
Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virus Discovery
2.3. Screening and Sequencing
2.4. Sequence and Phylogenetic Analyses
2.5. Codon Usage and Nucleotide Frequency Analyses
2.6. Statistical Analyses
3. Results
3.1. Positivity Rates
3.2. Molecular Features of the Novel Chaphamaparvoviruses
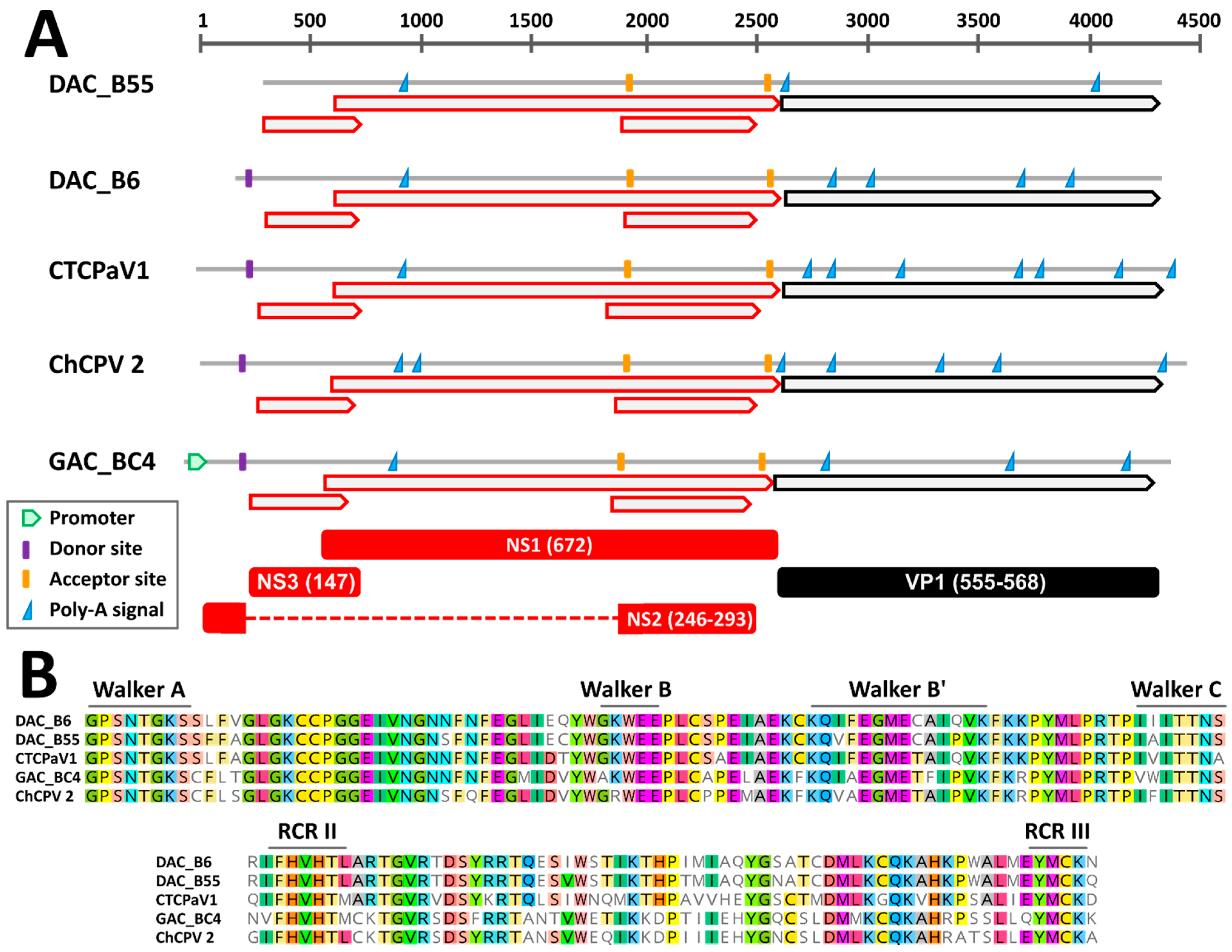
3.2.1. Genome Organization
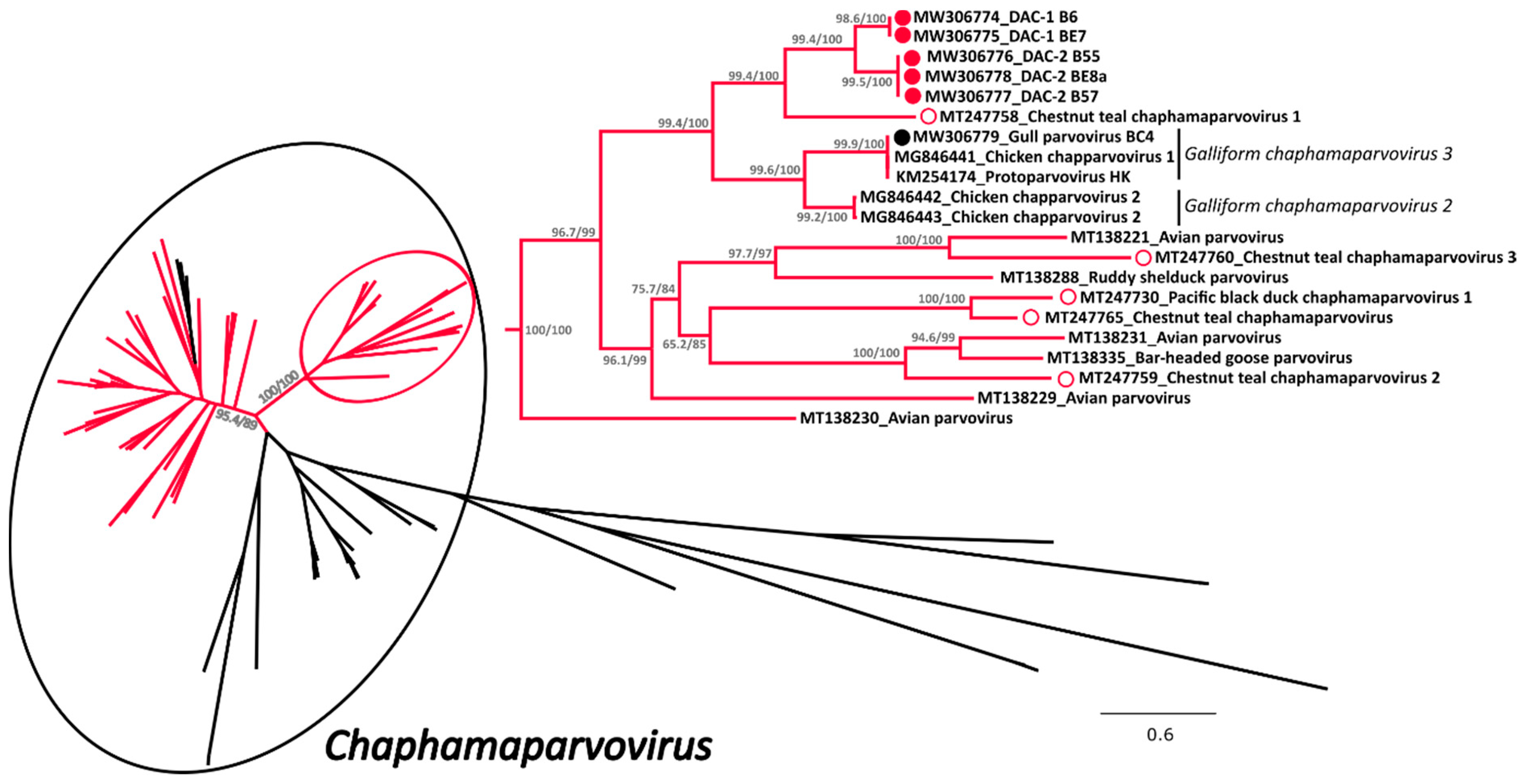
3.2.2. Phylogenetic Analyses
3.3. Molecular Features of the Novel Ambidensoviruses
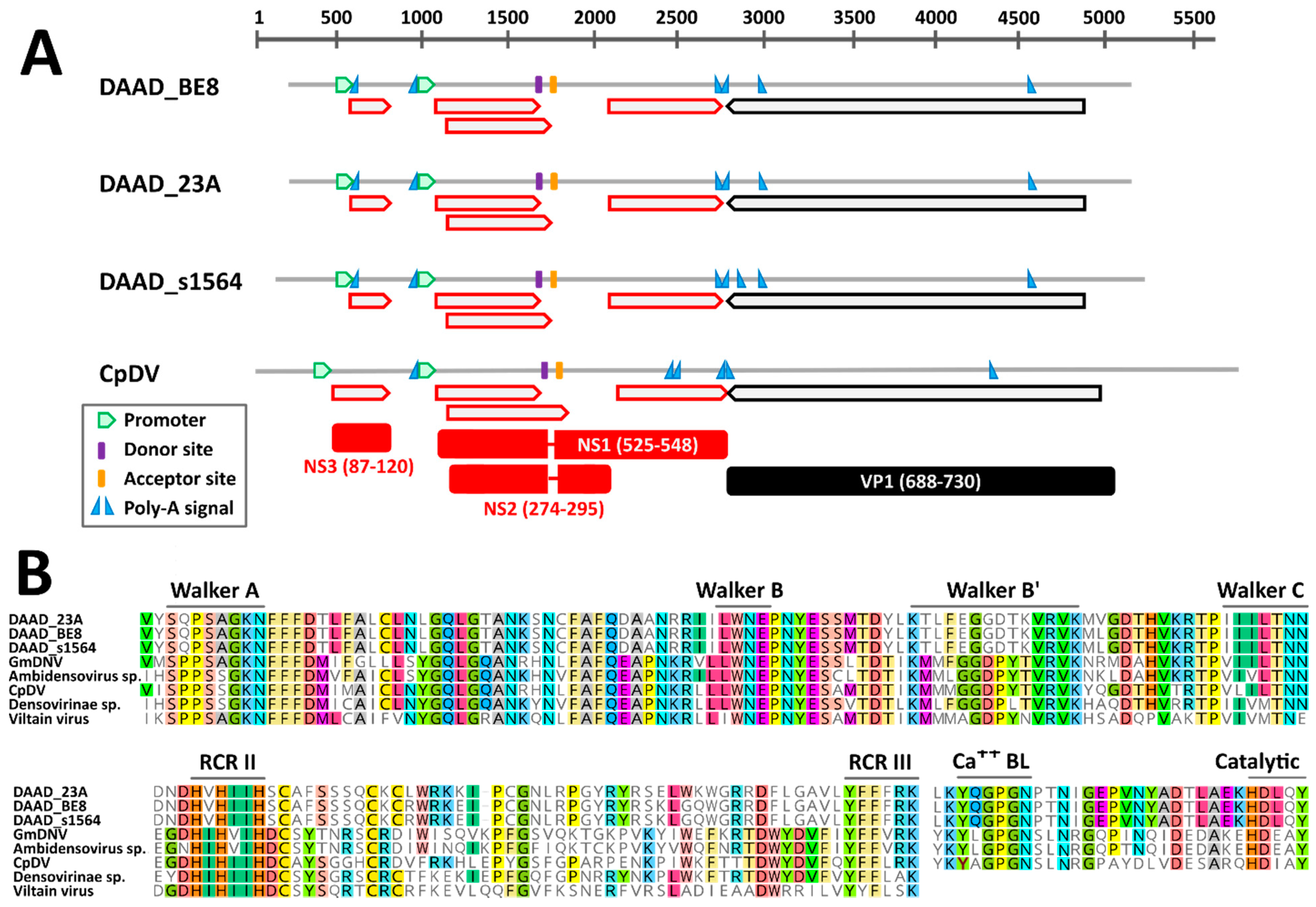
3.3.1. Genome Organization
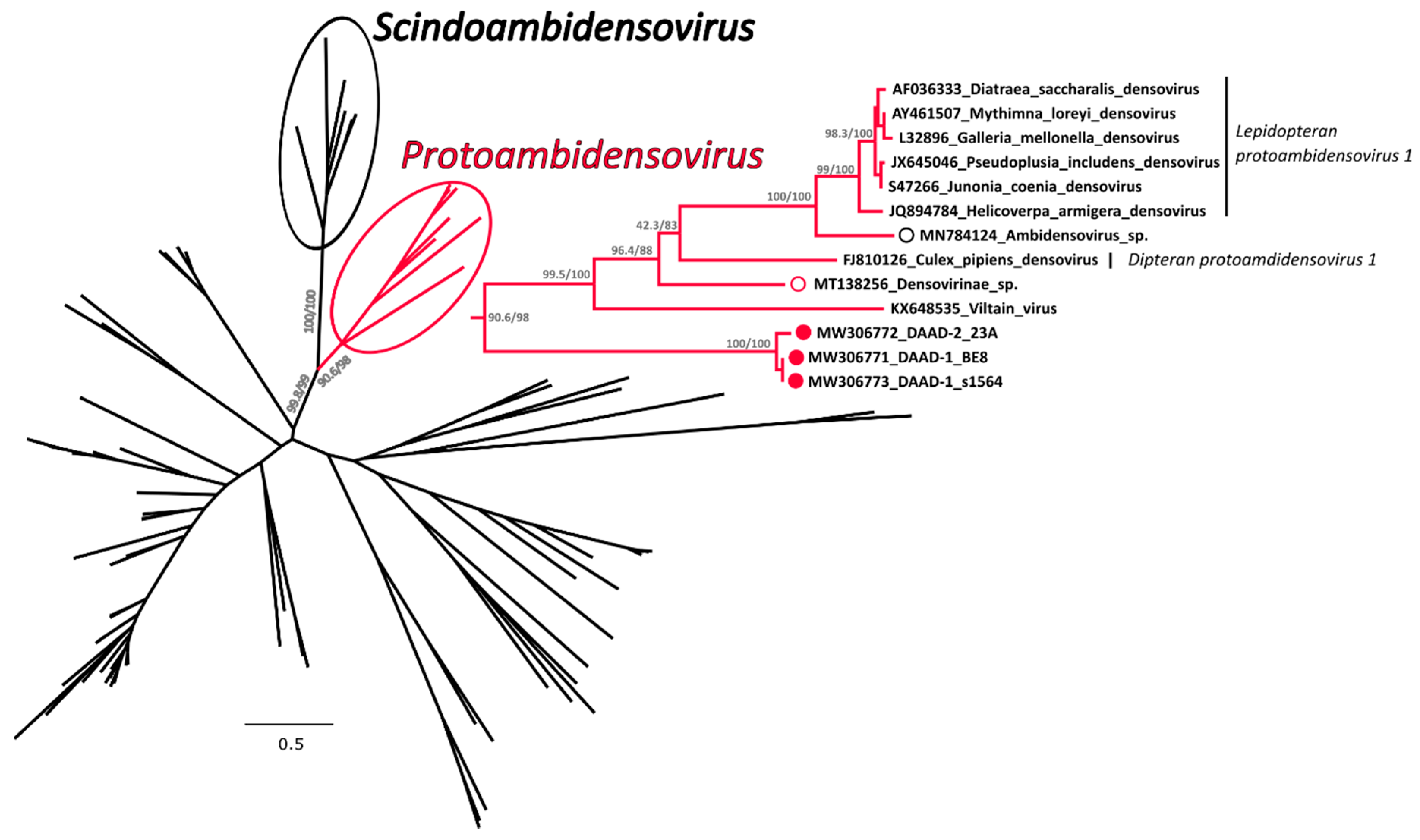
3.3.2. Phylogenetic Analysis
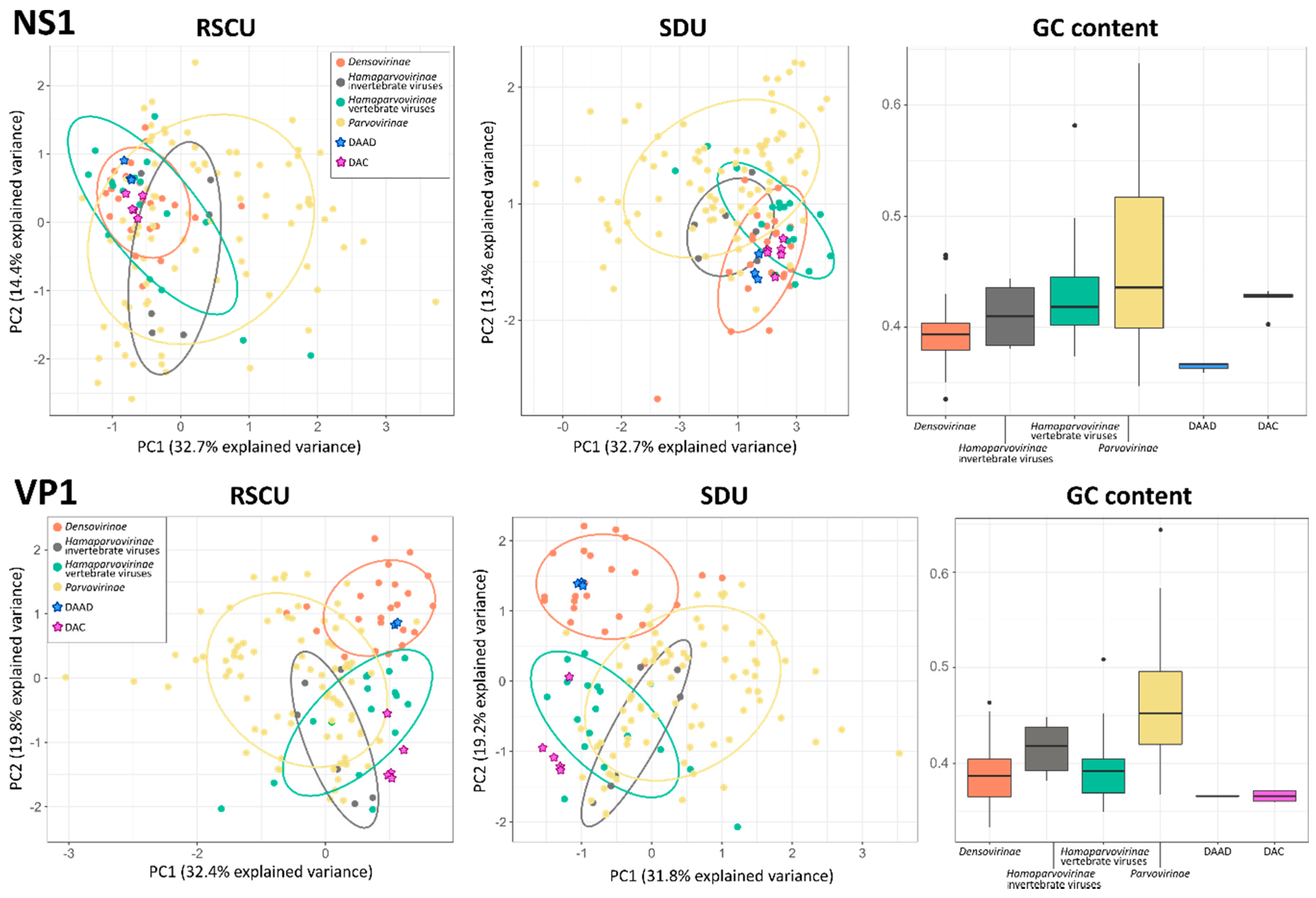
3.4. Codon Usage and Nucleotide Frequency Bias Analyses
4. Discussion
4.1. Duck-Associated Chapparvovirus (DAC) and Duck-Associated Ambidensovirus (DAAD) Are Novel Viral Species
