
Discovery and Genetic Characterization of Novel Paramyxoviruses Related to the Genus Henipavirus in Crocidura Species in the Republic of Korea

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Capture and RNA Extraction
2.3. Metagenomic Next-Generation Sequencing (NGS) Using Sequence-Independent, Single-Primer Amplification (SISPA)
2.4. NGS for Illumina MiSeq
2.5. NGS for Illumina HiSeq
2.6. Extraction of Paramyxoviral Genome Sequences from NGS Data
2.7. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Screening of Samples from Crocidura Species Individuals for Paramyxovirus
2.8. Rapid Amplification of cDNA Ends (RACE) PCR
2.9. Mitochondrial DNA (mtDNA) Analysis
2.10. Phylogenetic Analysis
2.11. Virus–Host Co-Divergence Analysis
2.12. Cell Lines
2.13. Virus Isolation
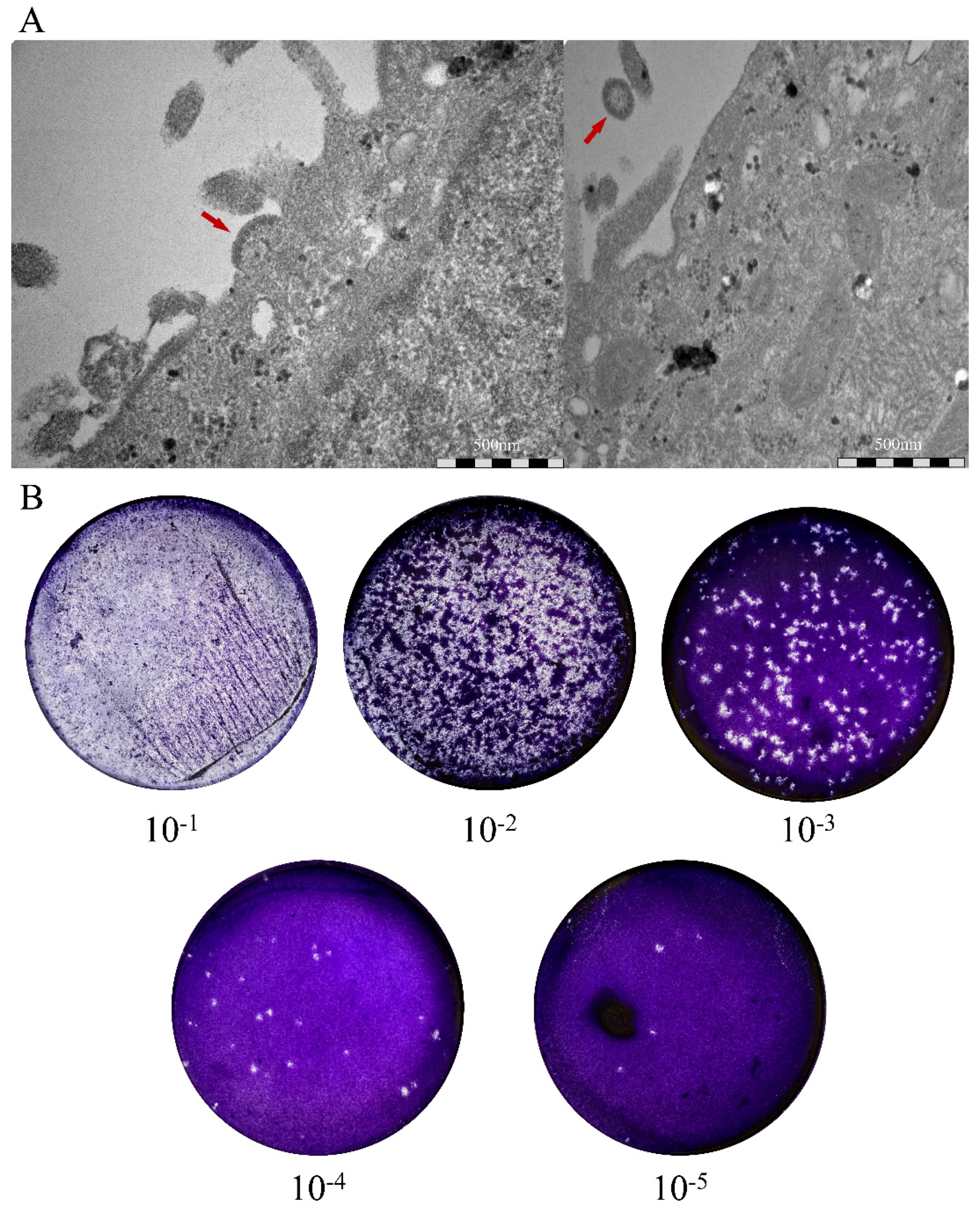
2.14. Plaque Assay
2.15. Electron Microscopy
2.16. In Vitro Infection
2.17. Statistical Analysis
3. Results
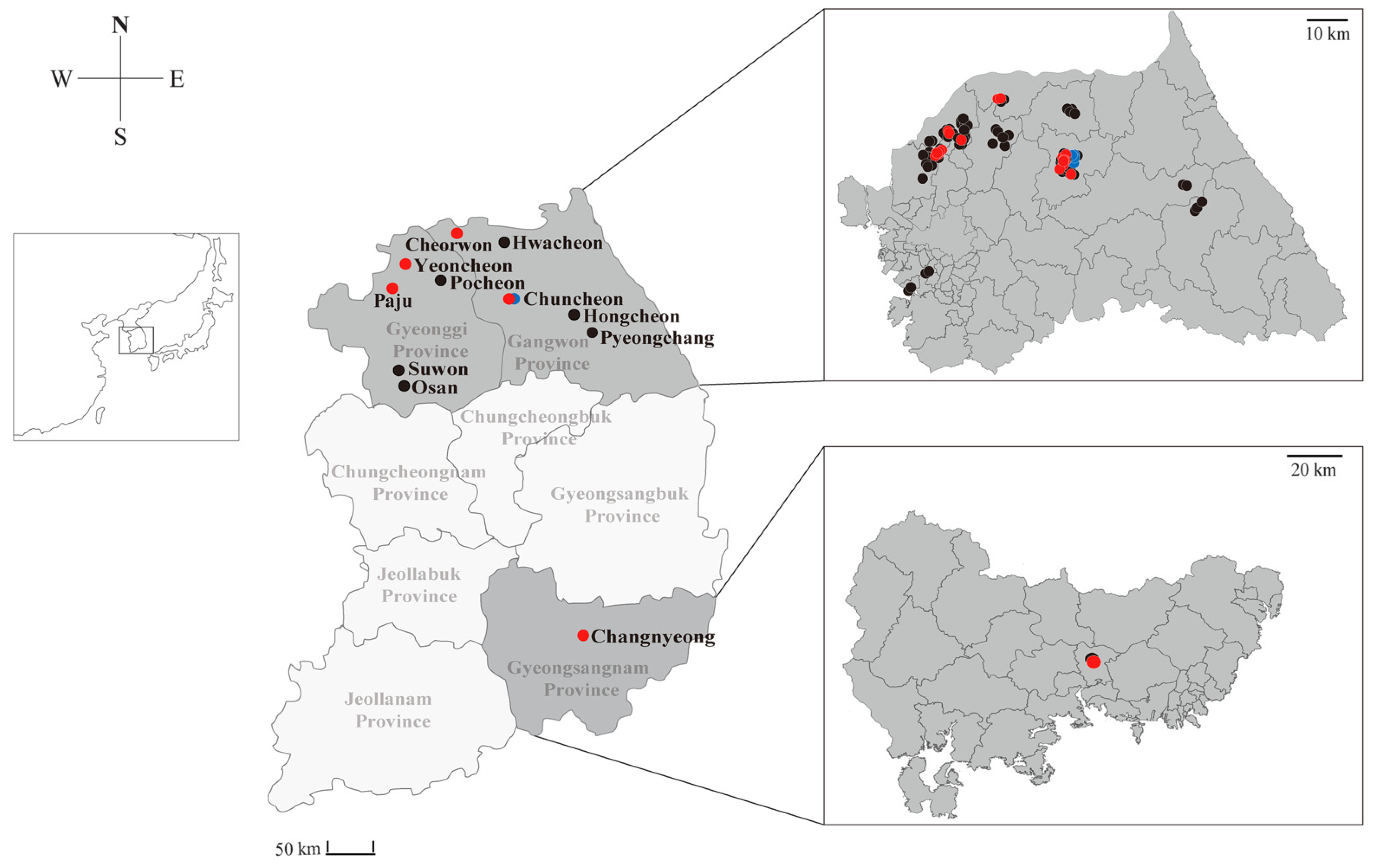
3.1. Discovery and Epidemiological Prevalence of Novel Henipaviruses in Shrews
3.2. Isolation of Novel Henipaviruses in Shrews
3.3. Nearly Whole-Genome Sequencing of Shrew-Borne Henipaviruses Using NGS
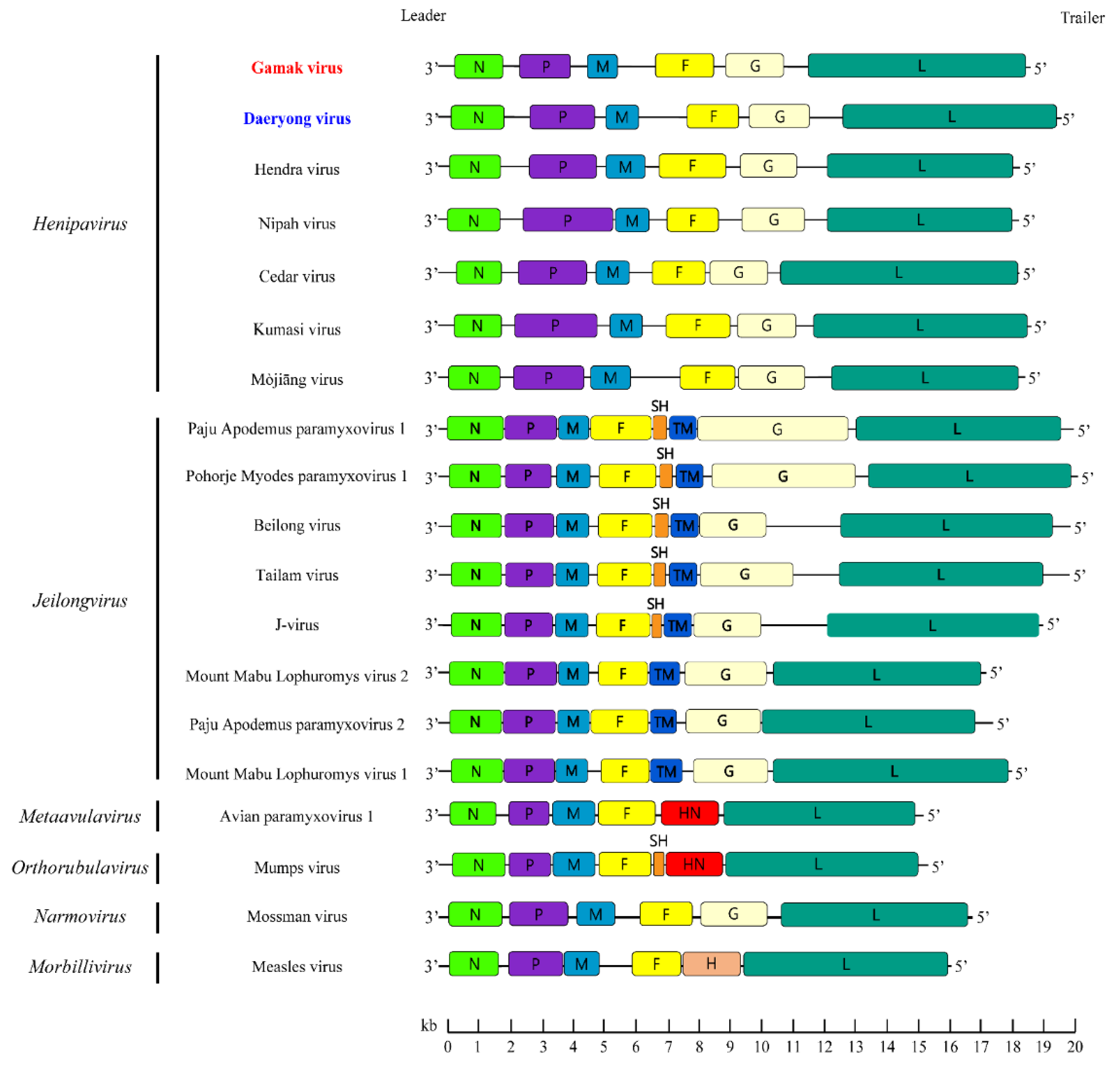
3.4. Genome Structure of GAKV and DARV
3.5. Phylogenetic Analysis
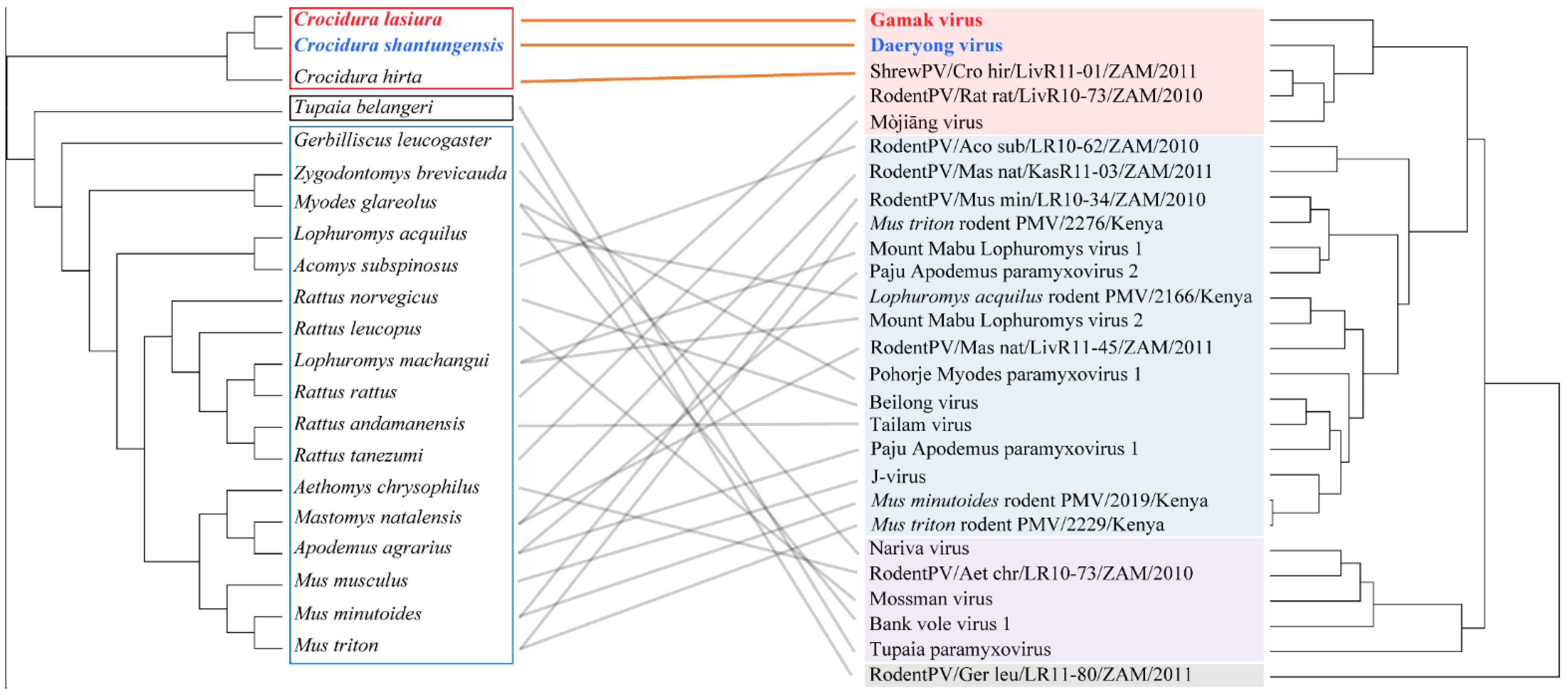
3.6. Co-Phylogeny of Paramyxovirus and Host
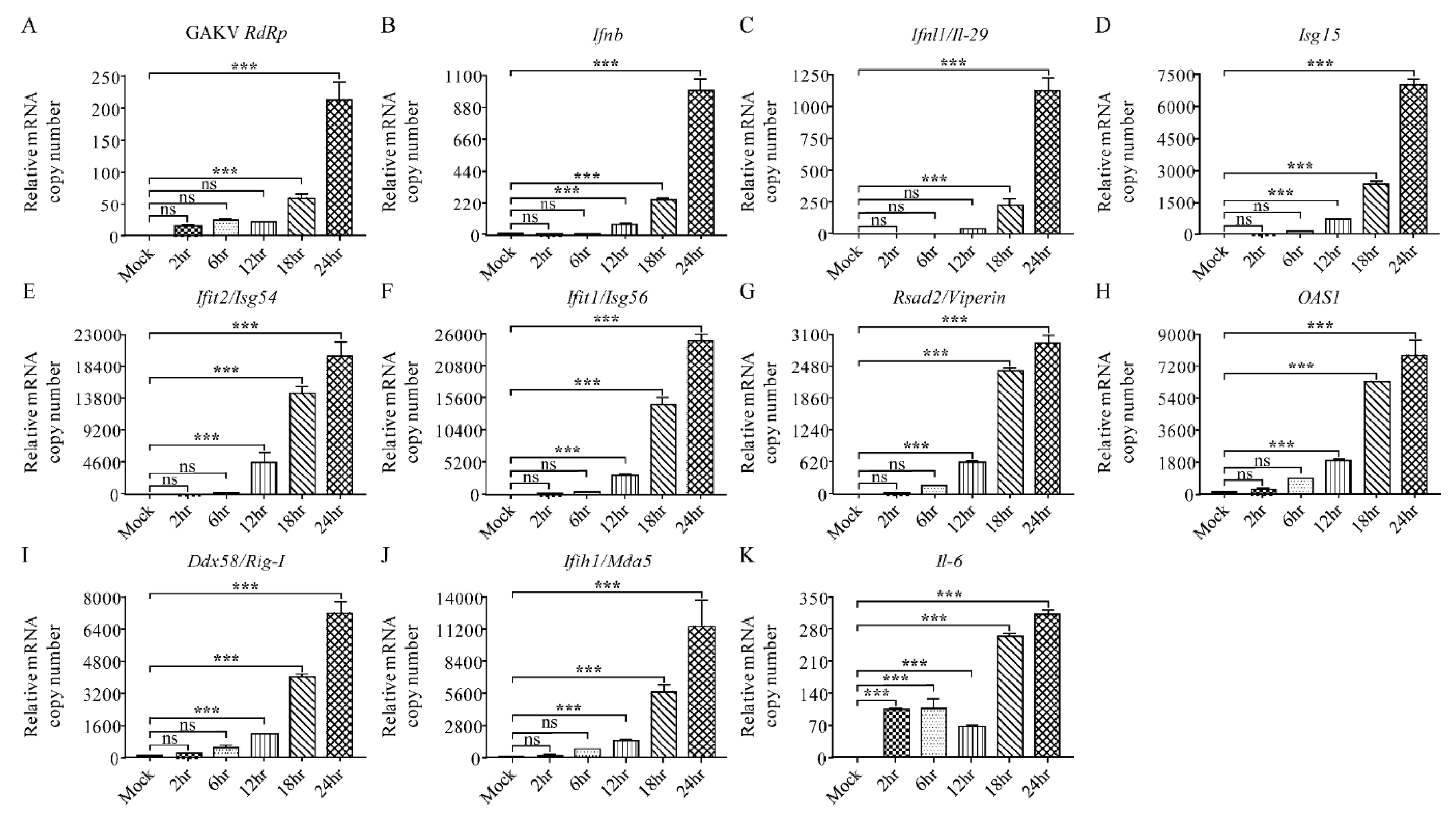
3.7. Characterization of Innate Immune Responses to GAKV in Human Lung Epithelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, L.H.; Latham, S.M.; Woolhouse, M.E. Risk factors for human disease emergence. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2001, 356, 983–989. [Google Scholar] [CrossRef]
- Woolhouse, M.E.; Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 2005, 11, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.K.; Hitchens, P.L.; Pandit, P.S.; Rushmore, J.; Evans, T.S.; Young, C.C.W.; Doyle, M.M. Global shifts in mammalian population trends reveal key predictors of virus spillover risk. Proc. R. Soc. B Boil. Sci. 2020, 287, 20192736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.K.; Hitchens, P.; Evans, T.S.; Goldstein, T.; Thomas, K.; Clements, A.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; Karesh, W.; et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 2015, 5, 14830. [Google Scholar] [CrossRef] [Green Version]
