
Prevalence of Hantaviruses Harbored by Murid Rodents in Northwestern Ukraine and Discovery of a Novel Puumala Virus Strain

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
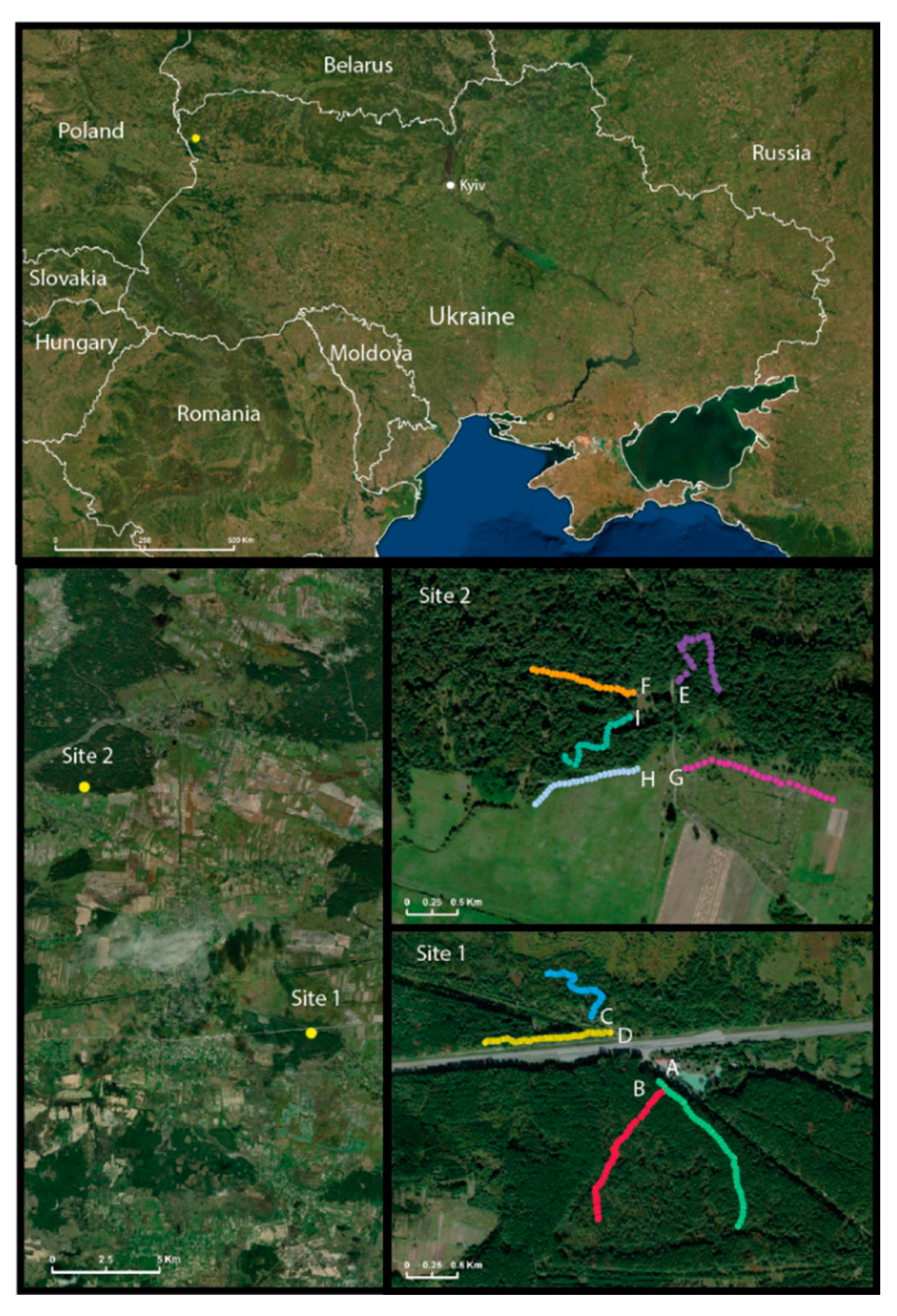
2.1. Small Mammal Collection
2.2. Immunofluorescence Assay (IFA)
2.3. Screening of Rodent Lung RNA by Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR) for PUUV and DOBV
2.4. Next-Generation Sequencing
2.5. Phylogenetic Analyses
2.6. Statistical Analyses
2.7. Ethics Statements
3. Results
3.1. Distribution of Small Mammal Species
3.2. Evidence and Distribution of Antibodies to Hantavirus by Line
3.3. Rodent Diversity within Each Line
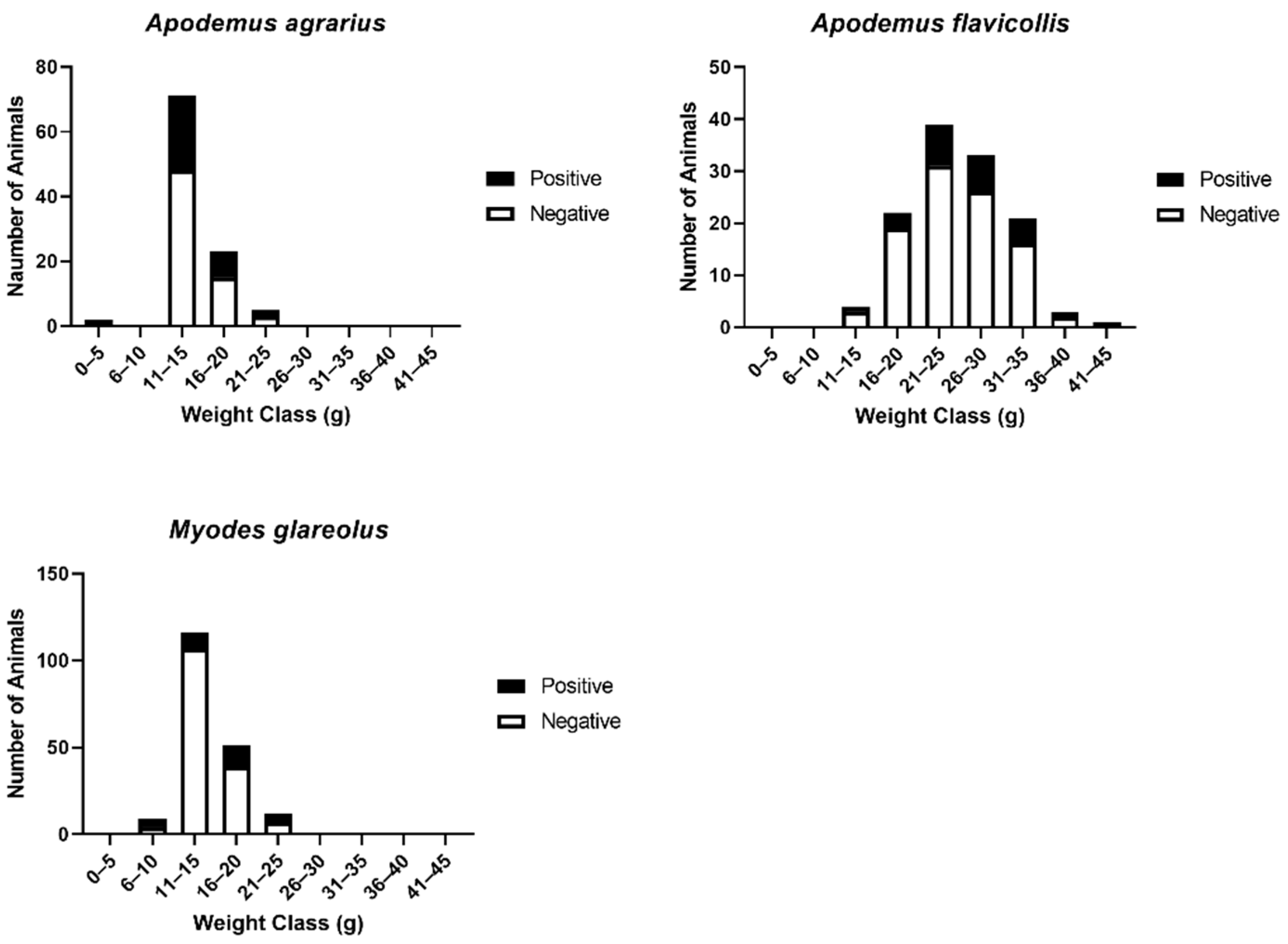
3.4. Association of Population Structure and Prevalence of Antibody to Hantavirus in Myodes glareolus and Apodemus Species
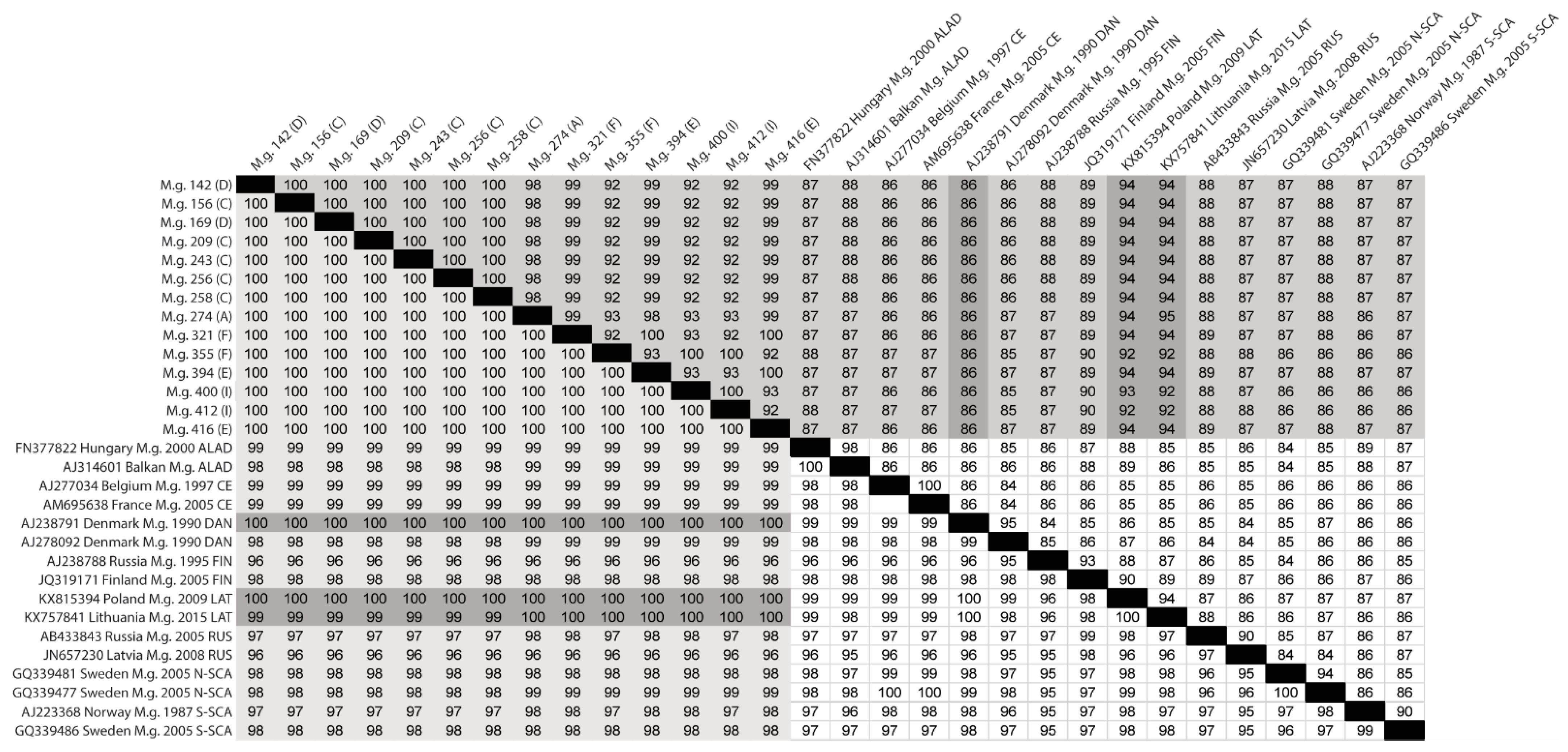
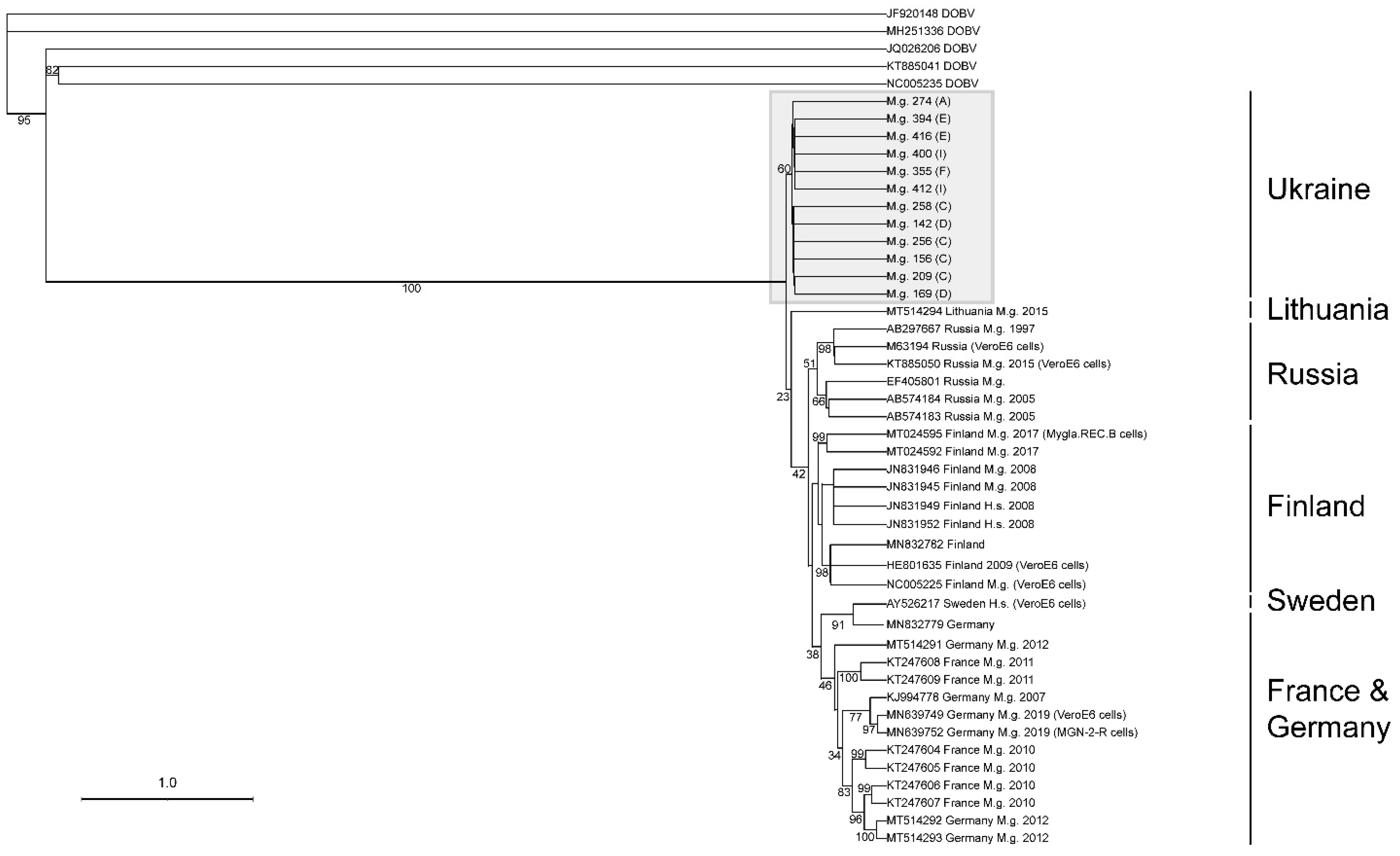
3.5. Next-Generation Sequencing and Phylogenetic Analyses of Viral Sequences Amplified from Myodes glareolus Lung
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Avsic-Zupanc, T.; Toney, A.; Anderson, K.; Chu, Y.K.; Schmaljohn, C. Genetic and antigenic properties of Dobrava virus: A unique member of the Hantavirus genus, family Bunyaviridae. J. Gen. Virol. 1995, 76 Pt 11, 2801–2808. [Google Scholar] [CrossRef]
- Avsic-Zupanc, T.; Nemirov, K.; Petrovec, M.; Trilar, T.; Poljak, M.; Vaheri, A.; Plyusnin, A. Genetic analysis of wild-type Dobrava hantavirus in Slovenia: Co-existence of two distinct genetic lineages within the same natural focus. J. Gen. Virol. 2000, 81, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Golovljova, I.; Vasilenko, V.; Prukk, T.; Brus Sjolander, K.; Plyusnin, A.; Lundkvist, A. Puumala and Dobrava hantaviruses causing hemorrhagic fever with renal syndrome in Estonia. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 968–969. [Google Scholar] [CrossRef] [PubMed]
