The Diversity and Distribution of Viruses Associated with Culex annulirostris Mosquitoes from the Kimberley Region of Western Australia

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
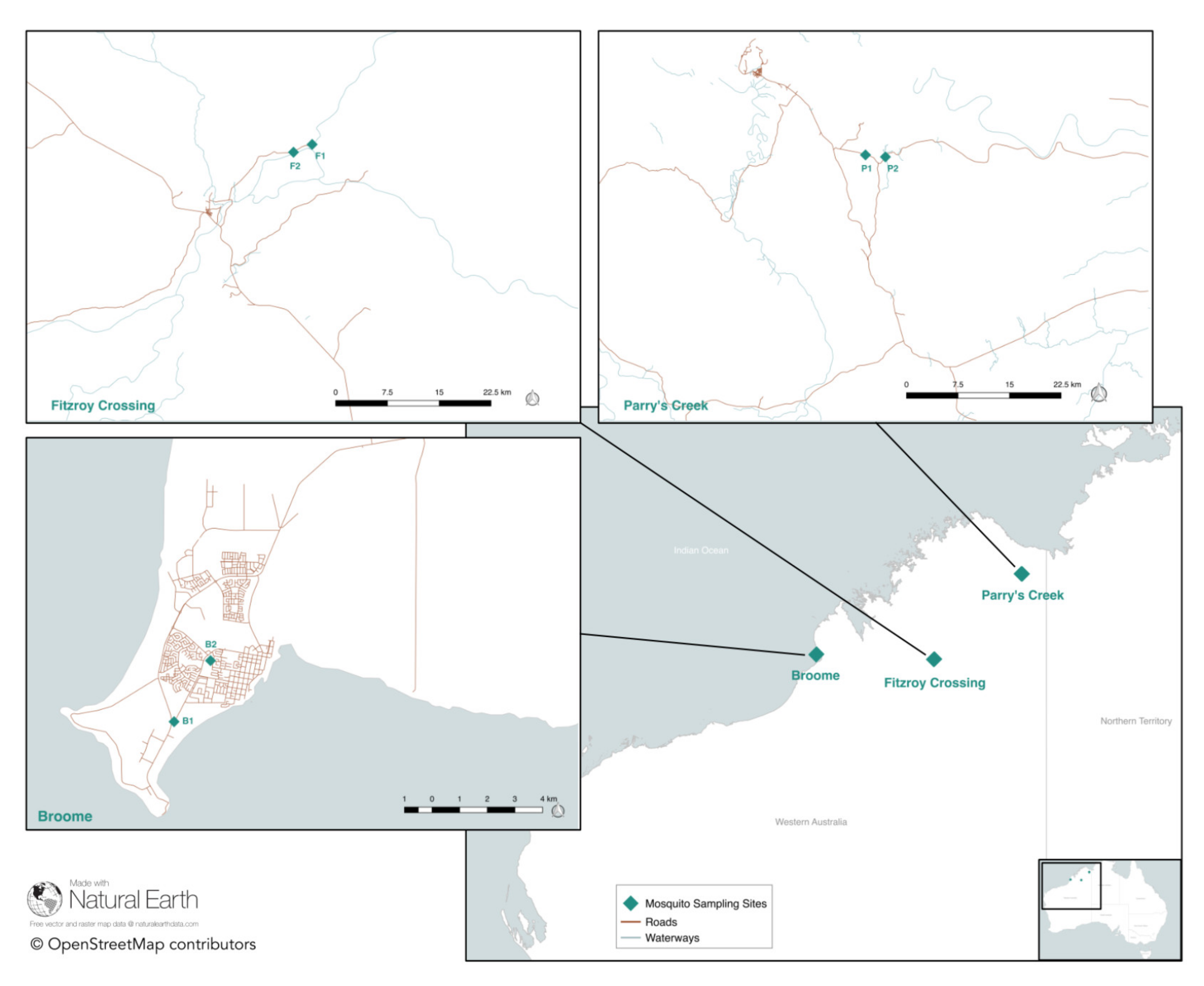
2.1. Mosquito Collection
2.2. Nucleic Acid Purification and High-Throughput Sequencing
2.3. PCR Screening
2.4. Phylogenetics
2.5. Accession Numbers
3. Results
3.1. Mosquito Collection
3.2. High-Throughput Sequencing
3.3. Virus Genomic Characterization
3.3.1. Positive-Sense ssRNA Viruses
Flavivirus
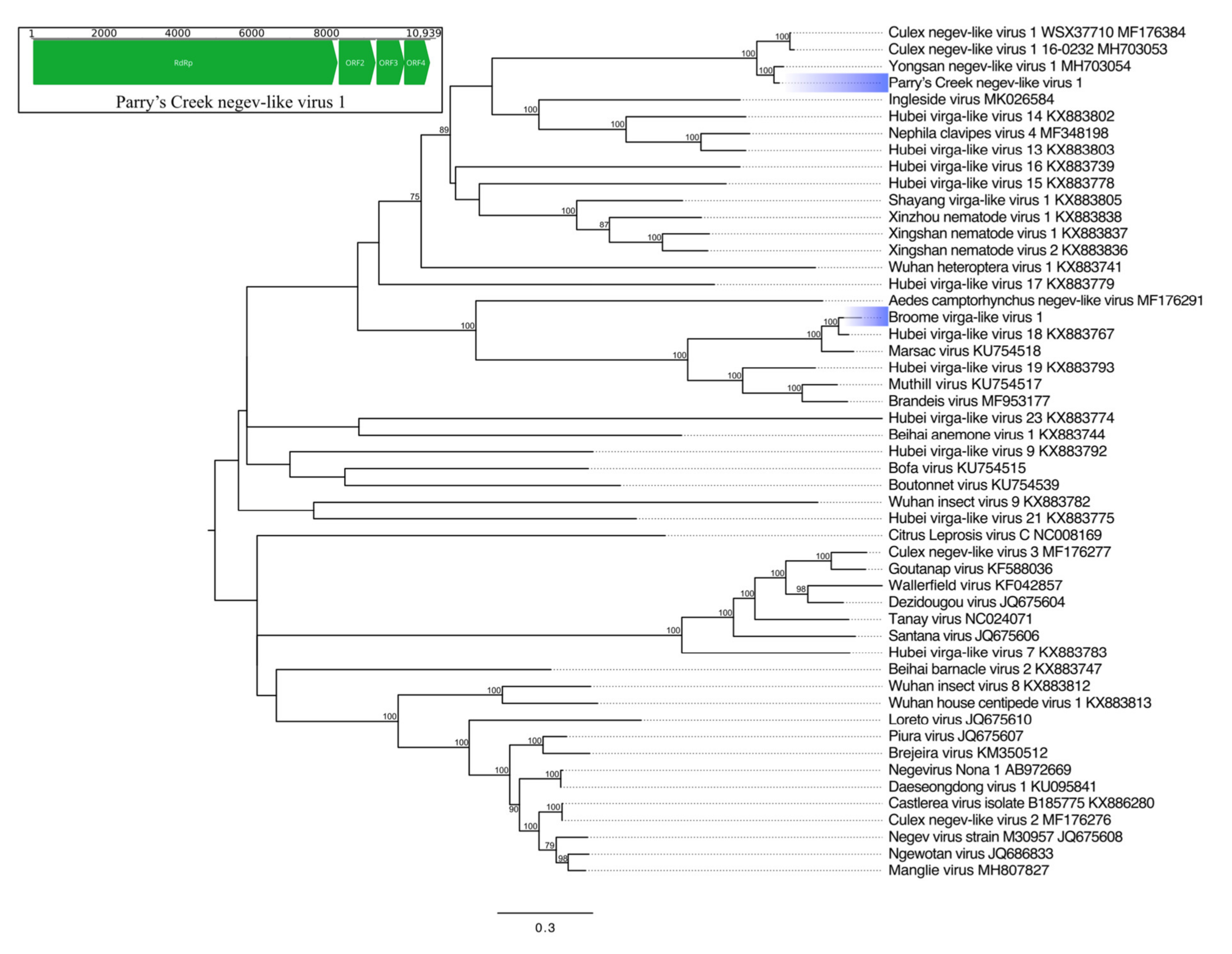
Nege- and Virgaviruses
Iflavirus
Luteo-Like Viruses
3.3.2. Negative-Sense ssRNA Viruses
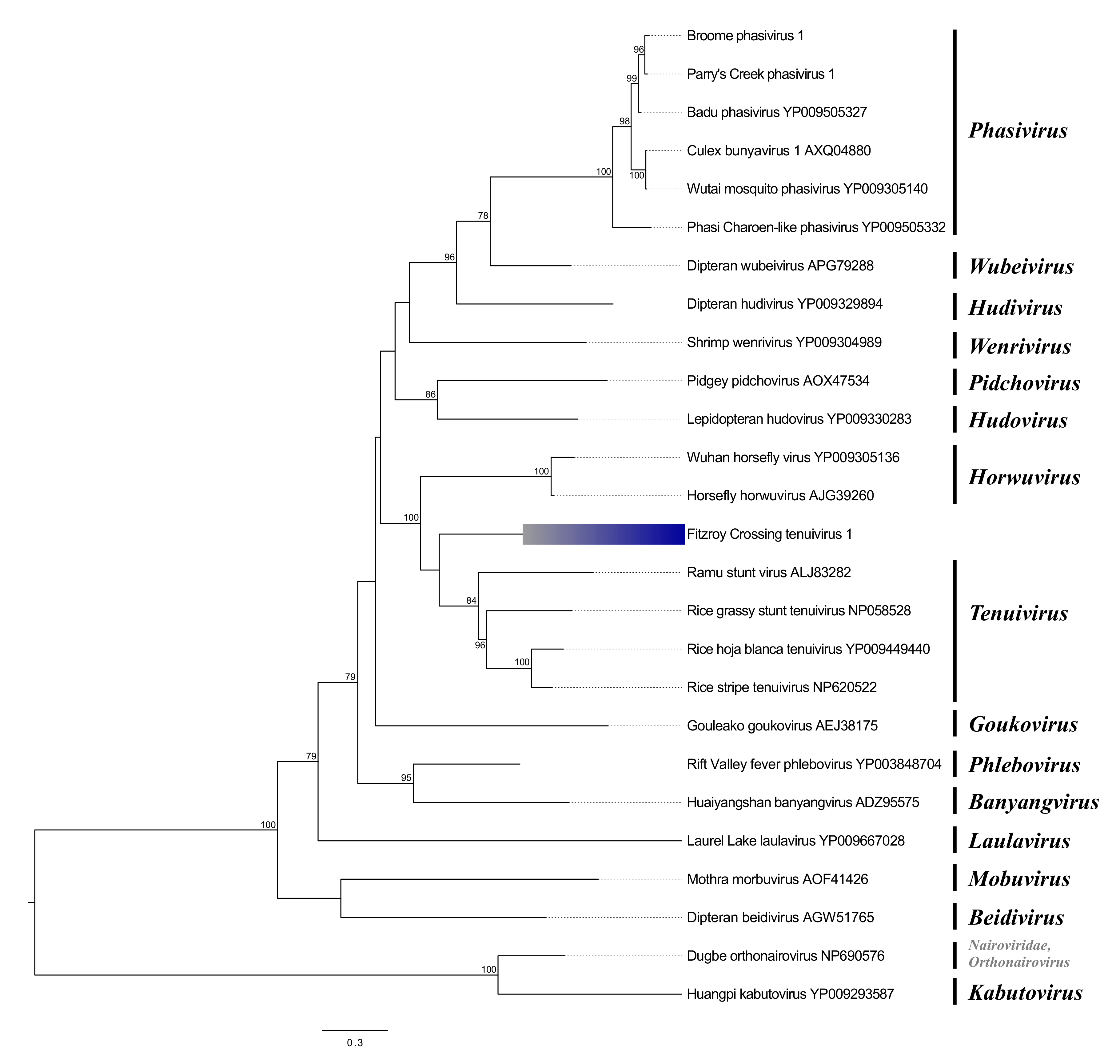
Phenuiviruses
Qinvirus
3.3.3. Double-Stranded RNA Viruses
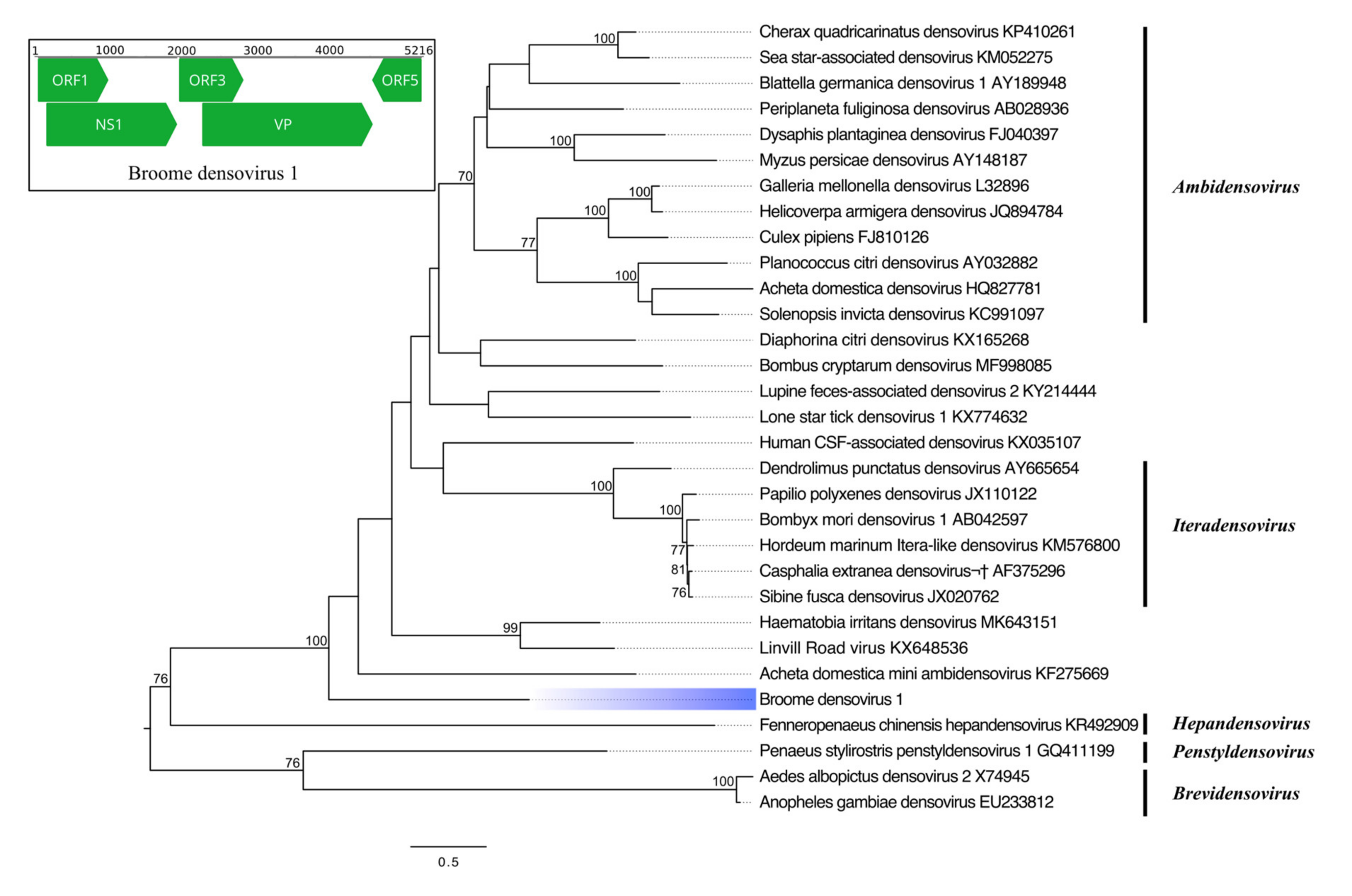
3.3.4. Single-Stranded DNA Virus
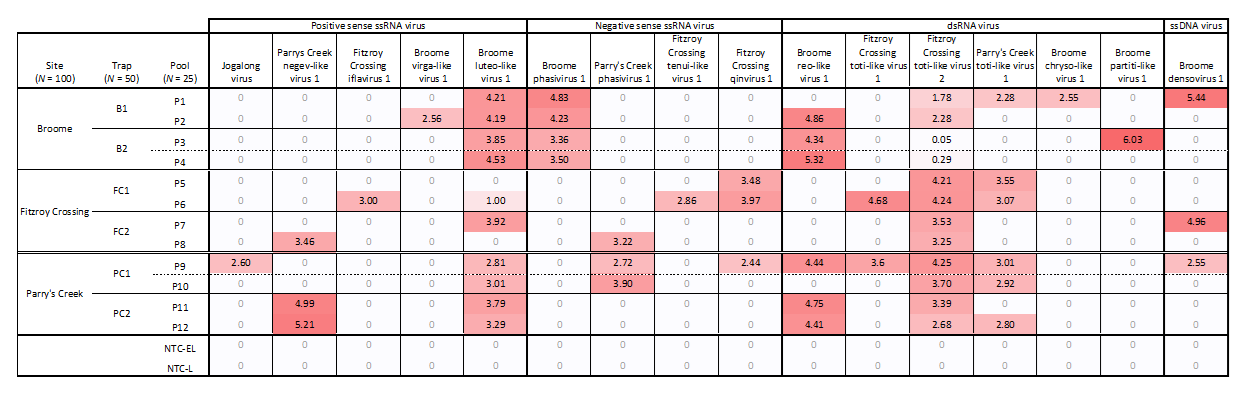