- Lessler, J.; Chaisson, L.H.; Kucirka, L.M.; Bi, Q.; Grantz, K.; Salje, H.; Carcelen, A.C.; Ott, C.T.; Sheffield, J.S.; Ferguson, N.M.; et al. Assessing the global threat from Zika virus. Science 2016, 353, aaf8160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martínez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N.; WHO Zika Causality Working Group. Zika Virus Infection as a Cause of Congenital Brain Abnormalities and Guillain–Barré Syndrome: Systematic Review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajero, O.; Vilas, V.D.R.; Wood, J.L.N.; Iacono, G.L. New methodologies for the estimation of population vulnerability to diseases: A case study of Lassa fever and Ebola in Nigeria and Sierra Leone. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.-K.; Cho, S.; Lee, S.-H.; No, J.S.; Lee, G.-Y.; Park, K.; Lee, D.; Jeong, S.T.; Song, J.-W. Genomic Epidemiology and Active Surveillance to Investigate Outbreaks of Hantaviruses. Front. Cell. Infect. Microbiol. 2021, 10, 532388. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Wilson, D.E.; Reeder, D.M. Mammal Species of the World A Taxonomic and Geographic Reference, 3rd ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Niller, H.H.; Angstwurm, K.; Rubbenstroth, D.; Schlottau, K.; Ebinger, A.; Giese, S.; Wunderlich, S.; Banas, B.; Forth, L.F.; Hoffmann, D.; et al. Zoonotic spillover infections with Borna disease virus 1 leading to fatal human encephalitis, 1999–2019: An epidemiological investigation. Lancet Infect. Dis. 2020, 20, 467–477. [Google Scholar] [CrossRef]
- Li, K.; Lin, X.-D.; Wang, W.; Shi, M.; Guo, W.-P.; Zhang, X.-H.; Xing, J.-G.; He, J.-R.; Wang, K.; Li, M.-H.; et al. Isolation and characterization of a novel arenavirus harbored by Rodents and Shrews in Zhejiang province, China. Virology 2015, 476, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Lin, X.-D.; Liao, Y.; Guan, X.-Q.; Guo, W.