
SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
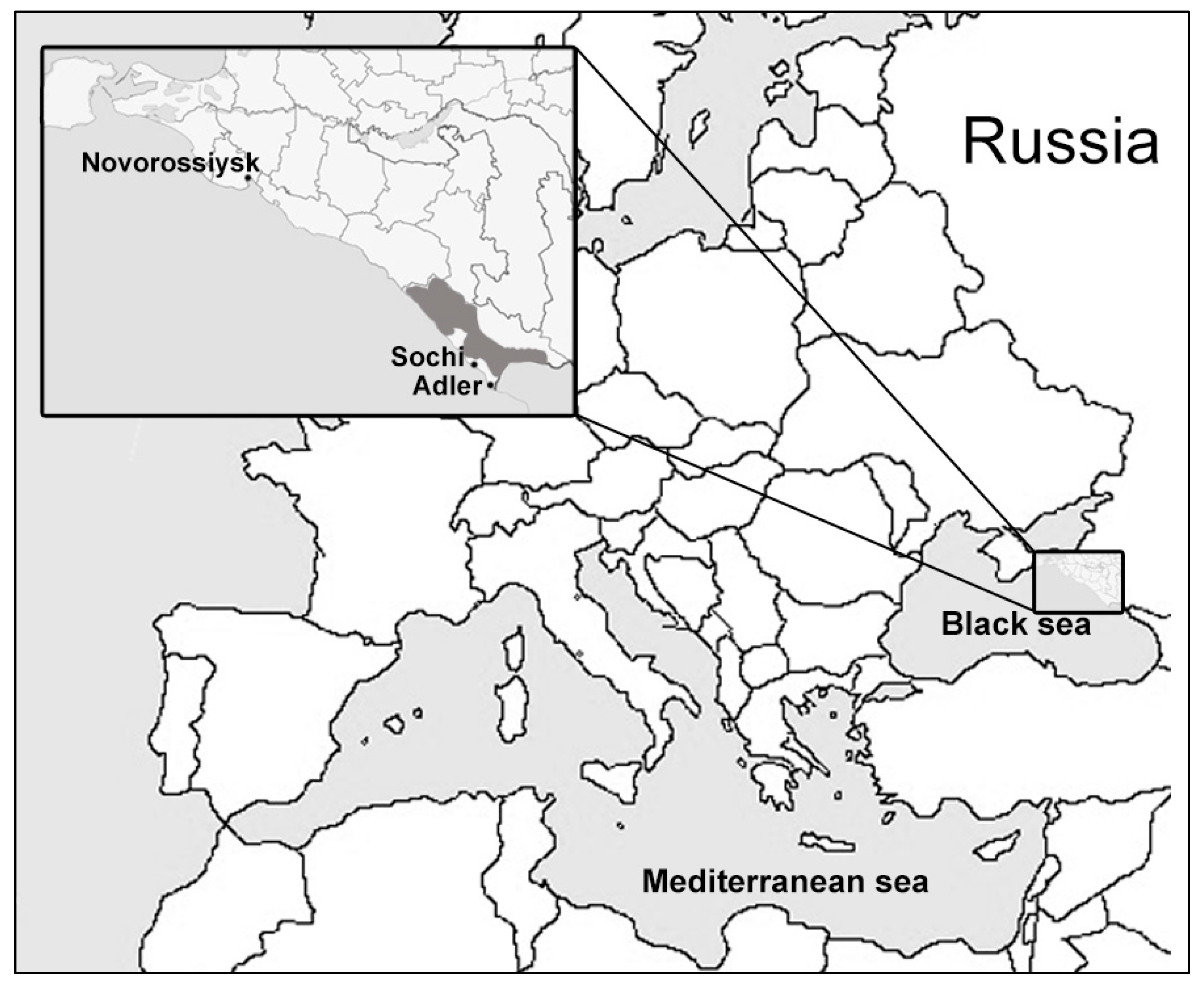
2.1. Sample Collection
2.2. Metagenomic Analysis
2.3. Genetic and Phylogenetic Analysis
2.4. RT-PCR Analysis
3. Results
3.1. Results of Sequencing of the Samples
3.2. Genetic and Phylogenetic Analysis
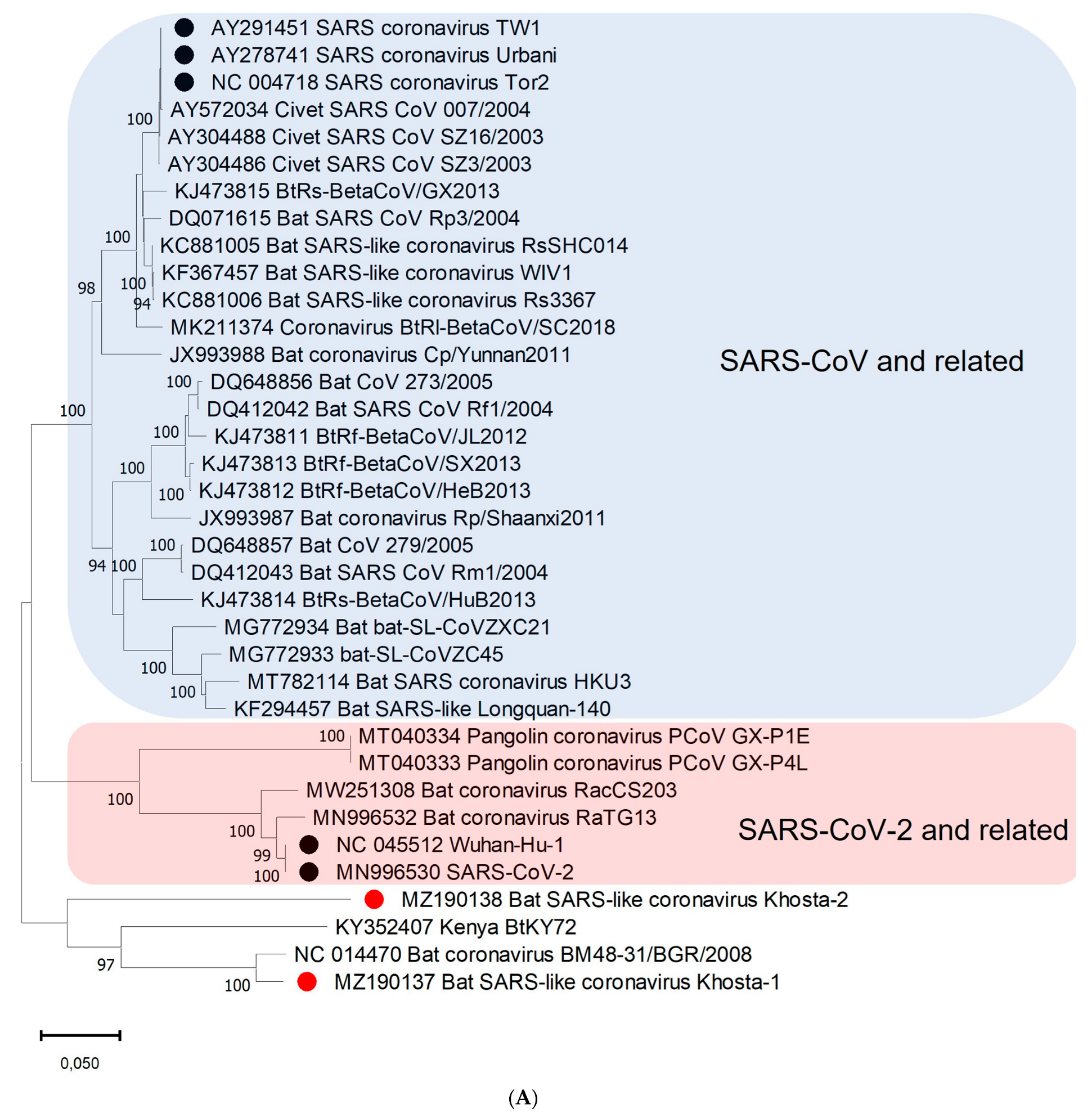
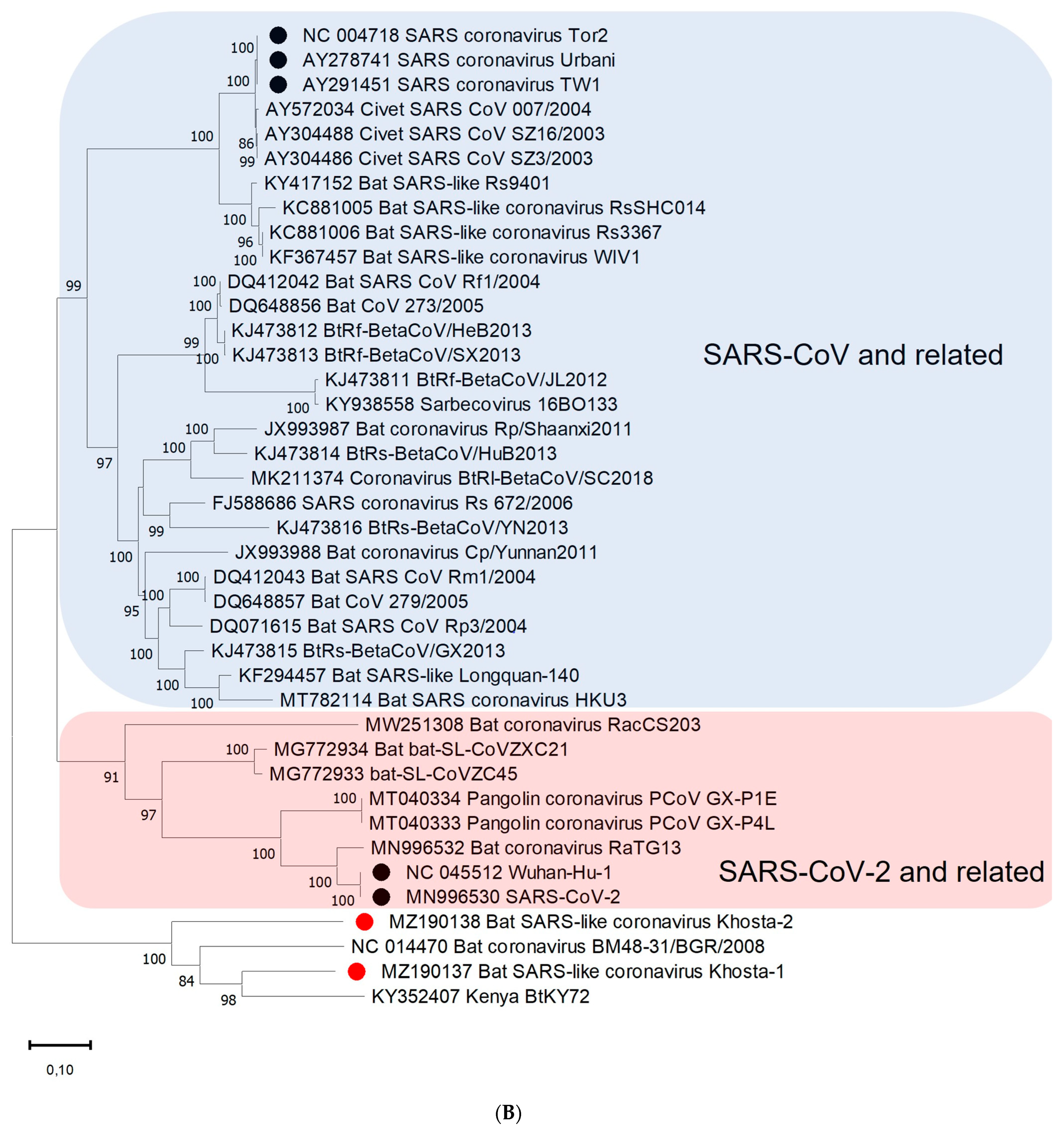
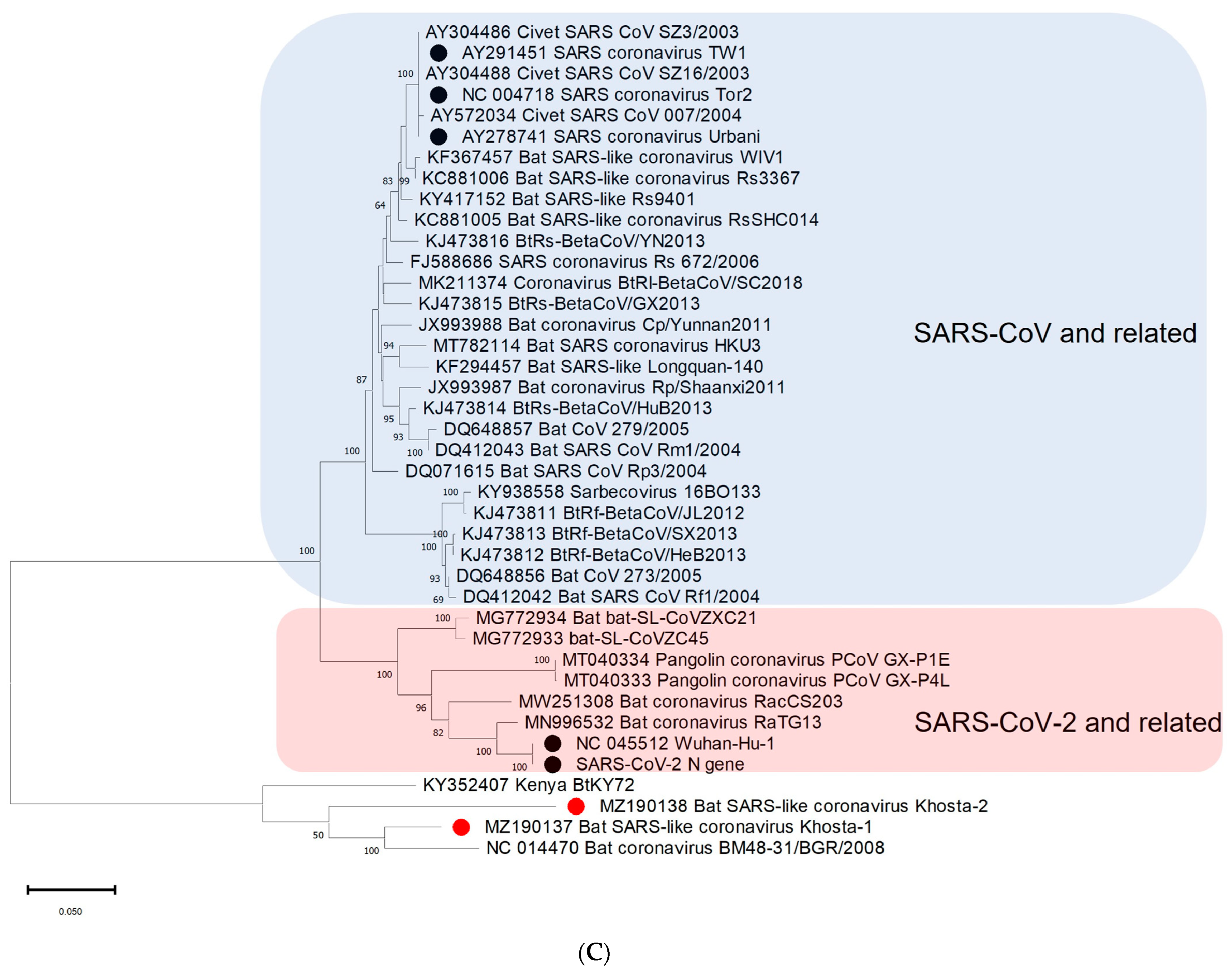
3.3. Phylogenetic Analysis
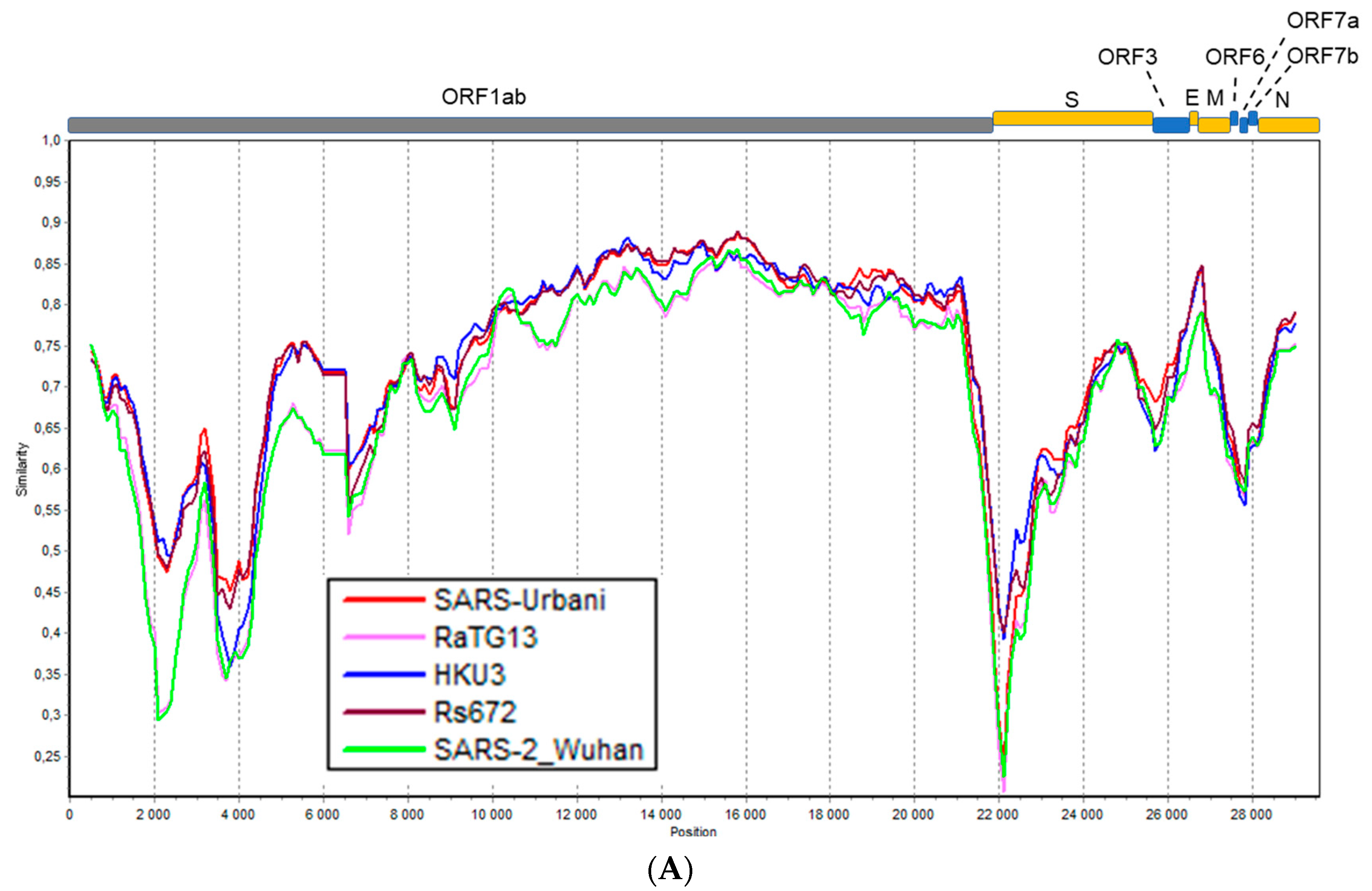
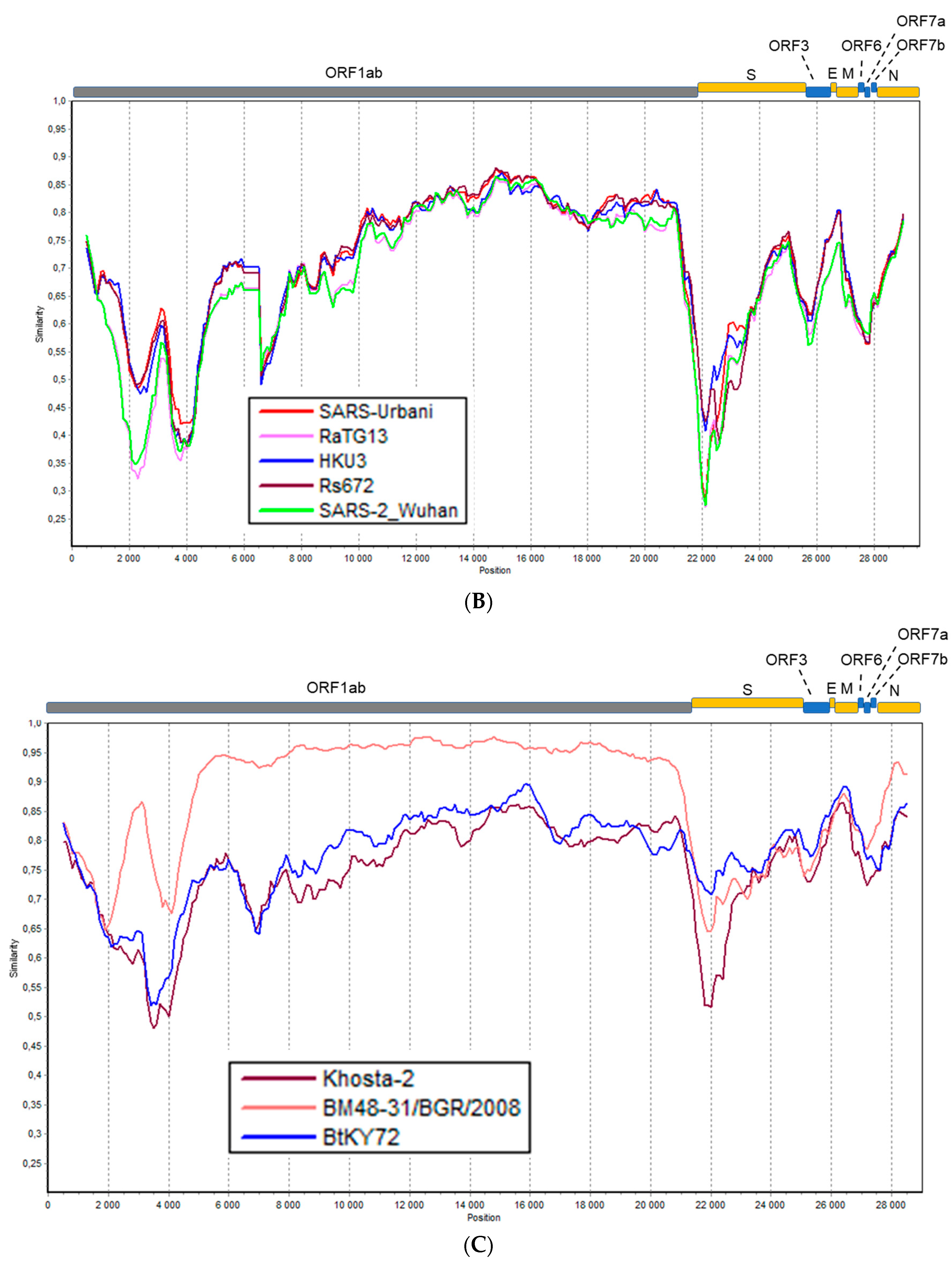
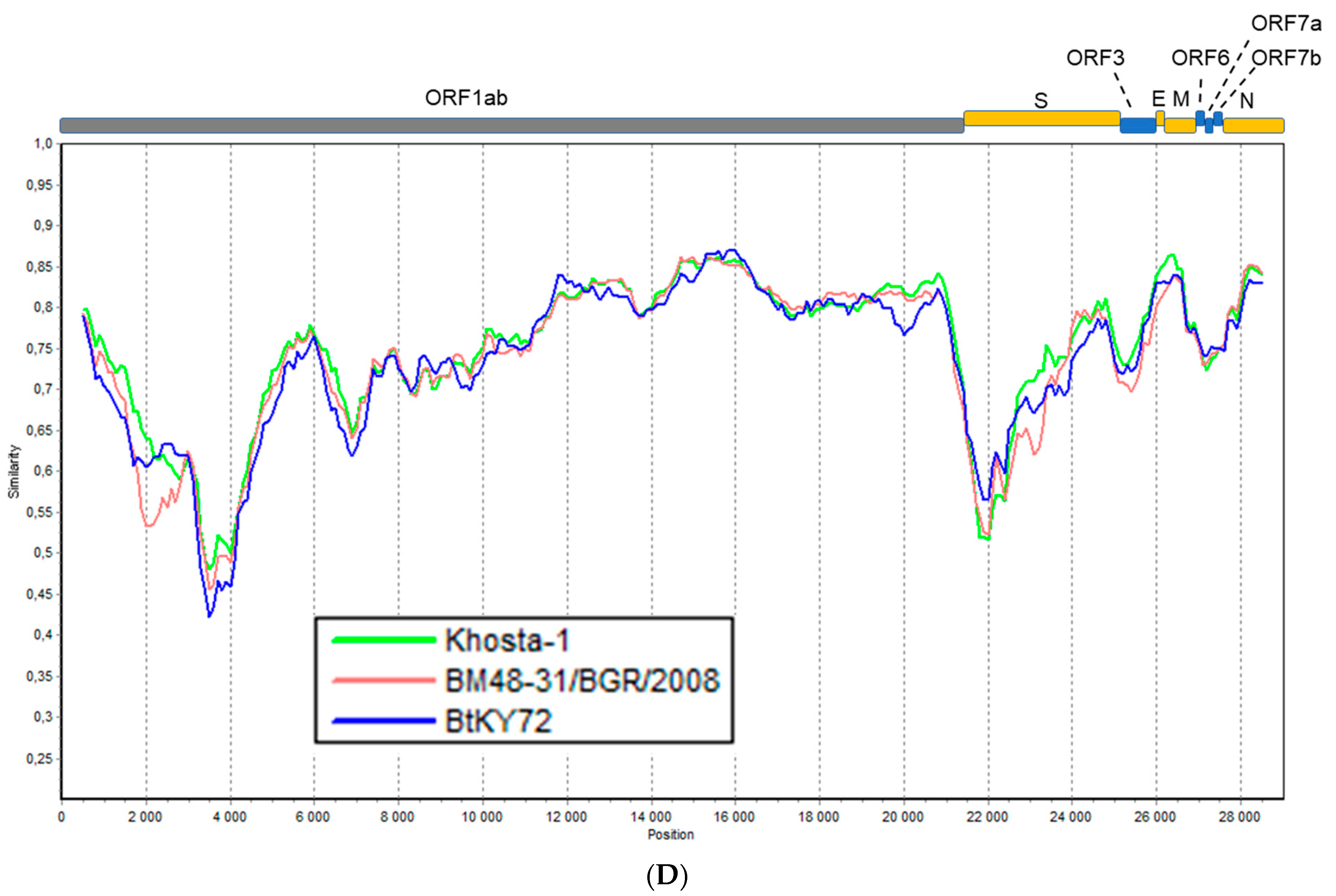
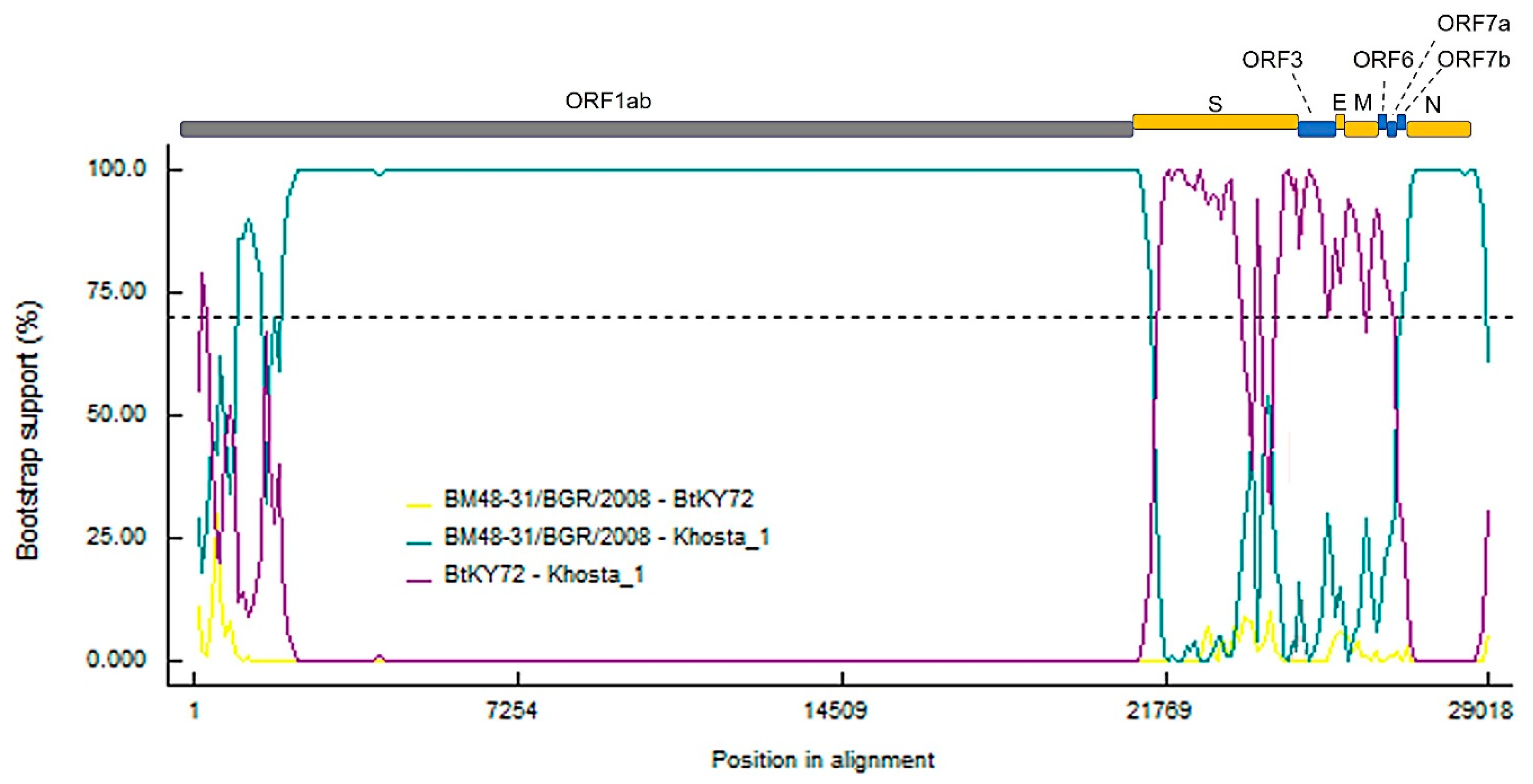
