Human Cytomegalovirus Mediates Unique Monocyte-to-Macrophage Differentiation through the PI3K/SHIP1/Akt Signaling Network

 , and
, and 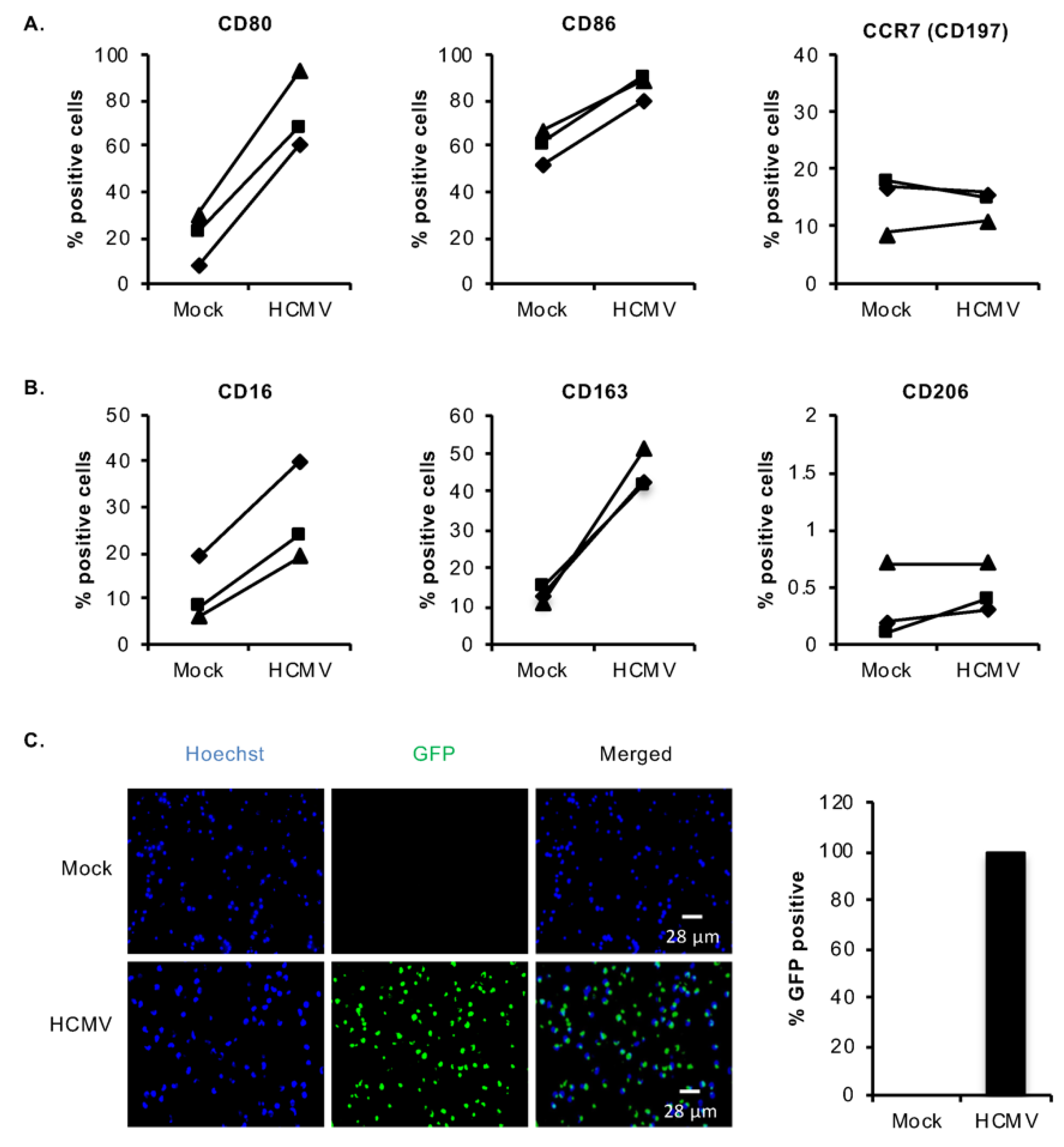{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Peripheral