-P.; Xing, J.-G.; Holmes, E.C.; Zhang, Y.-Z. Discovery of a Highly Divergent Coronavirus in the Asian House Shrew from China Illuminates the Origin of the Alphacoronaviruses. J. Virol. 2017, 91, e00764-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-W.; Baek, L.J.; Schmaljohn, C.S.; Yanagihara, R. Thottapalayam Virus, a Prototype Shrewborne Hantavirus. Emerg. Infect. Dis. 2007, 13, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Gu, S.H.; Bennett, S.N.; Arai, S.; Puorger, M.; Hilbe, M.; Yanagihara, R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol. J. 2007, 4, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-W.; Kang, H.J.; Song, K.-J.; Truong, T.T.; Bennett, S.N.; Arai, S.; Truong, N.U.; Yanagihara, R. Newfound Hantavirus in Chinese Mole Shrew, Vietnam. Emerg. Infect. Dis. 2007, 13, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Kang, H.J.; Gu, S.H.; Moon, S.S.; Bennett, S.N.; Song, K.-J.; Baek, L.J.; Kim, H.-C.; O’Guinn, M.L.; Chong, S.-T.; et al. Characterization of Imjin Virus, a Newly Isolated Hantavirus from the Ussuri White-Toothed Shrew (Crocidura lasiura). J. Virol. 2009, 83, 6184–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Gu, S.H.; Baek, L.J.; Tabara, K.; Bennett, S.N.; Oh, H.-S.; Takada, N.; Kang, H.J.; Tanaka-Taya, K.; Morikawa, S.; et al. Divergent ancestral lineages of newfound hantaviruses harbored by phylogenetically related crocidurine shrew species in Korea. Virology 2012, 424, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Lin, X.-D.; Huang, K.-Y.; Zhang, B.; Shi, M.; Guo, W.-P.; Wang, M.-R.; Wang, W.; Xing, J.-G.; Li, M.-H.; et al. Identification of novel and diverse rotaviruses in rodents and insectivores, and evidence of cross-species transmission into humans. Virology 2016, 494, 168–177. [Google Scholar] [CrossRef]
- Nie, F.-Y.; Tian, J.-H.; Lin, X.-D.; Yu, B.; Xing, J.-G.; Cao, J.-H.; Holmes, E.C.; Ma, R.Z.; Zhang, Y.-Z. Discovery of a highly divergent hepadnavirus in shrews from China. Virology 2019, 531, 162–170. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of Next-Generation Sequencing Technologies to Diagnostic Virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef] [Green Version]