- Jakab, F.; Horvath, G.; Ferenczi, E.; Sebok, J.; Varecza, Z.; Szucs, G. Detection of Dobrava hantaviruses in Apodemus agrarius mice in the Transdanubian region of Hungary. Virus Res. 2007, 128, 149–152. [Google Scholar] [CrossRef]
- Klempa, B.; Stanko, M.; Labuda, M.; Ulrich, R.; Meisel, H.; Kruger, D.H. Central European Dobrava Hantavirus isolate from a striped field mouse (Apodemus agrarius). J. Clin. Microbiol. 2005, 43, 2756–2763. [Google Scholar] [CrossRef] [Green Version]
- Klempa, B.; Tkachenko, E.A.; Dzagurova, T.K.; Yunicheva, Y.V.; Morozov, V.G.; Okulova, N.M.; Slyusareva, G.P.; Smirnov, A.; Kruger, D.H. Hemorrhagic fever with renal syndrome caused by 2 lineages of Dobrava hantavirus, Russia. Emerg. Infect. Dis. 2008, 14, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Klingstrom, J.; Hardestam, J.; Lundkvist, A. Dobrava, but not Saaremaa, hantavirus is lethal and induces nitric oxide production in suckling mice. Microbes Infect. 2006, 8, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Bojovic, B.; Antoniadis, A. Hantaviruses in Serbia and Montenegro. Emerg Infect. Dis. 2006, 12, 1015–1018. [Google Scholar] [CrossRef]
- Lähdevirta, J.; Savola, J.; Brummer-Korvenkontio, M.; Berndt, R.; Illikainen, R.; Vaheri, A. Clinical and serological diagnosis of Nephropathia epidemica, the mild type of haemorrhagic fever with renal syndrome. J. Infect. 1984, 9, 230–238. [Google Scholar] [CrossRef]