Blood Monocyte Isolation and Culture
2.2. Virus Preparation and Infection
2.3. Western Blot Analysis
2.4. Flow Cytometry
2.5. Microscopy
2.6. Statistical Analysis
3. Results
3.1. HCMV Infection of Monocytes Induces Select M1/M2 Macrophage Differentiation Markers
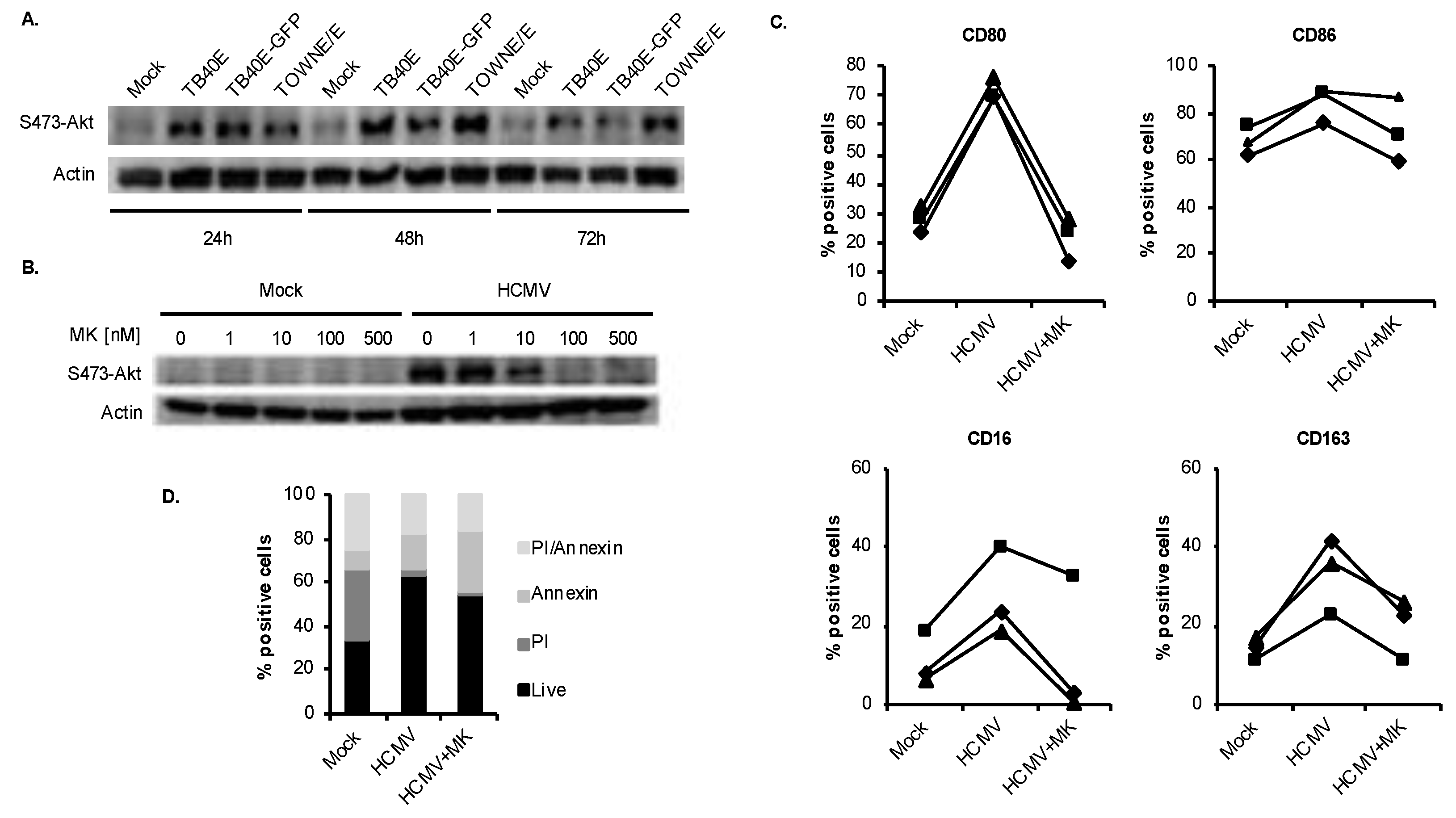
3.2. HCMV-Induced Akt Mediates Monocyte-to-Macrophage Differentiation
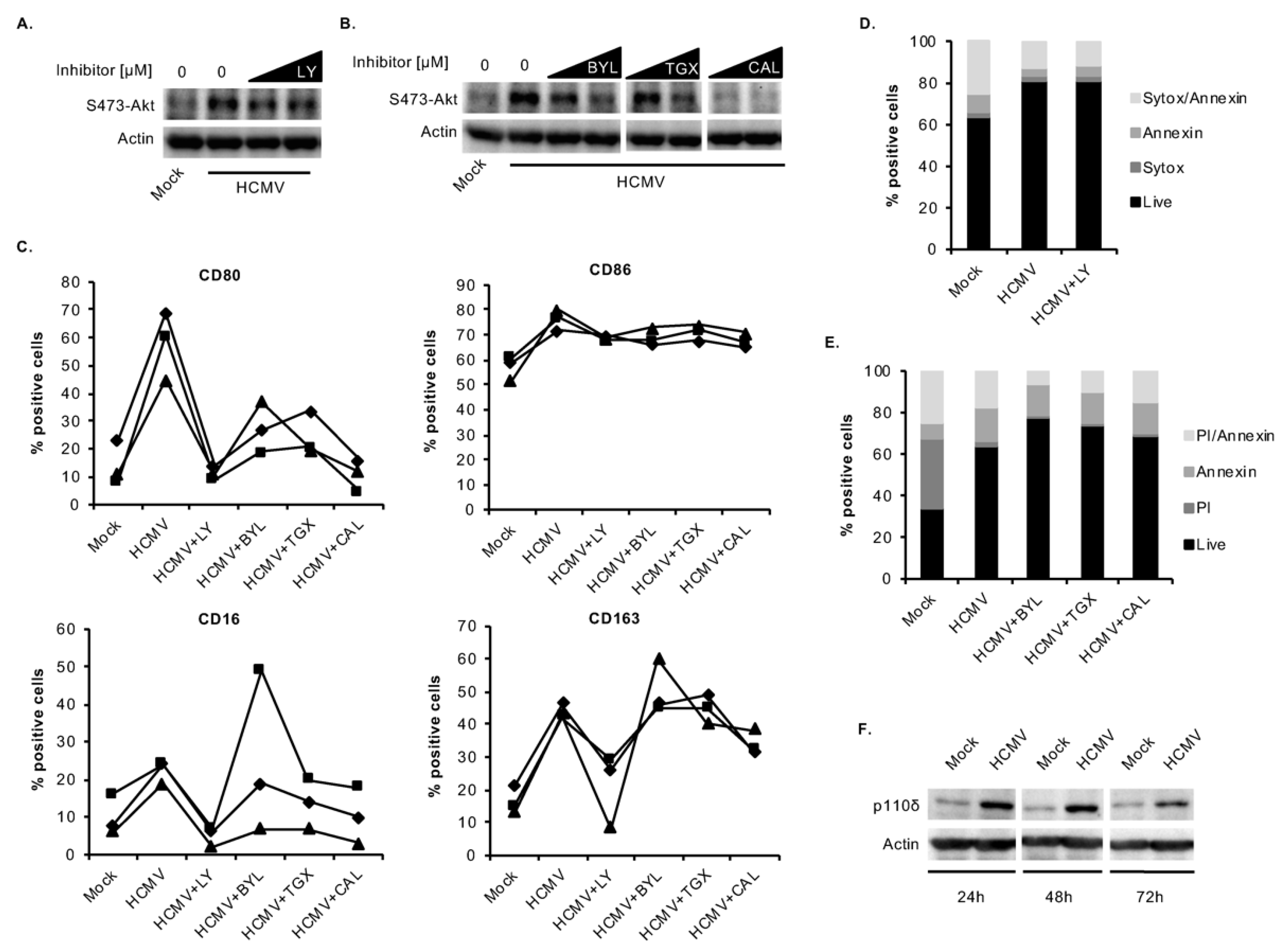