- Blomström, A.-L. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Veter. Q. 2011, 31, 107–114. [Google Scholar] [CrossRef]
- Belák, S.; Karlsson, O.E.; Blomström, A.-L.; Berg, M.; Granberg, F. New viruses in veterinary medicine, detected by metagenomic approaches. Veter. Microbiol. 2013, 165, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cárcer, D.A.; Angly, F.E.; Alcamí, A. Evaluation of viral genome assembly and diversity estimation in deep metagenomes. BMC Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral Diversity of House Mice in New York City. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef] [PubMed]
- Thibault, P.A.; Watkinson, R.E.; Moreira-Soto, A.; Drexler, J.F.; Lee, B. Zoonotic Potential of Emerging Paramyxoviruses: Knowns and Unknowns. Adv. Virus Res. 2017, 98, 1–55. [Google Scholar]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [Green Version]
- Vanmechelen, B.; Bletsa, M.; Laenen, L.; Lopes, A.R.; Vergote, V.; Beller, L.; Deboutte, W.; Korva, M.; Županc, T.A.; De Bellocq, J.G.; et al. Discovery and genome characterization of three new Jeilongviruses, a lineage of paramyxoviruses characterized by their unique membrane proteins. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; No, J.S.; Kim, K.; Budhathoki, S.; Park, K.; Lee, G.-Y.; Cho, S.; Kim, B.-H.; Cho, S.; Kim, J.; et al. Novel Paju Apodemus paramyxovirus 1 and 2, harbored by Apodemus agrarius in the Republic of Korea. Virology 2021, 562, 40–49. [Google Scholar] [CrossRef]
- Vanmechelen, B.; Meurs, S.; Zisi, Z.; de Bellocq, J.G.; Bletsa, M.; Lemey, P.; Maes, P. Genome Sequence of Ruloma Virus, a Novel Paramyxovirus Clustering Basally to Members of the Genus Jeilongvirus. Microbiol. Resour. Announc. 2021, 10, 18. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Nakagawa, S.; Mitsuhashi, S.; Ogawa, M.; Sugiyama, K.; Tamukai, K.; Koide, R.; Katayama, Y.; Nakano, T.; Makino, S.; et al. Molecular characterization of feline paramyxovirus in Japanese cat populations. Arch. Virol. 2019, 165, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Onyuok, S.O.; Hu, B.; Li, B.; Fan, Y.; Kering, K.; Ochola, G.O.; Zheng, X.