- Hjertqvist, M.; Klein, S.L.; Ahlm, C.; Klingstrom, J. Mortality rate patterns for hemorrhagic fever with renal syndrome caused by Puumala virus. Emerg. Infect. Dis. 2010, 16, 1584–1586. [Google Scholar] [CrossRef]
- Demchyshyna, I.V.; Glass, G.E.; Hluzd, O.A.; Kutseva, V.V.; Taylor, M.K.; Williams, E.P.; Kurpita, V.; Jonsson, C.B. Cocirculation of Two Orthohantavirus Species in Small Mammals of the Northwestern Ukraine. J. Wildl. Dis. 2020, 56, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Lozynskyi, I.; Shulgan, A.; Zarichna, O.; Ben, I.; Kessler, W.; Cao, X.; Nesterova, O.; Glass, G.E.; Spruill-Harrell, B.; Taylor, M.K.; et al. Seroprevalence of Old World Hantaviruses and Crimean Congo Hemorrhagic Fever Viruses in Human Populations in Northwestern Ukraine. Front. Cell. Infect. Microbiol. 2020, 10, 589464. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, C.B.; Chu, Y.K.; Ontiveros, S.P. Hantaan virus. In Molecular Detection of Human Viral Pathogens; Liu, D., Ed.; Taylor & Francis CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Chu, Y.K.; Owen, R.D.; Gonzalez, L.M.; Jonsson, C.B. The complex ecology of hantavirus in Paraguay. Am. J. Trop. Med. Hyg. 2003, 69, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.K.; Williams, E.P.; Wongsurawat, T.; Jenjaroenpun, P.; Nookaew, I.; Jonsson, C.B. Amplicon-Based, Next-Generation Sequencing Approaches to Characterize Single Nucleotide Polymorphisms of Orthohantavirus Species. Front. Cell Infect. Microbiol. 2020, 10, 565591. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Goeijenbier, M.; Verner-Carlsson, J.; Litjens, E.; Bos, W.J.; Pas, S.D.; Melo, M.M.; Koopmans, M.; Lundkvist, Å.; Reusken, C.B. Two clinical cases of renal syndrome caused by Dobrava/Saaremaa hantaviruses imported to the Netherlands from Poland and Belarus, 2012-2014. Infect. Ecol. Epidemiol. 2016, 6, 30548. [Google Scholar] [CrossRef] [PubMed]