3.4. Recombination Analysis
3.5. Analysis of Receptor-Binding Motif (RBM) and S1/S2 Cleavage Site of S Protein
3.6. PCR Testing
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: London, UK, 2012; pp. 806–828. [Google Scholar]
- Fan, Y.; Zhao, K.; Shi, Z.-L.; Zhou, P. Bat Coronaviruses in China. Viruses 2019, 11, 210. [Google Scholar] [CrossRef] [Green Version]
- Rihtarič, D.; Hostnik, P.; Steyer, A.; Grom, J.; Toplak, I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 2010, 155, 507–514. [Google Scholar] [CrossRef]
- Ar Gouilh, M.; Puechmaille, S.J.; Diancourt, L.; Vandenbogaert, M.; Serra-Cobo, J.; Lopez Roïg, M.; Brown, P.; Moutou, F.; Caro, V.; Vabret, A.; et al. SARS-CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old-world. Virology 2018, 517, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Balboni, A.; Palladini, A.; Bogliani, G.; Battilani, M. Detection of a virus related to betacoronaviruses in Italian greater horseshoe bats. Epidemiol. Infect. 2011, 139, 216–219. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic Characterization of Severe Acute Respiratory Syndrome-Related Coronavirus in European Bats and Classification of Coronaviruses Based on Partial RNA-Dependent RNA Polymerase Gene Sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.F.; Shi, Z.; Zhang, S.; Field, H.; Daszak, P.; Eaton, B.T. Review of bats and SARS. Emerg. Infect. Dis. 2006, 12, 1834–1840. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482–485. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Receptor recognition and cross-species infections of SARS coronavirus. Antivir. Res. 2013, 100, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Holmes, E.C.; Goldstein, S.A.; Rasmussen, A.L.; Robertson, D.L.; Crits-Christoph, A.; Wertheim, J.O.; Anthony, S.J.; Barclay, W.S.; Boni, M.F.; Doherty, P.C.; et al. The origins of SARS-CoV-2: A critical review. Cell 2021, 184, 4848–4856. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Mishra, A.; Sood, R.; Srivastava, P.; Tripathi, M.; Zhang, K.Y.J.; Bhatia, S. Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species. Viruses 2021, 13, 1847. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 2021, 184, 4380–4391.e14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Hu, D.; Zhu, C.; Ai, L.; He, T.; Wang, Y.; Ye, F.; Yang, L.; Ding, C.; Zhu, X.; Lv, R.; et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Chia, W.N.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Delaune, D.; Hul, V.; Karlsson, E.A.; Hassanin, A.; Ou, T.P.; Baidaliuk, A.; Gámbaro, F.; Prot, M.; Tu, V.T.; Chea, S.; et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Pasteur, I.; Vongphayloth, K.; Salazar, E.B.; Munier, S.; Bonomi, M. Coronaviruses with a SARS-CoV-2-like receptor-binding domain allowing ACE2-mediated entry into human cells isolated from bats of Indochinese peninsula. Available online: https://www.researchsquare.com/article/rs-871965/v1 (accessed on 14 November 2021).