3.4. Virus Distribution and Prevalence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mackenzie, J.S.; Williams, D.T. The zoonotic flaviviruses of southern, south-eastern and eastern Asia, and Australasia: The potential for emergent viruses. Zoonoses Public Health 2009, 56, 338–356. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Lindsay, M.D.; Coelen, R.J.; Broom, A.K.; Hall, R.A.; Smith, D.W. Arboviruses causing human disease in the Australasian zoogeographic region. Arch. Virol. 1994, 136, 447–467. [Google Scholar] [PubMed]
- Staples, J.E.; Breiman, R.F.; Powers, A.M. Chikungunya fever: An epidemiological review of a re-emerging infectious disease. Clin. Infect. Dis. 2009, 49, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Tesh, R.B.; Popov, V.L.; Widen, S.G.; Wood, T.G.; Forrester, N.L.; Gonzalez, J.P.; Saluzzo, J.F.; Alkhovsky, S.; Lam, S.K.; et al. Exploiting the legacy of the arbovirus hunters. Viruses 2019, 11, 471. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in Western Australia. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of >12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Rasic, G.; Darbro, J.; Krause, L.; Poo, Y.S.; Filipovic, I.; Parry, R.; Asgari, S.; Devine, G.; Suhrbier, A. Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 2018, 8, 4690. [Google Scholar] [CrossRef] [Green Version]
- Johansen, C.A.; Susai, V.; Hall, R.A.; Mackenzie, J.S.; Clark, D.C.; May, F.J.; Hemmerter, S.; Smith, D.W.; Broom, A.K. Genetic and phenotypic differences between isolates of Murray Valley encephalitis virus in Western Australia, 1972–2003. Virus Genes 2007, 35, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Liehne, P.F. An Atlas of the Mosquitoes of Western Australia; Health Department of Western Australia: Perth, WA, Australia, 1991. [Google Scholar]
- Quan, P.L.; Williams, D.T.; Johansen, C.A.; Jain, K.; Petrosov, A.; Diviney, S.M.; Tashmukhamedova, A.; Hutchison, S.K.; Tesh, R.B.; Mackenzie, J.S.; et al. Genetic characterization of K13965, a strain of Oak Vale virus from Western