- Maftei, I.D.; Segall, L.; Panculescu-Gatej, R.; Ceianu, C.; Covic, A. Hantavirus infection--hemorrhagic fever with renal syndrome: The first case series reported in Romania and review of the literature. Int. Urol. Nephrol. 2012, 44, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Mikhaĭlenko, A.G.; Tkachenko, E.A.; Smirnov, I.; Malkin, A.E.; Chebotar, I.F. [Results of serologic screening for hemorrhagic fever with kidney syndrome in Moldova]. Vopr. Virusol. 1994, 39, 260–262. [Google Scholar]
- Faludi, G.; Ferenczi, E. Serologically verified hantavirus infections in Hungary. Acta Microbiol. Immunol. Hung. 1995, 42, 419–426. [Google Scholar]
- Bilcíková, M.; Gresíková, M.; Valaseková, T.; Schréter, I.; Carnická, A. [The incidence and clinical picture of hemorrhagic fever with renal syndrome in East Slovakia: The eastern and western type]. Bratisl. Lek. Listy 1989, 90, 852–856. [Google Scholar]
- Tkachenko, E.; Ishmukhametov, A.; Dzagurova, T.; Bernshtein, A.; Morozov, V.; Siniugina, A.; Kurashova, S.; Balkina, A.; Tkachenko, P.; Kruger, D.; et al. Hemorrhagic Fever with Renal Syndrome, Russia. Emerg. Infect. Dis. J. 2019, 25, 2325. [Google Scholar] [CrossRef]
- Michalski, A.; Niemcewicz, M.; Bielawska-Drózd, A.; Nowakowska, A.; Gaweł, J.; Pitucha, G.; Joniec, J.; Zielonka, K.; Marciniak-Niemcewicz, A.; Kocik, J. Surveillance of hantaviruses in Poland: A study of animal reservoirs and human hantavirus disease in Subcarpathia. Vector Borne Zoonotic Dis. 2014, 14, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; No, J.S.; Kim, W.K.; Gajda, E.; Perec-Matysiak, A.; Kim, J.A.; Hildebrand, J.; Yanagihara, R.; Song, J.W. Molecular Epidemiology and Genetic Diversity of Orthohantaviruses in Small Mammals in Western Poland. Am. J. Trop. Med. Hyg. 2020, 103, 193–199. [Google Scholar] [CrossRef]
- Ali, H.S.; Drewes, S.; Sadowska, E.T.; Mikowska, M.; Groschup, M.H.; Heckel, G.; Koteja, P.; Ulrich, R.G. First molecular evidence for Puumala hantavirus in Poland. Viruses 2014, 6, 340–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidyuk, Y.; Shamsutdinov, A.; Kabwe, E.; Ismagilova, R.; Martynova, E.; Belyaev, A.; Shuralev, E.; Trifonov, V.; Savitskaya, T.; Isaeva, G.; et al. Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia. Pathogens 2020, 9, 540. [Google Scholar] [CrossRef] [PubMed]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Capture Lines | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | ||
| Apodemus agrarius | 4 | 0 | 11 | 20 | 3 | 0 | 21 | 1 | 2 | 62 |
| Apodemus flavicollis | 19 | 16 | 18 | 42 | 7 | 13 | 2 | 0 | 6 | 123 |
| Microtus arvalis | 0 | 0 | 0 | 2 | 0 | 0 | 3 | 0 | 1 | 6 |
| Microtus subterraneus | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 |
| Mus musculus | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Muscardinus avellanarius | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Mustela nivales | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 2 |
| Myodes glareolus | 9 | 24 | 51 | 39 | 24 | 25 | 2 | 0 | 17 | 191 |
| Sorex minutus | 5 | 0 | 10 | 15 | 3 | 1 | 1 | 0 | 0 | 35 |
| Total | 40 | 40 | 92 | 120 | 37 | 39 | 29 | 1 | 26 | 424 |
| Species | Capture Lines | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | ||