- Murakami, S.; Kitamura, T.; Suzuki, J.; Sato, R.; Aoi, T.; Fujii, M.; Matsugo, H.; Kamiki, H.; Ishida, H.; Takenaka-Uema, A.; et al. Detection and Characterization of Bat Sarbecovirus Phylogenetically Related to SARS-CoV-2, Japan. Emerg. Infect. Dis. 2020, 26, 3025–3029. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasllieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greeneugh, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Conceicao, C.; Thakur, N.; Human, S.; Kelly, J.T.; Logan, L.; Bialy, D.; Bhat, S.; Stevenson-Leggett, P.; Zagrajek, A.K.; Hollinghurst, P.; et al. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 2020, 18. [Google Scholar] [CrossRef]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.P.; Pfenning, A.R.; Zhao, H.; et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef]
- Ren, W.; Qu, X.; Li, W.; Han, Z.; Yu, M.; Zhou, P.; Zhang, S.-Y.; Wang, L.-F.; Deng, H.; Shi, Z. Difference in Receptor Usage between Severe Acute Respiratory Syndrome (SARS) Coronavirus and SARS-like Coronavirus of Bat Origin. J. Virol. 2008, 82, 1899–1907. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Feng, Y.; Chen, H.; Luk, H.K.H.; Yang, W.-H.; Li, K.S.M.; Zhang, Y.-Z.; Huang, Y.; Song, Z.-Z.; Chow, W.-N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.Y.; Wang, N.; Zhang, W.; Hu, B.; Li, B.; Zhang, Y.Z.; Zhou, J.H.; Luo, C.M.; Yang, X.L.; Wu, L.J.; et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 2016, 31, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Bat Species | Number of Samples Collected | Khosta-1 Virus-Positive Samples (% *) | Khosta-2 Virus-Positive Samples (% *) | |||
|---|---|---|---|---|---|---|---|
| Oral Swabs | Feces | Oral Swabs | Feces | Oral Swabs | Feces | ||
| Basement of the building at Research Institute of Medical Primatology (43°26′06.3″ N 39°59′26.4″ E) | Lesser horseshoe bat (R. hipposideros) | 27 | 24 | 0 | 0 | 1 (3.7%) | 2 (8.3%) |
| Mediterranean horseshoe bat (R. euryale) | 1 | 1 | 0 | 0 | 0 | 0 | |
| Museinaya cave (43°33′34.3″ N 39°53′46.2″ E) | Greater horseshoe bat (R. ferrumequinum) | 4 | 2 | 0 | 0 | 0 | 0 |
| Lesser horseshoe bat (R. hipposideros) | 3 | 2 | 0 | 0 | 0 | 0 | |
| Khosta 1 cave (43°33′49.5″ N 39°53′57.2″ E) | Greater horseshoe bat (R. ferrumequinum) | 21 | 13 | 0 | 1 (7.7%) | 0 | 0 |
| Common bent-wing bat Miniopterus schreibersii | 3 | 1 | 0 | 0 | 0 | 0 | |
| Kolokolnaya cave (43°33′08.3″ N, 39°56′02.4″ E) | Greater horseshoe bat (R. ferrumequinum) | 36 | 24 | 4 (11%) | 15 (62.5%) | 0 | 0 |
| Mediterranean horseshoe bat (R. euryale) | 2 | 0 | 0 | 0 | 0 | 0 | |
| Partizanskaya cave (43°37′38.86″ N, 39°54′46.06″ E) | Greater horseshoe bat (R. ferrumequinum) | 2 | 1 | 0 | 0 | 0 | 0 |
| Lesser horseshoe bat (R. hipposideros) | 5 | 3 | 0 | 1 (33%) | 0 | 0 | |
| Attic of house (44°0′57.51″ N, 39°15′3.63″ E) | Lesser horseshoe bat (R. hipposideros) | 6 | 4 | 0 | 0 | 0 | 0 |
| Krasnoaleksandrovskaya cave (44°0′57.21″ N, 39°21′49.68″ E) | Greater horseshoe bat (R. ferrumequinum) | 1 | 0 | 0 | 0 | 0 | 0 |
| Lesser horseshoe bat (R. hipposideros) | 6 | 0 | 0 | 0 | 0 | 0 | |
| Myotis bat Myotis spp. | 3 | 0 | 0 | 0 | 0 | 0 | |