Australia. Virus Res. 2011, 160, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broom, A.K.; Hall, R.A.; Johansen, C.A.; Oliveira, N.; Howard, M.A.; Lindsay, M.D.; Kay, B.H.; Mackenzie, J.S. Identification of Australian arboviruses in inoculated cell cultures using monoclonal antibodies in ELISA. Pathology 1998, 30, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.A.; Williams, S.H.; Melville, L.F.; Nicholson, J.; Hall, R.A.; Bielefeldt-Ohmann, H.; Prow, N.A.; Chidlow, G.R.; Wong, S.; Sinha, R.; et al. Characterization of Fitzroy River virus and serologic evidence of human and animal infection. Emerg. Infect. Dis. 2017, 23, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvey, L.A.; Johansen, C.A.; Broom, A.K.; Antao, C.; Lindsay, M.D.; Mackenzie, J.S.; Smith, D.W. Rainfall and sentinel chicken seroconversions predict human cases of Murray Valley encephalitis in the north of Western Australia. BMC Infect. Dis. 2014, 14, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broom, A.K.; Lindsay, M.D.; Johansen, C.A.; Wright, A.E.; Mackenzie, J.S. Two possible mechanisms for survival and initiation of Murray Valley encephalitis virus activity in the Kimberley region of Western Australia. Am. J. Trop. Med. Hyg. 1995, 53, 95–99. [Google Scholar] [CrossRef]
- Cowled, C.; Palacios, G.; Melville, L.; Weir, R.; Walsh, S.; Davis, S.; Gubala, A.; Lipkin, W.I.; Briese, T.; Boyle, D. Genetic and epidemiological characterization of Stretch Lagoon orbivirus, a novel orbivirus isolated from Culex and Aedes mosquitoes in northern Australia. J. Gen. Virol. 2009, 90, 1433–1439. [Google Scholar] [CrossRef]
- Rohe, D.; Fall, R.P. A miniature battery powered CO2 baited light trap for mosquito borne encephalitis surveillance. Bull. Soc. Vector Ecol. 1979, 4, 24–27. [Google Scholar]
- Team QD. QGIS geographic information system. Open source geospatial foundation project. Available online: http://qgis.osgeo.org (accessed on 1 December 2019).