4.2. Potential Hosts and Epidemiology of DAC and DAAD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the family Parvoviridae: A revised taxonomy independent of the canonical approach based on host association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Canuti, M.; Söderlund-Venermo, M.; Eis-Huebinger, A.M.; Ogliastro, M.; Harrach, B. Create three new genera and 19 new species (Piccovirales: Parvoviridae). In ICTV Taxonomy Proposal 2020; International Committee on Taxonomy of Viruses (ICTV), 2020. [Google Scholar]
- Mietzsch, M.; Pénzes, J.J.; Agbandje-McKenna, M. Twenty-five years of structural parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small does not mean simple. Ann. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapgate, S.S.; Kumanan, K.; Vijayarani, K.; Barbuddhe, S.B. Avian parvovirus: Classification, phylogeny, pathogenesis and diagnosis. Avian Pathol. 2018, 47, 536–545. [Google Scholar] [CrossRef]
- Bossis, I.; Chiorini, J.A. Cloning of an Avian adeno-associated virus (AAAV) and generation of recombinant AAAV particles. J. Virol. 2003, 77, 6799–6810. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; Vo, N.P.; Boros, Á.; Pankovics, P.; Reuter, G.; Li, O.T.W.; Wang, C.; Deng, X.; Poon, L.L.M.; Delwart, E. The viruses of wild pigeon droppings. PLoS ONE 2013, 8, e72787. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The fecal virome of red-crowned cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef]
- Chang, W.-S.; Li, C.-X.; Hall, J.; Eden, J.