-S.; Obanda, V.; Ommeh, S.; Yang, X.-L.; et al. Molecular Detection and Genetic Characterization of Novel RNA Viruses in Wild and Synanthropic Rodents and Shrews in Kenya. Front. Microbiol. 2019, 10, 2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, J.Y.; Jeong, D.G.; Yoon, S.-W.; Kim, J.H.; Choi, Y.G.; Kang, S.-Y.; Kim, H.K. Isolation and characterization of novel bat paramyxovirus B16-40 potentially belonging to the proposed genus Shaanvirus. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.S.; Noh, J.Y.; Lo, V.T.; Choi, Y.G.; Yoon, S.-W.; Jeong, D.G.; Kim, H.K. The Epidemiological Characteristics of the Korean Bat Paramyxovirus between 2016 and 2019. Microorganisms 2020, 8, 844. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Rota, P.A.; Rollin, P. A review of Nipah and Hendra viruses with an historical aside. Virus Res. 2011, 162, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Clayton, B.A. Nipah virus: Transmission of a zoonotic paramyxovirus. Curr. Opin. Virol. 2017, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.K.; Rota, P.A. The emergence of Nipah virus, a highly pathogenic paramyxovirus. J. Clin. Virol. 2008, 43, 396–400. [Google Scholar] [CrossRef]
- Sharma, V.; Kaushik, S.; Kumar, R.; Yadav, J.P.; Kaushik, S. Emerging trends of Nipah virus: A review. Rev. Med. Virol. 2019, 29, e2010. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Muleya, W.; Ishii, A.; Orba, Y.; Hang’Ombe, B.M.; Mweene, A.S.; Moonga, L.; Thomas, Y.; Kimura, T.; Sawa, H. Molecular epidemiology of paramyxoviruses in Zambian wild rodents and shrews. J. Gen. Virol. 2014, 95, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, L.; Yang, F.; Ren, X.; Jiang, J.; Dong, J.; Sun, L.; Zhu, Y.; Zhou, H.; Jin, Q. Novel Henipa-like Virus, Mojiang Paramyxovirus, in Rats, China, 2012. Emerg. Infect. Dis. 2014, 20, 1064–1066. [Google Scholar] [CrossRef]