- Mainroads Western Australia. Open data, maps and apps. Available online: http://portal-mainroads.opendata.arcgis.com (accessed on 1 December 2019).
- Australian Government data portal. An easy way to find, explore and reuse Australia’s public data. Available online: http://www.data.gov.au (accessed on 1 December 2019).
- Madeley, C.F.; Lennette, D.A.; Halonen, P. Specimen collection and transport. In Laboratory Diagnosis of Infectious Diseases: Principles and Practice; Lennette, E.H., Halonen, P., Murphy, F.A., Eds.; Springer: New York, NY, USA, 1988; Volume 2, p. 7. [Google Scholar]
- Chidlow, G.; Harnett, G.; Williams, S.; Levy, A.; Speers, D.; Smith, D.W. Duplex real-time reverse transcriptase PCR assays for rapid detection and identification of pandemic (H1N1) 2009 and seasonal influenza A/H1, A/H3, and B viruses. J. Clin. Microbiol. 2010, 48, 862–866. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Kim, K.W.; Allen, D.W.; Briese, T.; Couper, J.J.; Barry, S.C.; Colman, P.G.; Cotterill, A.M.; Davis, E.A.; Giles, L.C.; Harrison, L.C.; et al. Distinct gut virome profile of pregnant women with type 1 diabetes in the ENDIA study. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Dimmic, M.W.; Rest, J.S.; Mindell, D.P.; Goldstein, R.A. rtREV: An amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J. Mol. Evol. 2002, 55, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Government of Western Australia. Medical Entomology: 2017/2018 surveillance program annual report. Perth, Western Australia: Department of Health. 2018. Available online: https://ww2.health.wa.gov.au/Articles/J_M/Medical-Entomology-Annual-Report (accessed on 1 May 2020).
- Williams, S.H.; Levy, A.; Yates, R.A.; Somaweera, N.; Neville, P.J.; Nicholson, J.; Lindsay, M.D.A.; Mackenzie, J.S.; Jain, K.; Imrie, A.; et al. Discovery of Jogalong virus, a novel hepacivirus identified in a Culex annulirostris (Skuse) mosquito from the Kimberley region of Western Australia. PLoS ONE 2020, 15, e0227114. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; et al. Negevirus: A proposed new taxon of insect-specific viruses with wide geographic distribution. J. Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [Green Version]
- Kondo, H.; Chiba, S.; Maruyama, K.; Andika, I.B.; Suzuki, N. A novel insect-infecting virga/nege-like virus group and its pervasive endogenization into insect genomes. Virus Res. 2019, 262, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Lenz, O.; Pribylova, J.; Franova, J.; Koloniuk, I.; Spak, J. Identification and characterization of a new member of the genus Luteovirus from cherry. Arch. Virol. 2017, 162, 587–590. [Google Scholar] [CrossRef]
- Hobson-Peters, J.; Warrilow, D.; McLean, B.J.; Watterson, D.; Colmant, A.M.; van den Hurk, A.F.; Hall-Mendelin, S.; Hastie, M.L.; Gorman, J.J.; Harrison, J.J.; et al. Discovery and characterisation of a new insect-specific bunyavirus from Culex mosquitoes captured in northern Australia. Virology 2016, 489, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hajano, J.U.; Wang, X. New insights on the transmission mechanism of tenuiviruses by their vector insects. Curr. Opin. Virol. 2018, 33, 13–17. [Google Scholar] [CrossRef]