| Apodemus agrarius | 2 (4) | 0 | 3 (11) | 3 (20) | 1 (3) | 0 | 11 (21) | 1 (1) | 2 | 21 |
| Apodemus flavicollis | 3 (19) | 0 | 5 (18) | 8 (42) | 3 (7) | 4 (13) | 0 | 0 | 2 (6) | 25 |
| Microtus arvalis | 0 | 0 | 0 | 1 (2) | 0 | 0 | 0 | 0 | 0 | 1 |
| Microtus subterraneus | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mus musculus | 1 (1) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Muscardinus avellanarius | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mustela nivales | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Myodes glareolus | 2 (9) | 0 | 14 (51) | 4 (39) | 5 (24) | 3 (25) | 0 | 0 | 2 (17) | 30 |
| Sorex minutus | 0 | 0 | 1 (10) | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Total | 8 | 0 | 23 | 16 | 9 | 7 | 11 | 1 | 4 | 79 |
| Species | IFA Reciprocal Titers | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1:32 | 1:64 | 1:128 | 1:256 | 1:512 | 1:1024 | 1:2048 | 1:4096 | 1:8192 | |
| Apodemus agrarius | 0 | 5 | 5 | 3 | 1 | 3 | 1 | 2 | 1 |
| Apodemus flavicollis | 1 | 6 | 5 | 1 | 7 | 4 | 1 | 1 | 1 |
| Microtus arvalis | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Mus musculus | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Myodes glareolus | 0 | 1 | 7 | 9 | 2 | 4 | 3 | 2 | 3 |
| Sorex minutus | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Line | A | B | C | D | E | F | G | I |
|---|---|---|---|---|---|---|---|---|
| HV-Negative | 32 | 40 | 69 | 104 | 28 | 32 | 18 | 22 |
| HV-Positive | 8 | 0 | 23 | 16 | 9 | 7 | 11 | 4 |
| Total No. Rodents | 40 | 40 | 92 | 120 | 37 | 39 | 29 | 26 |
| Proportion of HV | 0.2 | 0.0 | 0.3 | 0.1 | 0.2 | 0.2 | 0.4 | 0.2 |
