
High-Throughput Next-Generation Sequencing Respiratory Viral Panel: A Diagnostic and Epidemiologic Tool for SARS-CoV-2 and Other Viruses

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Ethics
2.2. Samples
2.3. Laboratory Processes
2.3.1. RNA Extraction and RT-PCR for SARS-CoV-2
2.3.2. Next-Generation Sequencing
2.3.3. Sequence Data Analysis
2.3.4. Performance Metric Evaluation
2.3.5. Limit of Detection and Reproducibility Studies
2.3.6. Phylogenetic Clustering of Genomes
3. Results
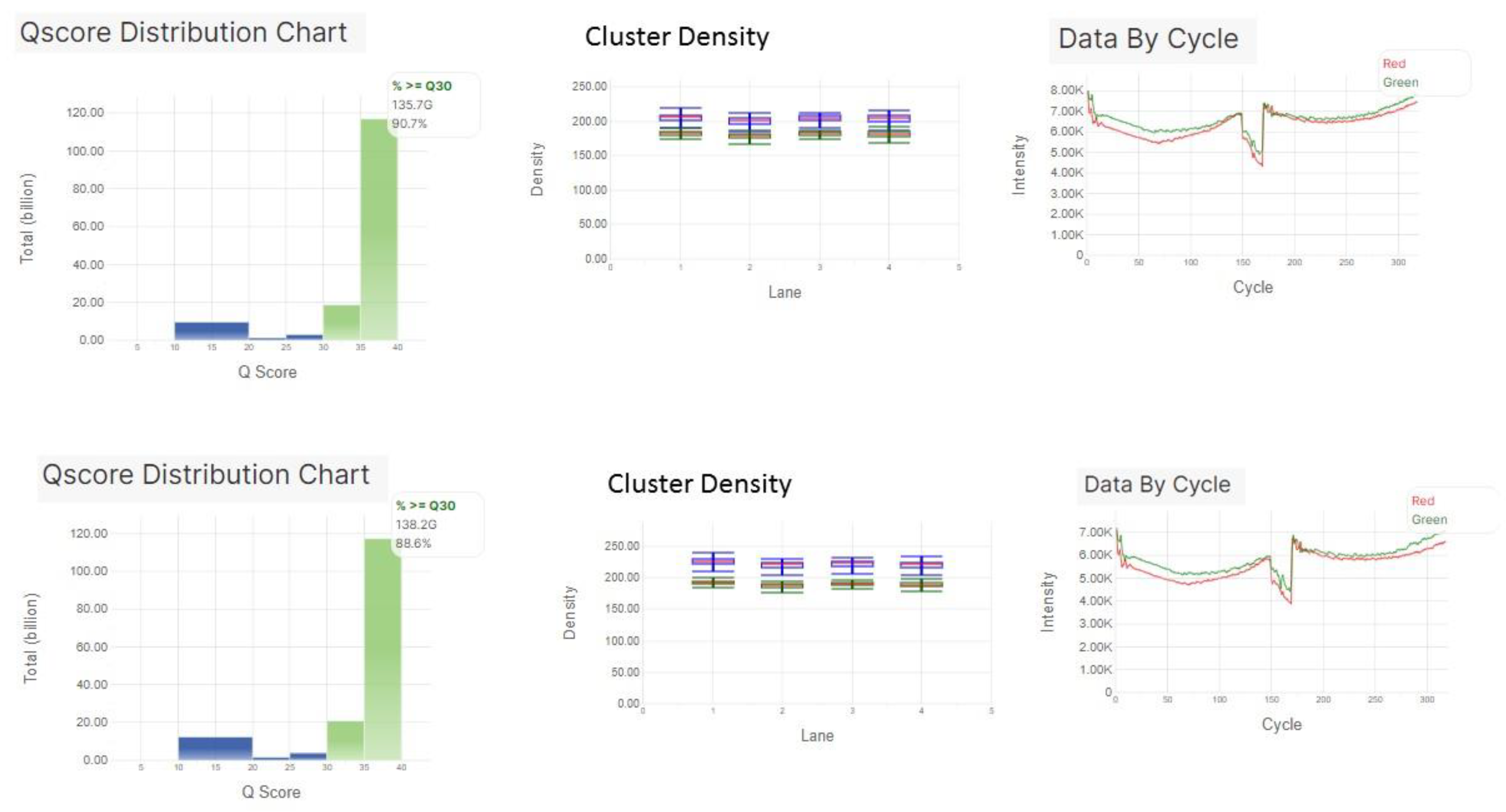