-S.; Hyndman, T.H.; Holmes, E.C.; Rose, K. Meta-transcriptomic discovery of a divergent circovirus and a chaphamaparvovirus in captive reptiles with proliferative respiratory syndrome. Viruses 2020, 12, 1073. [Google Scholar] [CrossRef]
- Du, J.; Wang, W.; Chan, J.F.-W.; Wang, G.; Huang, Y.; Yi, Y.; Zhu, Z.; Peng, R.; Hu, X.; Wu, Y.; et al. Identification of a novel ichthyic parvovirus in marine species in Hainan Island, China. Front. Microbiol. 2019, 10, 2815. [Google Scholar] [CrossRef] [Green Version]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal virome analysis of wild animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel circular single-stranded DNA virus from turkey faeces. Arch. Virol. 2014, 159, 2161–2164. [Google Scholar] [CrossRef] [PubMed]
- Lima, D.A.; Cibulski, S.P.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.R.; Cerva, C.; Junqueira, D.M.; Mayer, F.Q.; et al. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. 2019, 261, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Kwon, Y.-K.; Jang, I.; Bae, Y.-C. Viral metagenomic analysis of chickens with runting-stunting syndrome in the Republic of Korea. Virol. J. 2020, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, H.; Liu, X.; Li, Y.; Chen, J.; Zhang, J.; Wang, X.; Shen, S.; Wang, H.; Deng, F.; et al. Genomic and transcriptional analyses of novel parvoviruses identified from dead peafowl. Virology 2020, 539, 80–91. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci. Rep. 2020, 10, 12800. [Google Scholar] [CrossRef]
- de Souza, W.M.; Romeiro, M.F.; Fumagalli, M.J.; Modha, S.; de Araujo, J.; Queiroz, L.H.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.R.; Gifford, R.J. Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J. Gen. Virol. 2017, 98, 225–229. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Canuti, M.; Munro, H.J.; Robertson, G.J.; Kroyer, A.N.K.; Roul, S.; Ojkic, D.; Whitney, H.G.; Lang, A.S. New insight into avian papillomavirus ecology and evolution from characterization of novel wild bird papillomaviruses. Front. Microbiol. 2019, 10, 701. [Google Scholar] [CrossRef]
- Canuti, M.; Kroyer, A.N.K.; Ojkic, D.; Whitney, H.G.; Robertson, G.J.; Lang, A.S. Discovery and characterization of novel RNA viruses in aquatic North American wild birds. Viruses 2019, 11, 768. [Google Scholar] [CrossRef] [Green Version]
- Roul, S.; Robertson, G.; Lang, A.S. Assessing the Impact of Low-Pathogenicity Avian Influenza Virus on the Health of American Black Ducks (Anas rubripes). Unpublished.