- Rissanen, I.; Ahmed, A.A.; Azarm, K.; Beaty, S.; Hong, P.; Nambulli, S.; Duprex, W.P.; Lee, B.; Bowden, T.A. Idiosyncratic Mòjiāng virus attachment glycoprotein directs a host-cell entry pathway distinct from genetically related henipaviruses. Nat. Commun. 2017, 8, 16060. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.C.; Yan, L.; Dang, H.; Xu, K.; Epstein, J.; Veesler, D.; Broder, C. Functional Analysis of the Fusion and Attachment Glycoproteins of Mojiang Henipavirus. Viruses 2021, 13, 517. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-C.; Chain, P.S.G. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinform. 2014, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Chern, S.-W.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and Broadly Reactive Reverse Transcription-PCR Assays To Detect Novel Paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochrome b gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [Green Version]
- Meerburg, B.G.; Singleton, G.; Kijlstra, A. Rodent-borne diseases and their risks for public health. Crit. Rev. Microbiol. 2009, 35, 221–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cai, C.-L.; Chun-Lin, C.; Zhang, W.; Zhu, Y.; Chen, W.-H.; Zhuo, F.; Shi, Z.-L.; Yang, X.-L. Detection and characterization of three zoonotic viruses in wild rodents and shrews from Shenzhen city, China. Virol. Sin. 2017, 32, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceição-Neto, N.; Godinho, R.; Álvares, F.; Yinda, C.K.; Deboutte, W.; Zeller, M.; Laenen, L.; Heylen, E.; Roque, S.; Petrucci-Fonseca, F.; et al. Viral gut metagenomics of sympatric wild and domestic canids, and monitoring of viruses: Insights from an endangered wolf population. Ecol. Evol. 2017, 7, 4135–4146. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, F.; Silveira, C.B.; Gregoracci, G.; Thompson, C.C.; Edwards, R.; Brussaard, C.P.D.; Dutilh, B.E.; Thompson, F.L. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017, 8, 15955. [Google Scholar] [CrossRef]
- He, W.; Gao, Y.; Wen, Y.; Ke, X.; Ou, Z.; Li, Y.; He, H.; Chen, Q. Detection of Virus-Related Sequences Associated with Potential Etiologies of Hepatitis in Liver Tissue Samples From Rats, Mice, Shrews, and Bats. Front. Microbiol. 2021, 12, 653873. [Google Scholar] [CrossRef]