3.1. Sequencing Performance
3.2. Performance Metric Evaluation/Analytical Performance
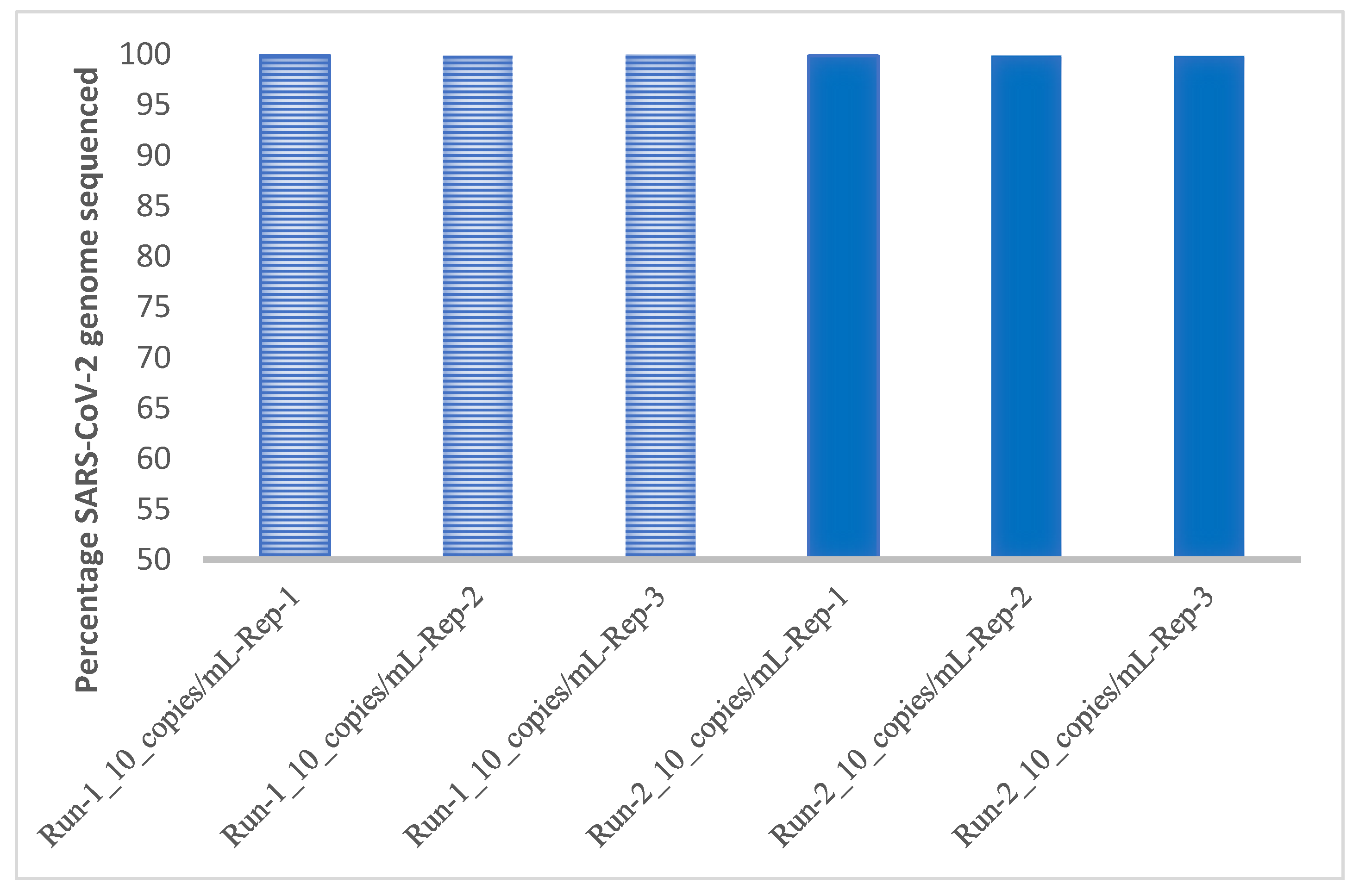
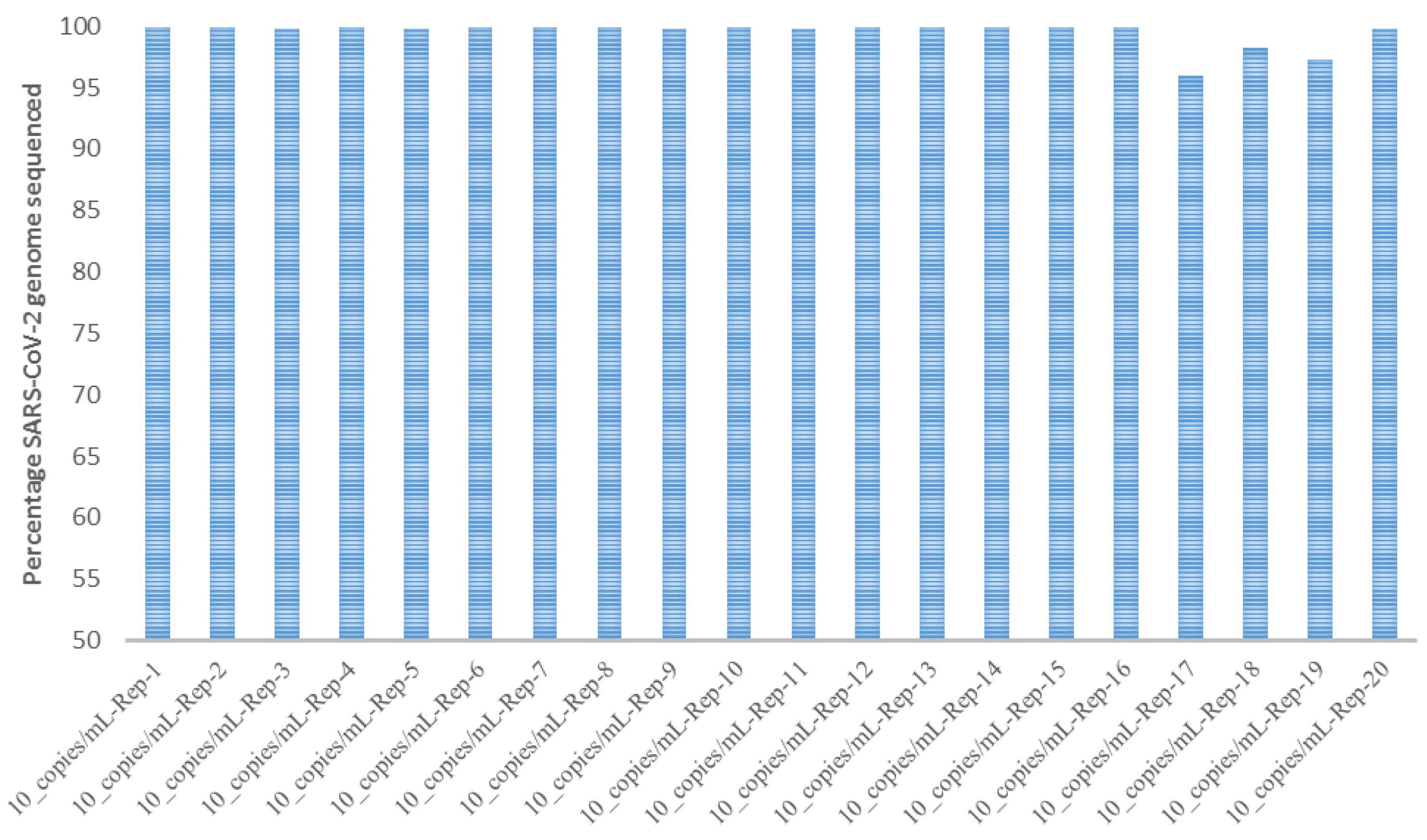
3.3. Limit of Detection and Reproducibility Studies
3.4. Co-Circulating Viruses
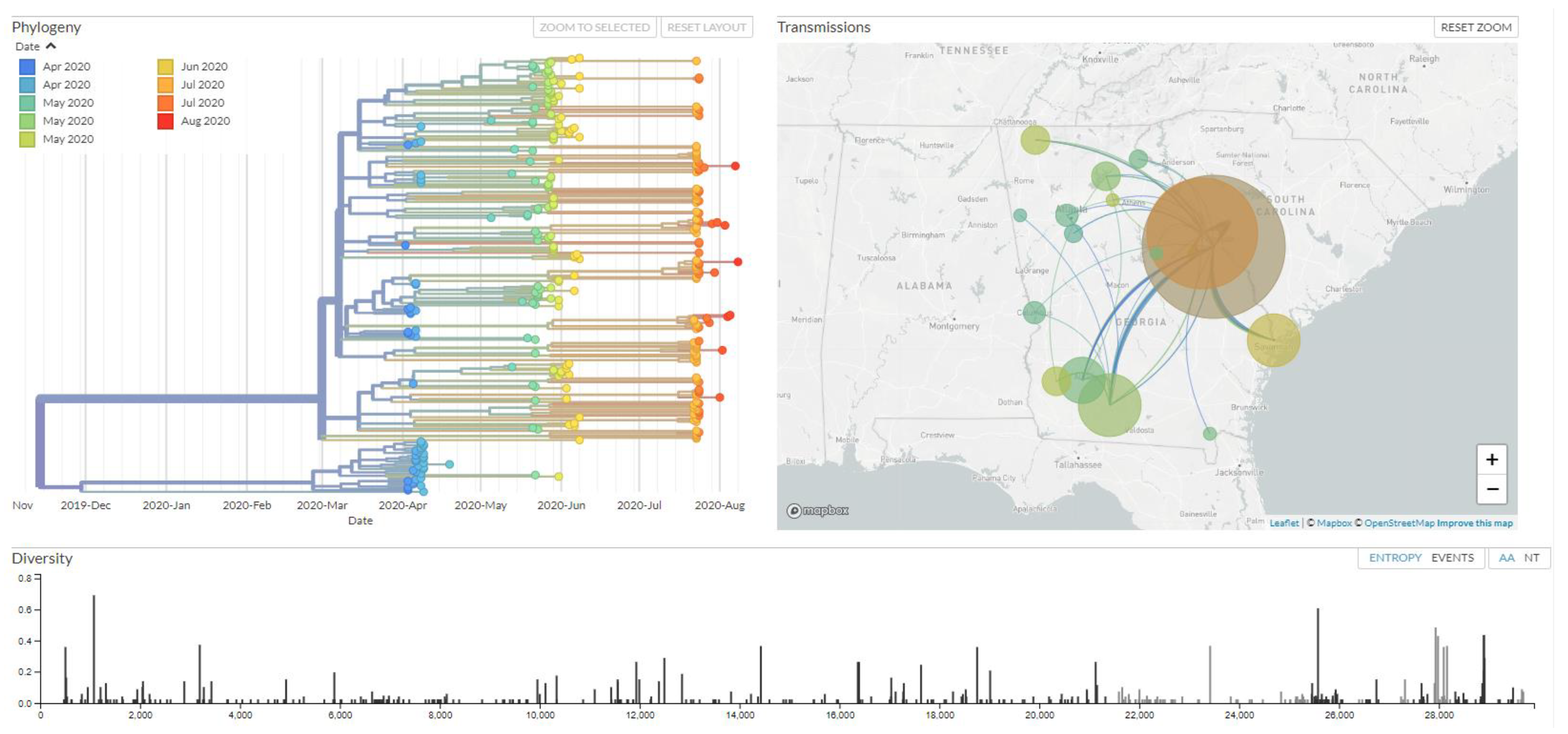
3.5. Phylogenetic Clustering of Genomes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 2 May 2021).
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Genom. Epidemiol. December 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 2 May 2021).
