–5121. [Google Scholar] [CrossRef]
- Paul, A.; Sarkar, A.; Saha, S.; Maji, A.; Janah, P.; Kumar Maity, T. Synthetic and computational efforts towards the development of peptidomimetics and small-molecule SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. 2021, 46, 116301. [Google Scholar] [CrossRef]
- Yarovaya, O.I.; Salakhutdinov, N.F. Mono- and sesquiterpenes as a starting platform for the development of antiviral drugs. Russ. Chem. Rev. 2021, 90, 488–510. [Google Scholar] [CrossRef]
- Merarchi, M.; Dudha, N.; Das, B.C.; Garg, M. Natural products and phytochemicals as potential anti-SARS-CoV-2 drugs. Phyther. Res. 2021, 35, 5384–5396. [Google Scholar] [CrossRef]
- Majnooni, M.B.; Fakhri, S.; Bahrami, G.; Naseri, M.; Farzaei, M.H.; Echeverría, J. Alkaloids as Potential Phytochemicals against SARS-CoV-2: Approaches to the Associated Pivotal Mechanisms. Evidence-Based Complement. Altern. Med. 2021, 2021, 6632623. [Google Scholar] [CrossRef]
- Macedo, D.C.S.; Almeida, F.J.F.; Wanderley, M.S.O.; Ferraz, M.S.; Santos, N.P.S.; López, A.M.Q.; Santos-Magalhães, N.S.; Lira-Nogueira, M.C.B. Usnic acid: From an ancient lichen derivative to promising biological and nanotechnology applications. Phytochem. Rev. 2021, 20, 609–630. [Google Scholar] [CrossRef]
- Campanella, L.; Delfini, M.; Ercole, P.; Iacoangeli, A.; Risuleo, G. Molecular characterization and action of usnic acid: A drug that inhibits proliferation of mouse polyomavirus in vitro and whose main target is RNA transcription. Biochimie 2002, 84, 329–334. [Google Scholar] [CrossRef]
- Sokolov, D.N.; Zarubaev, V.V.; Shtro, A.A.; Polovinka, M.P.; Luzina, O.A.; Komarova, N.I.; Salakhutdinov, N.F.; Kiselev, O.I. Anti-viral activity of (−)- and (+)-usnic acids and their derivatives against influenza virus A(H1N1)2009. Bioorg. Med. Chem. Lett. 2012, 22, 7060–7064. [Google Scholar] [CrossRef]
- Shtro, A.; Zarubaev, V.; Luzina, O.; Sokolov, D.; Salakhutdinov, N. Derivatives of usnic acid inhibit broad range of influenza viruses and protect mice from lethal influenza infection. Antivir. Chem. Chemother. 2015, 24, 92–98. [Google Scholar] [CrossRef]
- Shtro, A.A.; Zarubaev, V.V.; Luzina, O.A.; Sokolov, D.N.; Kiselev, O.I.; Salakhutdinov, N.F. Novel derivatives of usnic acid effectively inhibiting reproduction of influenza A virus. Bioorg. Med. Chem. 2014, 22, 6826–6836. [Google Scholar] [CrossRef]
- Gupta, A.; Sahu, N.; Singh, A.P.; Singh, V.K.; Singh, S.C.; Upadhye, V.J.; Mathew, A.T.; Kumar, R.; Sinha, R.P. Exploration of Novel Lichen Compounds as Inhibitors of SARS-CoV-2 Mpro: Ligand-Based Design, Molecular Dynamics, and ADMET Analyses. Appl. Biochem. Biotechnol. 2022. [Google Scholar] [CrossRef]
- Prateeksha, G.; Rana, T.S.; Asthana, A.K.; Singh, B.N.; Barik, S.K. Screening of cryptogamic secondary metabolites as putative inhibitors of SARS-CoV-2 main protease and ribosomal binding domain of spike glycoprotein by molecular docking and molecular dynamics approaches. J. Mol. Struct. 2021, 1240, 130506. [Google Scholar] [CrossRef]