- Pettersson, J.H.; Shi, M.; Eden, J.S.; Holmes, E.C.; Hesson, J.C. Meta-transcriptomic comparison of the RNA viromes of the mosquito vectors Culex pipiens and Culex torrentium in northern Europe. Viruses 2019, 11, 1033. [Google Scholar] [CrossRef] [Green Version]
- Liehne, P.F.; Anderson, S.; Stanley, N.F.; Liehne, C.G.; Wright, A.E.; Chan, K.H.; Leivers, S.; Britten, D.K.; Hamilton, N.P. Isolation of Murray Valley encephalitis virus and other arboviruses in the Ord River Valley 1972-1976. Aust. J. Exp. Biol. Med. Sci. 1981, 59, 347–356. [Google Scholar] [CrossRef]
- Atoni, E.; Wang, Y.; Karungu, S.; Waruhiu, C.; Zohaib, A.; Obanda, V.; Agwanda, B.; Mutua, M.; Xia, H.; Yuan, Z. Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses 2018, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.L.; Reeves, W.C. Statistical estimation of virus infection rates in mosquito vector populations. Am. J. Hyg. 1962, 75, 377–391. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trap Code | Trap Location | GPS (Latitude) | GPS (Longitude) | Trap Set Date | Total Trapped Cx. annulirostris |
|---|---|---|---|---|---|
| B1 | Adjacent to caravan park, Broome | −17.97763161 | 122.2125935 | 9 April 2018 | 188 * |
| B2 | Cemetery, Broome | −17.95775700 | 122.2244490 | 10 April 2018 | 405 * |
| FC1 | Rotunda, Fitzroy Crossing | −18.10542047 | 125.7016503 | 6 April 2018 | 135 |
| FC2 | Floodway, Fitzroy Crossing | −18.11554849 | 125.6773817 | 6 April 2018 | 170 |
| PC1 | Jogalong Billabong, Parry’s Creek | −15.59154698 | 128.2619530 | 26 March 2018 | 1971 * |
| PC2 | Mangrove, Parry’s Creek | −15.59409900 | 128.2880270 | 26 March 2018 | 17,687 * |
| Agent | Target Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Product Size (nt) |
|---|---|---|---|---|
| Jogalong virus | NS5B | CAGGTCCCTATTCTTACACGG | TCTGGTAACCGAGGTGTTGC | 337 |
| Broome phasivirus 1 | RdRp | TTCAGATGGATTAAACCTGGCG | CTAGATCTTCTTGCCACTTCAGC | 269 |
| Parry’s Creek phasivirus 1 | RdRp | CCAGACTGTTAGCAGCATCAATC | TCAATTCCTCTTGCCTGGAGAG | 225 |
| Fitzroy Crossing tenuivirus 1 | RdRp | CTGGCATTGCCGGATTATCC | CTAGGCTTGAAATGACTCCAGG | 351 |
| Parry’s Creek negev-like virus 1 | RdRp | AACTGCAGAGGGTGATATCGG | ATAGCATCGCCGCTCTTCC | 204 |
| Fitzroy Crossing iflavirus 1 | Polyprotein | GTTGCAATACTACCAACGGCTC | CAAACCCACCATCGTGGTC | 240 |
| Virus Name | Family | BLAST Region | BLAST Length (aa) | Most Similar Viral Sequence | Coverage (%) | BLAST Identity (%) | E-Value |
|---|---|---|---|---|---|---|---|
| Jogalong virus | Flaviviridae | Polyprotein | 2941 | Bald eagle hepacivirus | 91 | 40.32 | 0 |
| Parry’s Creek negev-like virus 1 | Hepe-Virga * | RdRp | 2742 * | Yongsan negev-like virus 1 | 99 * | 87.71 | 0 |
| Broome virga-like virus 1 | Hepe-Virga * | RdRp | 2417 | Hubei virga-like virus 18 | 100 | 81.81 | 0 |
| Fitzroy Crossing iflavirus 1 | Iflaviridae | Polyprotein | 2905 | Hubei arthropod virus 1 | 96 | 47.74 | 0 |