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Science Brief: Emerging SARS-CoV-2 Variants. Updated 28 January 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-emerging-variants.html (accessed on 2 May 2021).
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging SARS-CoV-2 variants and impact in global vaccination programs against sars-cov-2/covid-19. Vaccines 2021, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Influenza Collaborators. Mortality, morbidity, and hospitalisations due to influenza lower respiratory tract infections, 2017: An analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2019, 7, 69–89. [Google Scholar] [CrossRef] [Green Version]
- Xing, Q.; Li, G.J.; Xing, Y.H.; Chen, T.; Li, W.J.; Ni, W.; Deng, K.; Gao, R.Q.; Chen, C.Z.; Gao, Y.; et al. Precautions are needed for COVID-19 patients with co-infection of common respiratory pathogens. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Rothrock, A.N.; Swetland, S.; Andris, H.; Davis, P.; Rothrock, S.G. Viral and atypical respiratory co-infections in COVID-19: A systematic review and meta-analysis. J. Am. Coll. Emerg. Physicians Open 2020, 1, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Quinn, J.; Pinsky, B.; Shah, N.H.; Brown, I. Rates of co-infection between SARS-CoV-2 and other respiratory pathogens. JAMA 2020, 323, 2085–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazra, A.; Collison, M.; Pisano, J.; Kumar, M.; Oehler, C.; Ridgway, J.P. Coinfections with SARS-CoV-2 and other respiratory pathogens. Infect. Control Hosp. Epidemiol. 2020, 41, 1228–1229. [Google Scholar] [CrossRef] [PubMed]
- Peddu, V.; Shean, R.C.; Xie, H.; Shrestha, L.; Perchetti, G.A.; Minot, S.S.; Roychoudhury, P.; Huang, M.L.; Nalla, A.; Reddy, S.B.; et al. Metagenomic analysis reveals clinical SARS-CoV-2 infection and bacterial or viral superinfection and colonization. Clin. Chem. 2020, 66, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Van Tan, L.; Man, D.N.; Hang, V.T.; Khanh, P.N.; Xuan, T.C.; Phong, N.T.; Tu, T.N.; Hien, T.T.; Hung, L.M.; Truong, N.T.; et al. SARS-CoV-2 and co-infections detection in nasopharyngeal throat swabs of COVID-19 patients by metagenomics. J. Infect. 2020, 81, e175–e177. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. In Vitro Diagnostics EUAs. Available online: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas (accessed on 2 May 2021).
- Nextstrain Build for SARS-COV-2 in, GA. Available online: https://nextstrain.org/community/Bahl-Lab-IOB/SARS_CoV2_Augusta_Edu@main (accessed on 2 May 2021).
- Licastro, D.; Rajasekharan, S.; Dal Monego, S.; Segat, L.; D’Agaro, P.; Marcello, A. Isolation and full-length genome characterization of SARS-CoV-2 from COVID-19 cases in Northern Italy. J. Virol. 2020, 94, e00543-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J.; et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fu, A.; Hu, B.; Tong, Y.; Liu, R.; Liu, Z.; Gu, J.; Xiang, B.; Liu, J.; Jiang, W.; et al. Nanopore targeted sequencing for the accurate and comprehensive detection of SARS-CoV-2 and other respiratory viruses. Small 2020, 16, 2002169. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.D.; Sordillo, E.M.; Gitman, M.R.; Paniz Mondolfi, A.E. Coinfection in SARS-CoV-2 infected patients: Where are influenza virus and rhinovirus/enterovirus? J. Med Virol. 2020, 92, 1699–1700. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Virus |