- Coban, M.A.; Morrison, J.; Maharjan, S.; Hernandez Medina, D.H.; Li, W.; Zhang, Y.S.; Freeman, W.D.; Radisky, E.S.; Le Roch, K.G.; Weisend, C.M.; et al. Attacking COVID-19 Progression Using Multi-Drug Therapy for Synergetic Target Engagement. Biomolecules 2021, 11, 787. [Google Scholar] [CrossRef]
- Oh, E.; Wang, W.; Park, K.-H.; Park, C.; Cho, Y.; Lee, J.; Kang, E.; Kang, H. (+)-Usnic acid and its salts, inhibitors of SARS-CoV-2, identified by using in silico methods and in vitro assay. Sci. Rep. 2022, 12, 13118. [Google Scholar] [CrossRef] [PubMed]
- Kutney, J.P.; Sanchez, I.H.; Yee, T. Studies in the usnic acid series. II. The condensation of (+)-usnic acid with hydroxylamine. Can. J. Chem. 1976, 54, 3713–3720. [Google Scholar] [CrossRef]
- Luzina, O.A.; Sokolov, D.N.; Shernyukov, A.V.; Salakhutdinov, N.F. Synthesis of aurones based on usninic acid. Chem. Nat. Compd. 2012, 48, 385–391. [Google Scholar] [CrossRef]
- Luzina, O.A.; Sokolov, D.N.; Komarova, N.I.; Salakhutdinov, N.F. Synthesis of sulfides based on (+)-usninic acid. Chem. Nat. Compd. 2014, 50, 266–271. [Google Scholar] [CrossRef]
- Sokolov, D.N.; Luzina, O.A.; Polovinka, M.P.; Salakhutdinov, N.F. Reduction of (+)-usninic acid and its pyrazole derivative by sodium borohydride. Chem. Nat. Compd. 2011, 47, 203–205. [Google Scholar] [CrossRef]
- Bruno, M.; Trucchi, B.; Burlando, B.; Ranzato, E.; Martinotti, S.; Akkol, E.K.; Süntar, I.; Keleş, H.; Verotta, L. (+)-Usnic acid enamines with remarkable cicatrizing properties. Bioorganic Med. Chem. 2013, 21, 1834–1843. [Google Scholar] [CrossRef]
- Zakharova, O.; Luzina, O.; Zakharenko, A.; Sokolov, D.; Filimonov, A.; Dyrkheeva, N.; Chepanova, A.; Ilina, E.; Ilyina, A.; Klabenkova, K.; et al. Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors. Bioorg. Med. Chem. 2018, 26, 4470–4480. [Google Scholar] [CrossRef]
- Shibata, S.; Takahashi, K.; Tanaka, Y. Decomposition of Usnic Acid. V. Pyrolysis of Dihydrousnic Acid.(2). some Observations on Dihydrousnic Acid. Pharm. Bull. 1956, 4, 65–67. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef]
- Schrodinger Small Molecule Drug Discovery Suite; Schrödinger, LLC: New York, NY, USA, 2016.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; dos Santos, M.S.; et al. SARS-CoV-2 can recruit a heme metabolite to evade antibody immunity. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Kurt Yilmaz, N.; Thompson, P.R.; Schiffer, C.A. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Khamitov, E.M.; Gureev, M.A.; Yarovaya, O.I.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakov, D.N.; Maksyutov, R.A.; Salakhutdinov, N.F. Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses 2022, 14, 119. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Li, H.; Cheng, C.; Shi, S.; Wu, Y.; Gao, Y.; Liu, Z.; Liu, M.; Li, Z.; Huo, L.; Pan, X.; et al. Identification, optimization, and biological evaluation of 3-O-β-chacotriosyl ursolic acid derivatives as novel SARS-CoV-2 entry inhibitors by targeting the prefusion state of spike protein. Eur. J. Med. Chem. 2022, 238, 114426. [Google Scholar] [CrossRef]