- Alkhovsky, S.; Butenko, A.; Eremyan, A.; Shchetinin, A. Genetic characterization of bank vole virus (BaVV), a new paramyxovirus isolated from kidneys of bank voles in Russia. Arch. Virol. 2017, 163, 755–759. [Google Scholar] [CrossRef]
- Jackson, A.P.; Charleston, M.A. A Cophylogenetic Perspective of RNA–Virus Evolution. Mol. Biol. Evol. 2004, 21, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Vanmechelen, B.; Zisi, Z.; Gryseels, S.; de Bellocq, J.G.; Vrancken, B.; Lemey, P.; Maes, P.; Bletsa, M. Phylogenomic Characterization of Lopma Virus and Praja Virus, Two Novel Rodent-Borne Arteriviruses. Viruses 2021, 13, 1842. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Herbreteau, V.; Henttonen, H.; Yoshimatsu, K.; Gonzalez, J.-P.; Suputtamongkol, Y.; Hugot, J.-P. Hantavirus Coevolution with Their Rodent Hosts. Encycl. Infect. Dis. 2006, 243–264. [Google Scholar] [CrossRef]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, 22, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plyusnin, A.; Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100MYR. Virus Res. 2014, 187, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.-W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsden, C.; Holmes, E.; Charleston, M. Hantavirus Evolution in Relation to Its Rodent and Insectivore Hosts: No Evidence for Codivergence. Mol. Biol. Evol. 2008, 26, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripp, R.A.; Oshansky, C.; Alvarez, R. Cytokines and respiratory syncytial virus infection. Proc. Am. Thorac. Soc. 2005, 2, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.-F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Genet. 2006, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- León, A.J.; Borisevich, V.; Boroumand, N.; Seymour, R.; Nusbaum, R.; Escaffre, O.; Xu, L.; Kelvin, D.J.; Rockx, B. Host gene expression profiles in ferrets infected with genetically distinct henipavirus strains. PLoS Negl. Trop. Dis. 2018, 12, e0006343. [Google Scholar] [CrossRef]
- Ikegame, S.; Carmichael, J.C.; Wells, H.; Furler, R.L.; Acklin, J.A.; Chiu, H.-P.; Oguntuyo, K.Y.; Cox, R.M.; Patel, A.R.; Kowdle, S.; et al. Zoonotic potential of a novel bat morbillivirus. bioRxiv 2021, arXiv:2021.09.17.460143. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Species | Province | Location | Number of Samples | Positivity for Paramyxovirus (%) |