|---|---|
| 1 | Human coronavirus 229E |
| 2 | Human coronavirus NL63 |
| 3 | Human coronavirus OC43 |
| 4 | Human Corona virus HKU1 |
| 5 | SARS-CoV-2 |
| 6 | Human adenovirus B1 |
| 7 | Human adenovirus C2 |
| 8 | Human adenovirus E4 |
| 9 | Human bocavirus 1 (primate bocaparvovirus 1 isolate st2) |
| 10 | Human bocavirus 2c PK isolate PK-5510 |
| 11 | Human bocavirus 3 |
| 12 | Human parainfluenza virus 1 |
| 13 | Human parainfluenza virus 2 |
| 14 | Human parainfluenza virus 3 |
| 15 | Human parainfluenza virus 4a |
| 16 | Human metapneumovirus (CAN97-83) |
| 17 | Respiratory syncytial virus (type A) |
| 18 | Respiratory syncytial virus 9320 (type B) |
| 19 | Influenza A virus (A/Puerto Rico/B/1934(H1N1)) |
| 20 | Influenza A virus (A/Korea/426/1968(H2N2)) |
| 21 | Influenza A virus (A/New York/392/2004(H3N2)) |
| 22 | Influenza A virus (A/goose/Guangdong/1/1996(H5N1)) |
| 23 | Human bocavirus 4 NI strain HBoV4-NI-385 |
| 24 | KI polyomavirus Stockholm 60 |
| 25 | WU polyomavirus |
| 26 | Human parechovirus 1 picoBank/HPeV1/A |
| 27 | Human parechovirus 6 |
| 28 | Human rhinovirus A89 |
| 29 | Human rhinovirus C (strain 024) |
| 30 | Human rhinovirus B14 |
| 31 | Human enterovirus C10 strain AK11 |
| 32 | Human enterovirus C109 isolate NCA08-4327 |
| 33 | Influenza A virus (A/Zhejiang/DTIDZJU01/2013(H7N9)) |
| 34 | Influenza A virus (A/Hong Kong/1073/99(H9N2)) |
| 35 | Influenza A virus (A/Texas/5020120/99(H3N2)) |
| 36 | Influenza A virus (A/Michigan/45/2015(H1N1)) |
| 37 | Influenza B virus (B/Lee/1940) |
| 38 | Influenza B virus (B/Wisconsin/60/2008) |
| 39 | Influenza B virus (B/Brisbane 60/2008) |
| 40 | Influenza B virus (B/Colorado/60/2017) |
| 41 | Influenza B virus (B/Washington/02/2019) |
| Performance Criterion | Percentage (%) |
|---|---|
| Positive percentage agreement (PPA) = TP/(TP + FN) | 95.98 |
| Negative percentage agreement (NPA) = TN/(TN + FP) | 85.96 |
| Accuracy = TP + TN/All Results | 97.48 |
| False negative rate (FNR) = FN/(FN + TP) | 4.02 |
| False positive rate (FPR) = FP/(FP + TN) | 14.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahajpal, N.S.; Mondal, A.K.; Njau, A.; Petty, Z.; Chen, J.; Ananth, S.; Ahluwalia, P.; Williams, C.; Ross, T.M.; Chaubey, A.; et al. High-Throughput Next-Generation Sequencing Respiratory Viral Panel: A Diagnostic and Epidemiologic Tool for SARS-CoV-2 and Other Viruses. Viruses 2021, 13, 2063. https://doi.org/10.3390/v13102063
Sahajpal NS, Mondal AK, Njau A, Petty Z, Chen J, Ananth S, Ahluwalia P, Williams C, Ross TM, Chaubey A, et al. High-Throughput Next-Generation Sequencing Respiratory Viral Panel: A Diagnostic and Epidemiologic Tool for SARS-CoV-2 and Other Viruses. Viruses. 2021; 13(10):2063. https://doi.org/10.3390/v13102063
Chicago/Turabian StyleSahajpal, Nikhil S., Ashis K. Mondal, Allan Njau, Zachary Petty, Jiani Chen, Sudha Ananth, Pankaj Ahluwalia, Colin Williams, Ted M. Ross, Alka Chaubey, and et al. 2021. "High-Throughput Next-Generation Sequencing Respiratory Viral Panel: A Diagnostic and Epidemiologic Tool for SARS-CoV-2 and Other Viruses" Viruses 13, no. 10: 2063. https://doi.org/10.3390/v13102063