- Musarrat, F.; Chouljenko, V.; Dahal, A.; Nabi, R.; Chouljenko, T.; Jois, S.D.; Kousoulas, K.G. The anti-HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV-2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID-19 infections. J. Med. Virol. 2020, 92, 2087–2095. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Domingo, P.; de Benito, N. Alpha variant SARS-CoV-2 infection: How it all starts. EBioMedicine 2021, 74, 103703. [Google Scholar] [CrossRef]
- Twohig, K.A.; Nyberg, T.; Zaidi, A.; Thelwall, S.; Sinnathamby, M.A.; Aliabadi, S.; Seaman, S.R.; Harris, R.J.; Hope, R.; Lopez-Bernal, J.; et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: A cohort study. Lancet Infect. Dis. 2021, 3099, 1–9. [Google Scholar] [CrossRef]
- Vanmechelen, B.; Logist, A.; Wawina-Bokalanga, T.; Verlinden, J.; Martí-Carreras, J.; Geenen, C.; Slechten, B.; Cuypers, L.; André, E.; Baele, G.; et al. Identification of the First SARS-CoV-2 Lineage B.1.1.529 Virus Detected in Europe. Microbiol. Resour. Announc. 2022, 11, 9–12. [Google Scholar] [CrossRef]
- Kudriavtsev, A.V.; Vakhrusheva, A.V.; Novoseletsky, V.N.; Bozdaganyan, M.E.; Shaitan, K.V.; Kirpichnikov, M.P.; Sokolova, O.S. Immune Escape Associated with RBD Omicron Mutations and SARS-CoV-2 Evolution Dynamics. Viruses 2022, 14, 1603. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzyme Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yan, S.; Wang, Y.; Li, M.; Xiao, Y.; Li, Y. Discovery of 4′-O-methylscutellarein as a potent SARS-CoV-2 main protease inhibitor. Biochem. Biophys. Res. Commun. 2022, 604, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Dunlap, T.L.; Dietz, B.M. Formation and biological targets of botanical o-quinones. Food Chem. Toxicol. 2018, 120, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhu, G.-H.; Zhang, Y.-N.; Hu, Q.; Wang, H.-N.; Yu, H.-N.; Qin, X.-Y.; Guan, X.-Q.; Xiang, Y.-W.; Tang, H.; et al. Flavonoids in Ampelopsis grossedentata as covalent inhibitors of SARS-CoV-2 3CLpro: Inhibition potentials, covalent binding sites and inhibitory mechanisms. Int. J. Biol. Macromol. 2021, 187, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, D.N.; Luzina, O.A.; Salakhutdinov, N.F. Usnic acid: Preparation, structure, properties and chemical transformations. Russ. Chem. Rev. 2012, 81, 747–768. [Google Scholar] [CrossRef]
- Araújo, H.D.A.d.; Silva, H.A.M.F.; Silva Júnior, J.G.d.; Albuquerque, M.C.P.d.A.; Coelho, L.C.B.B.; Aires, A.d.L. The Natural Compound Hydrophobic Usnic Acid and Hydrophilic Potassium Usnate Derivative: Applications and Comparisons. Molecules 2021, 26, 5995. [Google Scholar] [CrossRef]
- Yarovaya, O.I.; Shcherbakov, D.N.; Borisevich, S.S.; Sokolova, A.S.; Gureev, M.A.; Khamitov, E.M.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; et al. Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses 2022, 14, 1295. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Wuhan Lineages B a | Delta Lineage B.1.617.2 b | Omicron Lineage B.1.1.529 c | ||||
|---|---|---|---|---|---|---|---|
| CC50 d, µM | IC50 e, µM | SI f | IC50, µM | SI | IC50, µM | SI | |