|---|---|---|---|---|---|
| 2017 | Crocidura lasiura | Gangwon | Cheorwon | 4 | 2/4 (50.0) |
| Chuncheon | 17 | 5/17 (29.4) | |||
| Hwacheon | 5 | 0/5 | |||
| Gyeonggi | Paju | 11 | 6/11 (54.5) | ||
| Yeoncheon | 20 | 3/20 (15.0) | |||
| Gyeongsangnam | Changnyeong | 2 | 1/2 (50.0) | ||
| Crocidura shantungensis | Gangwon | Chuncheon | 8 | 5/8 (62.5) | |
| Gyeonggi | Paju | 2 | 0/2 | ||
| Yeoncheon | 2 | 0/2 | |||
| Subtotal | 71 | 22/71 (31.0) | |||
| 2018 | Crocidura lasiura | Gangwon | Chuncheon | 10 | 3/10 (30.0) |
| Hongcheon | 2 | 0/2 | |||
| Pyeongchang | 2 | 0/2 | |||
| Gyeonggi | Osan | 2 | 0/2 | ||
| Paju | 7 | 0/7 | |||
| Pocheon | 4 | 0/4 | |||
| Suwon | 2 | 0/2 | |||
| Yeoncheon | 6 | 0/6 | |||
| Crocidura shantungensis | Gangwon | Chuncheon | 1 | 1/1 (100) | |
| Pyeongchang | 1 | 0/1 | |||
| Gyeonggi | Paju | 4 | 0/4 | ||
| Pocheon | 2 | 0/2 | |||
| Yeoncheon | 1 | 0/1 | |||
| Subtotal | 44 | 4/44 (9.1) | |||
| Total | 115 | 26/115 (22.6) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Kim, K.; Kim, J.; No, J.S.; Park, K.; Budhathoki, S.; Lee, S.H.; Lee, J.; Cho, S.H.; Cho, S.; et al. Discovery and Genetic Characterization of Novel Paramyxoviruses Related to the Genus Henipavirus in Crocidura Species in the Republic of Korea. Viruses 2021, 13, 2020. https://doi.org/10.3390/v13102020
Lee S-H, Kim K, Kim J, No JS, Park K, Budhathoki S, Lee SH, Lee J, Cho SH, Cho S, et al. Discovery and Genetic Characterization of Novel Paramyxoviruses Related to the Genus Henipavirus in Crocidura Species in the Republic of Korea. Viruses. 2021; 13(10):2020. https://doi.org/10.3390/v13102020
Chicago/Turabian StyleLee, Seung-Ho, Kijin Kim, Jongwoo Kim, Jin Sun No, Kyungmin Park, Shailesh Budhathoki, Seung Ho Lee, Jingyeong Lee, Seung Hye Cho, Seungchan Cho, and et al. 2021. "Discovery and Genetic Characterization of Novel Paramyxoviruses Related to the Genus Henipavirus in Crocidura Species in the Republic of Korea" Viruses 13, no. 10: 2020. https://doi.org/10.3390/v13102020