- Verhoeven, J.T.P.; Canuti, M.; Munro, H.J.; Dufour, S.C.; Lang, A.S. ViDiT-CACTUS: An inexpensive and versatile library preparation and sequence analysis method for virus discovery and other microbiology applications. Can. J. Microbiol. 2018, 64, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Gertz, E.M.; Schäffer, A.A.; Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 2006, 13, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: http://www.r-project.org/index.html (accessed on 20 September 2020).
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Biological and Medical Physics, Biomedical Engineering; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytras, S.; Hughes, J. Synonymous dinucleotide usage: A codon-aware metric for quantifying dinucleotide representation in viruses. Viruses 2020, 12, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pénzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An ancient lineage of highly divergent parvoviruses infects both vertebrate and invertebrate hosts. Viruses 2019, 11, 525. [Google Scholar] [CrossRef] [Green Version]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal viral diversity of captive and wild tasmanian devils characterized using virion-enriched metagenomics and metatranscriptomics. J. Virol. 2019, 93, e00205-19. [Google Scholar] [CrossRef] [Green Version]
- Baquerizo-Audiot, E.; Abd-Alla, A.; Jousset, F.-X.; Cousserans, F.; Tijssen, P.; Bergoin, M. Structure and expression strategy of the genome of Culex pipiens densovirus, a mosquito densovirus with an ambisense organization. J. Virol. 2009, 83, 6863–6873. [Google Scholar] [CrossRef] [Green Version]
- George, U.; Simsek, C.; Faleye, T.O.C.; Arowolo, O.; Oragwa, A.; Adewumi, O.M.; Matthijnssens, J.; Adeniji, J.A. Genome sequences of novel members of previously described DNA and RNA virus families, isolated from feces of a drill monkey in Nigeria. Microbiol. Resour. Announc. 2020, 9, e00092-20. [Google Scholar] [CrossRef]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [Green Version]
- Wille, M.; Holmes, E.C. Wild birds as reservoirs for diverse and abundant gamma- and deltacoronaviruses. FEMS Microbiol. Rev. 2020, 44, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian influenza viruses in wild birds: Virus evolution in a multihost ecosystem. J. Virol. 2018, 92, e00433-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, E.F.; Hsieh, W.-T.; Kramer, J.A.; Breed, M.W.; Roelke-Parker, M.E.; Stephens-Devalle, J.; Pate, N.M.; Bassel, L.L.; Hollingshead, M.G.; Karim, B.O.; et al. Naturally acquired mouse kidney parvovirus infection produces a persistent interstitial nephritis in immunocompetent laboratory mice. Vet. Pathol. 2020, 57, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Lee, Q.; Padula, M.P.; Pinello, N.; Williams, S.H.; O’Rourke, M.B.; Fumagalli, M.J.; Orkin, J.D.; Song, R.; Shaban, B.; Brenner, O.; et al. Murine and related chapparvoviruses are nephro-tropic and produce novel accessory proteins in infected kidneys. PLoS Pathog. 