| 1 | 146.7 ± 22.1 | 10.9 ± 2.2 | 13 | 20.9 ± 3.4 | 7 | 3.7 ± 0.8 | 39 |
| 2 | 252.2 ± 31.0 | NA | - | NT | - | NT | - |
| 3 | 15.5 ± 2.8 | 9.2 ± 2.4 | 1.6 | 9.1 ± 1.8 | 1.7 | 3.5 ± 1.1 | 4.5 |
| 5a | 177.3 ± 18.2 | NA | - | NT | - | NT | - |
| 5b | 176.5 ± 22.6 | 76.5 ± 8.2 | 2 | NT | - | NT | - |
| 6 | 99.0 ± 12.1 | 65.3 ± 7.4 | 1,5 | NT | - | NT | - |
| 7 | 527.7 ± 16 | NA | – | NT | - | NT | - |
| 8 | 175.8 ± 19.9 | 44.3 ± 5.2 | 4 | 95 ± 8.7 | 2 | 47.0 ± 6.5 | 4 |
| 10 | 306.3 ± 28.6 | 25.3 ± 2.1 | 12 | 9.7 ± 1.4 | 31 | 17.5 ± 3.8 | 17 |
| 11 | 84.9 ± 12.1 | 21.0 ± 3.5 | 4 | NA | - | NA | - |
| 12 | 262 ± 32.0 | 91 ± 7.1 | 3 | 85 ± 11.2 | 3 | 60 ± 12.1 | 4 |
| 13 | 72.7 ± 14.6 | 6.7 ± 1.4 | 10 | 13.5 ± 2.1 | 5 | 6.6 ± 1.9 | 10 |
| 14 | 295.5 ± 11.9 | NA | - | NT | - | NT | - |
| Remdesivir | 710.9 ± 21.2 | 3.8 ± 0.42 | 186 | 2.1 ± 0.16 | 338 | 2.0 ± 0.13 | 356 |
| Ligand | Glide Score | Emodel | IFD Score |
|---|---|---|---|
| 10 | −11.568 | −72.557 | −664.659 |
| 1 | −9.782 | −67.242 | −662.940 |
| 6 | −9.408 | −70.692 | −663.973 |
| 5b | −9.300 | −89.025 | −662.874 |
| 2 | −9.152 | −49.980 | −663.230 |
| 12 | −9.029 | −70.173 | −662.796 |
| 8 | −9.017 | −73.237 | −661.496 |
| 3 | −8.736 | −67.297 | −663.686 |
| 5a | −8.619 | −83.363 | −665.003 |
| ML188 | −8.379 | −83.299 | −666.051 |
| 14 | −7.768 | −64.279 | −660.219 |
| 7 | −7.657 | −67.654 | −661.368 |
| 11 | −7.563 | −65.594 | −660.863 |
| 13 | −6.884 | −53.540 | −660.770 |
| Agent | CC50 a, µM | % Neutralization at 25 µM | IC50 b µM | SI c |
|---|---|---|---|---|
| 1 | >1000 | 12 | NT | - |
| 2 | >1000 | 55 | 30 ± 2.9 | 33 |
| 3 | 150 ± 18.5 | 65 | 32 ± 5.1 | 6 |
| 5a | >1000 | 0 | NT | - |
| 5b | >1000 | 41 | NT | - |
| 6 | 168 ± 21.2 | 40 | NT | - |
| 7 | 850 ± 22.6 | 12 | NT | - |
| 8 | 358 ± 31.1 | 0 | NT | - |
| 10 | 750 ± 36.6 | 45 | NT | - |
| 11 | 469 ± 24.2 | 78 | 22 ± 3.2 | 21 |
| 12 | >1000 | 53 | 28 ± 3.7 | 35 |
| 13 | 151 ± 14.9 | 95 | 5.28 ± 0.8 | 28 |
| 14 | >1000 | 15 | NT | - |
| Arbidol | 19.8 ± 4.6 | 58 | 12.6 ± 4.1 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filimonov, A.S.; Yarovaya, O.I.; Zaykovskaya, A.V.; Rudometova, N.B.; Shcherbakov, D.N.; Chirkova, V.Y.; Baev, D.S.; Borisevich, S.S.; Luzina, O.A.; Pyankov, O.V.; et al. (+)-Usnic Acid and Its Derivatives as Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses 2022, 14, 2154. https://doi.org/10.3390/v14102154
Filimonov AS, Yarovaya OI, Zaykovskaya AV, Rudometova NB, Shcherbakov DN, Chirkova VY, Baev DS, Borisevich SS, Luzina OA, Pyankov OV, et al. (+)-Usnic Acid and Its Derivatives as Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses. 2022; 14(10):2154. https://doi.org/10.3390/v14102154
Chicago/Turabian StyleFilimonov, Aleksandr S., Olga I. Yarovaya, Anna V. Zaykovskaya, Nadezda B. Rudometova, Dmitriy N. Shcherbakov, Varvara Yu. Chirkova, Dmitry S. Baev, Sophia S. Borisevich, Olga A. Luzina, Oleg V. Pyankov, and et al. 2022. "(+)-Usnic Acid and Its Derivatives as Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses" Viruses 14, no. 10: 2154. https://doi.org/10.3390/v14102154