| SHANNON (H) | 1.46 | 0.67 | 1.24 | 1.44 | 1.00 | 0.75 | 0.95 | 0.94 |
| A. agrarius | A. flavicollis | M. glareolus | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total No. | Total No. Ab Pos * | % Pos | Total No. | Total No. Ab Pos | % Pos | Total No. | Total No. Ab Pos | % Pos | |
| MALE | |||||||||
| Juvenile | 3 | 1 | 33.3 | 22 | 4 | 18.2 | 35 | 6 | 16.7 |
| Adult | 31 | 11 | 35.5 | 60 | 14 | 23.3 | 108 | 16 | 15.0 |
| FEMALE | |||||||||
| Juvenile | 2 | 0 | 0 | 7 | 1 | 14.3 | 1 | 0 | 0 |
| Adult | 26 | 9 | 34.6 | 33 | 6 | 18.2 | 47 | 8 | 17 |
| Total | 62 | 21 | 122 | 25 | 191 | 30 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.P.; Taylor, M.K.; Demchyshyna, I.; Nebogatkin, I.; Nesterova, O.; Khuda, I.; Chernenko, L.; Hluzd, O.A.; Kutseva, V.V.; Glass, G.E.; et al. Prevalence of Hantaviruses Harbored by Murid Rodents in Northwestern Ukraine and Discovery of a Novel Puumala Virus Strain. Viruses 2021, 13, 1640. https://doi.org/10.3390/v13081640
Williams EP, Taylor MK, Demchyshyna I, Nebogatkin I, Nesterova O, Khuda I, Chernenko L, Hluzd OA, Kutseva VV, Glass GE, et al. Prevalence of Hantaviruses Harbored by Murid Rodents in Northwestern Ukraine and Discovery of a Novel Puumala Virus Strain. Viruses. 2021; 13(8):1640. https://doi.org/10.3390/v13081640
Chicago/Turabian StyleWilliams, Evan P., Mariah K. Taylor, Iryna Demchyshyna, Igor Nebogatkin, Olena Nesterova, Iryna Khuda, Lyudmyla Chernenko, Oleksandra A. Hluzd, Vira V. Kutseva, Gregory E. Glass, and et al. 2021. "Prevalence of Hantaviruses Harbored by Murid Rodents in Northwestern Ukraine and Discovery of a Novel Puumala Virus Strain" Viruses 13, no. 8: 1640. https://doi.org/10.3390/v13081640