2020, 16, e1008262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceição-Neto, N.; Godinho, R.; Álvares, F.; Yinda, C.K.; Deboutte, W.; Zeller, M.; Laenen, L.; Heylen, E.; Roque, S.; Petrucci-Fonseca, F.; et al. Viral gut metagenomics of sympatric wild and domestic canids, and monitoring of viruses: Insights from an endangered wolf population. Ecol. Evol. 2017, 7, 4135–4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Messacar, K.; Dominguez, S.R.; da Costa, A.C.; Deng, X.; Delwart, E. A new densovirus in cerebrospinal fluid from a case of anti-NMDA-receptor encephalitis. Arch. Virol. 2016, 161, 3231–3235. [Google Scholar] [CrossRef]
- Fahsbender, E.; da-Costa, A.C.; Gill, D.E.; Milagres, F.A.d.P.; Brustulin, R.; Monteiro, F.J.C.; Rego, M.O.d.S.; Ribeiro, E.S.D.; Sabino, E.C.; Delwart, E. Plasma virome of 781 Brazilians with unexplained symptoms of arbovirus infection include a novel parvovirus and densovirus. PLoS ONE 2020, 15, e0229993. [Google Scholar] [CrossRef] [Green Version]
- Sirihongthong, T.; Jitobaom, K.; Phakaratsakul, S.; Boonarkart, C.; Suptawiwat, O.; Auewarakul, P. The relationship of codon usage to the replication strategy of parvoviruses. Arch. Virol. 2019, 164, 2479–2491. [Google Scholar] [CrossRef]
- Canuti, M.; van der Hoek, L. Virus discovery: Are we scientists or genome collectors? Trends Microbiol. 2014, 22, 229–231. [Google Scholar] [CrossRef]
- Canuti, M.; Deijs, M.; Jazaeri Farsani, S.M.; Holwerda, M.; Jebbink, M.F.; de Vries, M.; van Vugt, S.; Brugman, C.; Verheij, T.; Lammens, C.; et al. Metagenomic analysis of a sample from a patient with respiratory tract infection reveals the presence of a γ-papillomavirus. Front. Microbiol. 2014, 5, 347. [Google Scholar] [CrossRef]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | DAC 1 | DAAD 1 | |
|---|---|---|---|
| Ducks | Total, n = 123 | 7 (5.7%) | 26 (21.1%) |
| American black duck, n = 109 | 6 (27.3%) | 22 (20.2%) | |
| Mallard, n = 9 | 1 (11.1%) | 2 (22.2%) | |
| Northern pintail, n = 1 | 0 (0%) | 1 (100%) | |
| Hybrids, n = 4 | 0 (0%) | 1 (25%) | |
| Gulls | Total, n = 21 | 0 (0%) | 0 (0%) |
| Herring gull, n = 8 | 0 (0%) | 0 (0%) | |
| Iceland gull, n = 4 | 0 (0%) | 0 (0%) | |
| Ring-billed gull, n = 9 | 0 (0%) | 0 (0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canuti, M.; Verhoeven, J.T.P.; Munro, H.J.; Roul, S.; Ojkic, D.; Robertson, G.J.; Whitney, H.G.; Dufour, S.C.; Lang, A.S. Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses 2021, 13, 193. https://doi.org/10.3390/v13020193
Canuti M, Verhoeven JTP, Munro HJ, Roul S, Ojkic D, Robertson GJ, Whitney HG, Dufour SC, Lang AS. Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses. 2021; 13(2):193. https://doi.org/10.3390/v13020193
Chicago/Turabian StyleCanuti, Marta, Joost T. P. Verhoeven, Hannah J. Munro, Sheena Roul, Davor Ojkic, Gregory J. Robertson, Hugh G. Whitney, Suzanne C. Dufour, and Andrew S. Lang. 2021. "Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis" Viruses 13, no. 2: 193. https://doi.org/10.3390/v13020193