| Broome luteo-like virus 1 | Luteo-Sobemo * | RdRp | 372 | Culex mosquito virus 6 | 100 | 76.88 | 0 |
| Broome phasivirus 1 | Phenuiviridae | RdRp | 2182 | Badu phasivirus | 100 | 87.53 | 0 |
| Parry’s Creek phasivirus 1 | Phenuiviridae | RdRp | 2219 | Badu phasivirus | 99 | 88.27 | 0 |
| Fitzroy Crossing tenui-like virus 1 | Phenuiviridae | RdRp | 2842 | Rice grassy stunt virus | 78 | 32.98 | 0 |
| Fitzroy Crossing qinvirus 1 | Qinvirus * | RdRp | 1752 | Vinslov virus | 100 | 79.74 | 0 |
| Broome reo-like virus 1 | Reoviridae | RdRp | 1308 | Sanxia reo-like virus 1 | 88 | 36.76 | 9 × 10−177 |
| Fitzroy Crossing toti-like virus 1 | Toti-Chryso * | RdRp | 806 | Lindangsbacken virus | 100 | 79.16 | 0 |
| Fitzroy Crossing toti-like virus 2 | Toti-Chryso * | Hyp. protein 2 | 758 | Hubei toti-like virus 10 | 89 | 49.93 | 0 |
| Parry’s Creek toti-like virus 1 | Toti-Chryso * | RdRp | 1418 | Ahus virus | 91 | 75.48 | 0 |
| Broome chryso-like virus 1 | Chrysoviridae | RdRp | 1118 | Hubei chryso-like virus 1 | 100 | 88.46 | 0 |
| Broome partiti-like virus 1 | Partiti-Picobirna * | RdRp | 393 | Vespa velutina partiti-like virus 1 | 96 | 56.81 | 1 × 10−149 |
| Broome densovirus 1 | Parvoviridae | NS1 | 583 | Haematobia irritans densovirus | 86 | 28.04 | 2 × 10−53 |
| Site | Trap | Jogalong Virus | Parry’s Creek Negev-Like Virus 1 | Fitzroy Crossing Iflavirus 1 | Broome Phasivirus 1 | Parry’s Creek Phasivirus 1 | Fitzroy Crossing Tenui-Like Virus 1 |
|---|---|---|---|---|---|---|---|
| Broome | B1 | 0/50 | 0/50 | 0/50 | 35/50 (70%) | 0/50 | 0/50 |
| B2 | 0/50 | 1/50 (2%) | 0/50 | 24/50 (48%) | 0/50 | 0/50 | |
| Fitzroy Crossing | FC1 | 0/50 | 0/50 | 1/50 (2%) | 2/50 (4%) | 0/50 | 2/50 (4%) |
| FC2 | 0/50 | 1/50 (2%) | 0/50 | 0/50 | 1/50 (2%) | 0/50 | |
| Parry’s Creek | PC1 | 1/50 (2%) | 2/50 (4%) | 0/50 | 2/50 (4%) | 14/50 (28%) | 0/50 |
| PC2 | 0/50 | 3/50 (6%) | 0/50 | 1/50 (2%) | 2/50 (4%) | 0/50 | |
| Total | 1/300 (0.3%) | 7/300 (2.3%) | 1/300 (0.3%) | 64/300 (21.3%) | 17/300 (5.7%) | 2/300 (0.7%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, S.H.; Levy, A.; Yates, R.A.; Somaweera, N.; Neville, P.J.; Nicholson, J.; Lindsay, M.D.A.; Mackenzie, J.S.; Jain, K.; Imrie, A.; et al. The Diversity and Distribution of Viruses Associated with Culex annulirostris Mosquitoes from the Kimberley Region of Western Australia. Viruses 2020, 12, 717. https://doi.org/10.3390/v12070717
Williams SH, Levy A, Yates RA, Somaweera N, Neville PJ, Nicholson J, Lindsay MDA, Mackenzie JS, Jain K, Imrie A, et al. The Diversity and Distribution of Viruses Associated with Culex annulirostris Mosquitoes from the Kimberley Region of Western Australia. Viruses. 2020; 12(7):717. https://doi.org/10.3390/v12070717
Chicago/Turabian StyleWilliams, Simon H., Avram Levy, Rachel A. Yates, Nilusha Somaweera, Peter J. Neville, Jay Nicholson, Michael D. A. Lindsay, John S. Mackenzie, Komal Jain, Allison Imrie, and et al. 2020. "The Diversity and Distribution of Viruses Associated with Culex annulirostris Mosquitoes from the Kimberley Region of Western Australia" Viruses 12, no. 7: 717. https://doi.org/10.3390/v12070717