| Attic of house, Izmaylovka village (43°37′51.72″ N 39°49′45.38″ E) | Lesser horseshoe bat (R. hipposideros) | 0 | 2 | 0 | 0 | 0 | 0 |
| Total | 120 | 77 | 4 (4.6% **) | 17 (14.9% **) | 1 (0.89% **) | 2 (1.75% **) | |
| Protein | Viruses | Amino-Acid Identity (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bat SARS-CoV-like BGR/2008 (Bulgaria, 2008) | Bat SARS-CoV-like BtKY72 (Kenya, 2007) | Bat SARS-CoV-like (China, 2005–2016) * | Civet SARS-CoV-like SZ3 (China, 2003) | SARS-CoV Urbani (2003) | Bat SARS-CoV-2-like RaTG13 (China, 2013) | Pangolin SARS-CoV-2-like (China, 2017) | SARS-CoV-2 Wuhan-Hu-1 (2019) | Khosta-1 vs. Khosta-2 | ||
| ORF1a | Khosta-1 | 92.95 | 84.6 | 81.53–81.6 | 81.67 | 81.53 | 77.2 | 77.89 | 77.32 | 82 |
| Khosta-2 | 81.1 | 80.9 | 79.4–79.6 | 79.5 | 79.4 | 76.3 | 77.1 | 76.45 | ||
| ORF1b | Khosta-1 | 99.07 | 96.3 | 95.82–96.3 | 96.15 | 96.15 | 94.22 | 94.22 | 94.21 | 94.75 |
| Khosta-2 | 94.7 | 93.7 | 94.9–95.17 | 95.02 | 94.9 | 93.44 | 93.47 | 93.5 | ||
| S | Khosta-1 | 84.37 | 89.11 | 75.5–76.2 | 75.7 | 75.7 | 73.0 | 72.4 | 72.22 | 82 |
| Khosta-2 | 79.54 | 79.7 | 73.03–73.9 | 73.2 | 73.0 | 72.5 | 71.74 | 72.54 | ||
| S RBD | Khosta-1 | 81.3 | 90.0 | 77.1–78.5 | 78.0 | 76.7 | 74.0 | 74.2 | 72.2 | 80 |
| Khosta-2 | 74.9 | 80.0 | 64.3–75.4 | 75.3 | 75.9 | 67.5 | 68.5 | 69.0 | ||
| ORF3 | Khosta-1 | 85.98 | 86.7 | 66.8–72.3 | 70.8 | 70.8 | 65.1 | 66.2 | 64.7 | 81.8 |
| Khosta-2 | 77.9 | 82.22 | 67.9–69.34 | 67.15 | 67.5 | 64.5 | 65.8 | 63.27 | ||
| E | Khosta-1 | 89.47 | 98.7 | 87.0 | 87 | 87 | 93.42 | 93.42 | 93.42 | 94.7 |
| Khosta-2 | 88.16 | 94.74 | 90.7 | 90.7 | 90.7 | 89.5 | 89.5 | 89.5 | ||
| M | Khosta-1 | 95.0 | 97.29 | 91.86–92.31 | 92.31 | 91.86 | 88.24 | 87.73 | 88.13 | 91 |
| Khosta-2 | 90.9 | 90.5 | 88.7–89.6 | 90.5 | 89.6 | 87.3 | 87.27 | 87.0 | ||
| ORF6 | Khosta-1 | 68.25 | 63.0 | 49.21–52.38 | 49.21 | 49.21 | 50.82 | 50.82 | 50.82 | 58.73 |
| Khosta-2 | 58.1 | 58.1 | 44.4–47.6 | 46.03 | 46.03 | 46.7 | 46.7 | 46.7 | ||
| ORF7a | Khosta-1 | 69.7 | 70.6 | 58–59.7 | 61.34 | 61.34 | 58.5 | 59.32 | 58.5 | 73.5 |
| Khosta-2 | 63.25 | 70.34 | 58.2–59.26 | 60.0 | 60.0 | 60.0 | 58.3 | 59.13 | ||
| ORF7b | Khosta-1 | 86.05 | 81.4 | 71.8 | 71.8 | 71.8 | 61.5 | 71.8 | 74.4 | 70.7 |
| Khosta-2 | 71.4 | 73.1 | 64.2 | 64.2 | 64.2 | 64.2 | 64.2 | 64.2 | ||
| N | Khosta-1 | 96.64 | 92.6 | 88.36–88.9 | 89.1 | 89.1 | 87.9 | 87.6 | 87.4 | 91.85 |
| Khosta-2 | 91.13 | 90.21 | 85.75–86.73 | 86.5 | 86.5 | 85.5 | 86.4 | 85.24 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhovsky, S.; Lenshin, S.; Romashin, A.; Vishnevskaya, T.; Vyshemirsky, O.; Bulycheva, Y.; Lvov, D.; Gitelman, A. SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020. Viruses 2022, 14, 113. https://doi.org/10.3390/v14010113
Alkhovsky S, Lenshin S, Romashin A, Vishnevskaya T, Vyshemirsky O, Bulycheva Y, Lvov D, Gitelman A. SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020. Viruses. 2022; 14(1):113. https://doi.org/10.3390/v14010113
Chicago/Turabian StyleAlkhovsky, Sergey, Sergey Lenshin, Alexey Romashin, Tatyana Vishnevskaya, Oleg Vyshemirsky, Yulia Bulycheva, Dmitry Lvov, and Asya Gitelman. 2022. "SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020" Viruses 14, no. 1: 113. https://doi.org/10.3390/v14010113