3.3. HCMV Predominantly Uses the p110δ Isoform of PI3K to Mediate the M1/M2 Differentiation of Infected Monocytes
3.4. HCMV Engages SHIP1 to Stimulate Monocyte-to-Macrophage Differentiation
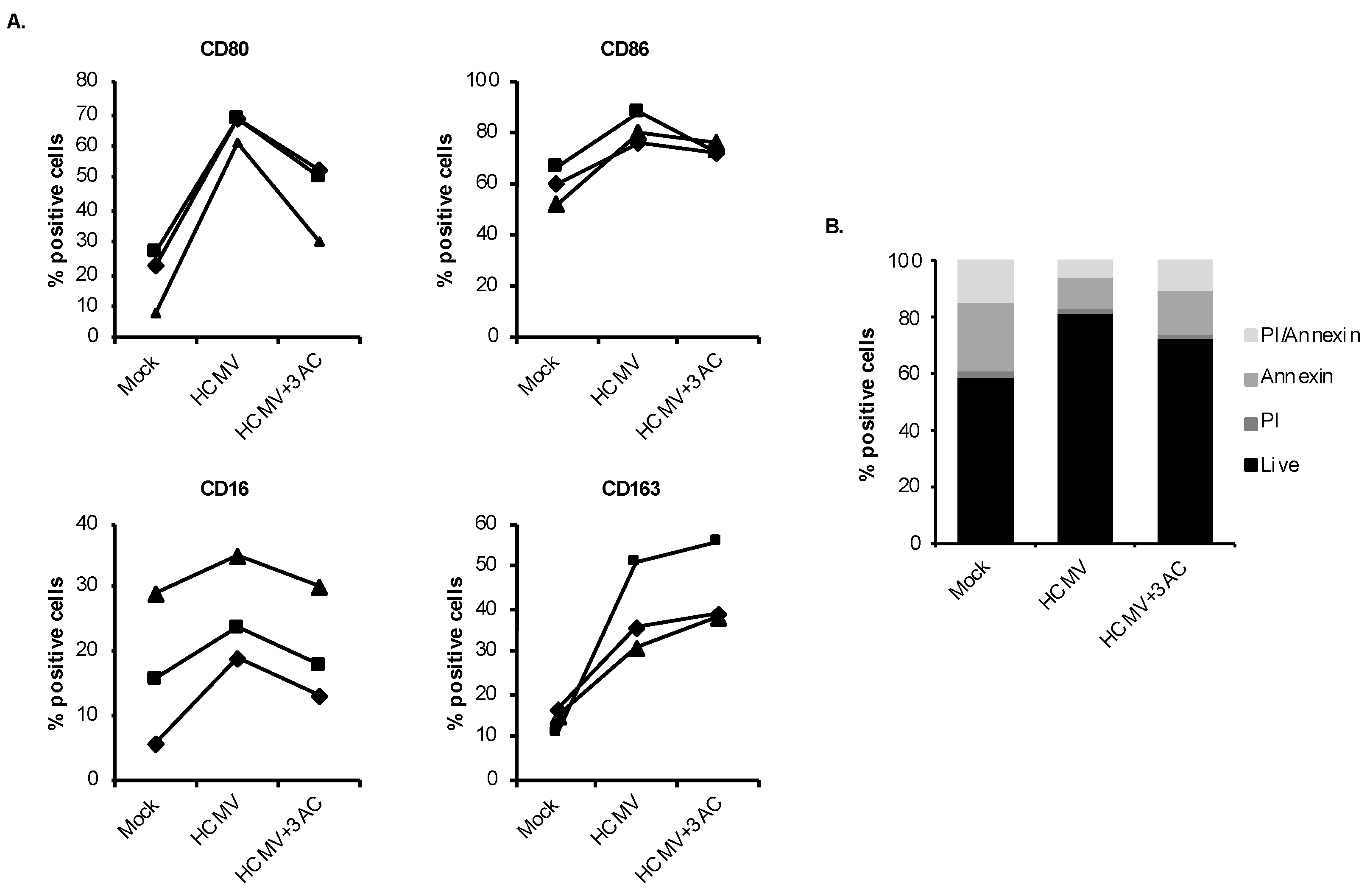
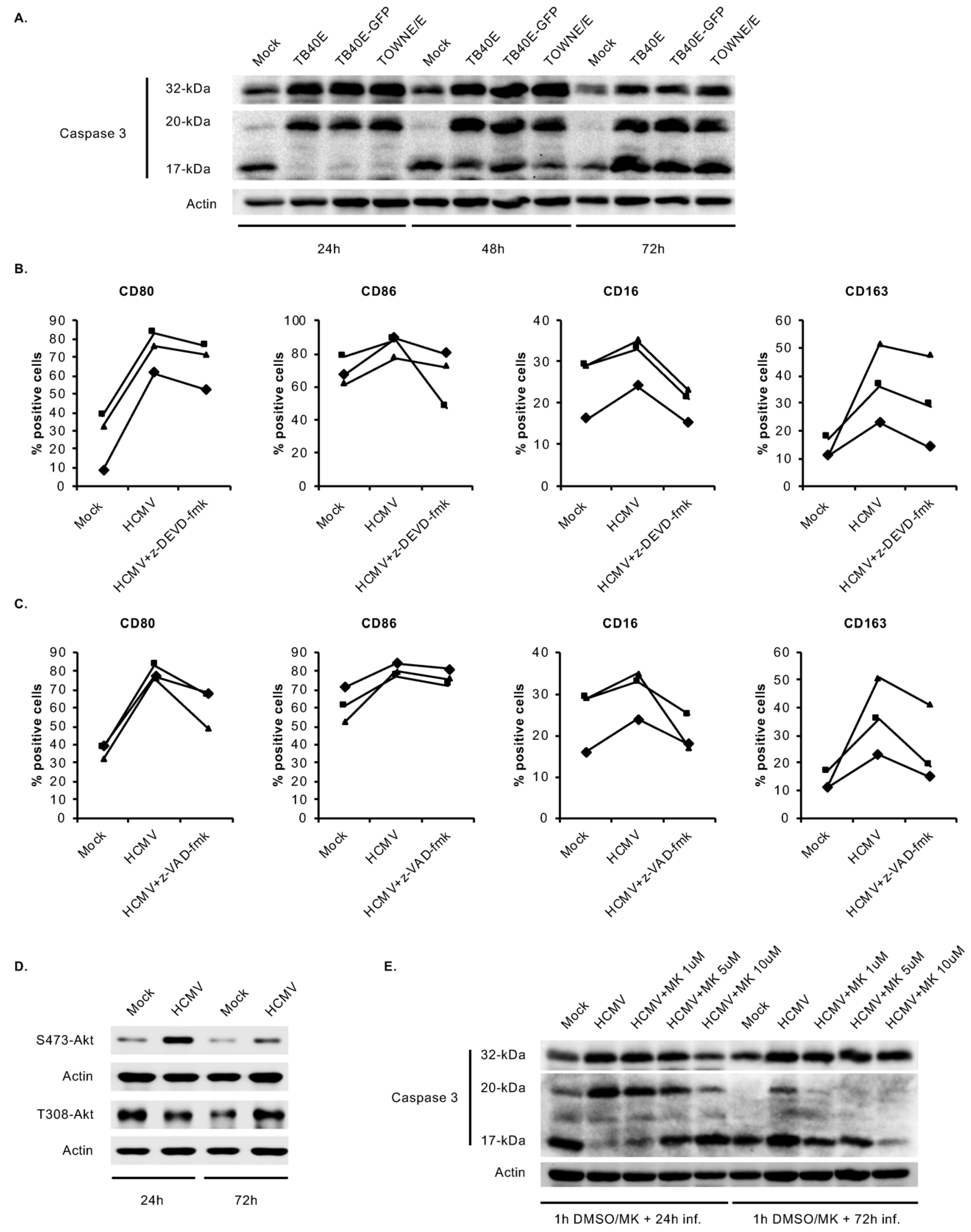
3.5. HCMV-Induced Caspase 3 Activation after the 48-h Viability Gate Is Required for the Unique M1/M2 Polarization of Infected Monocytes
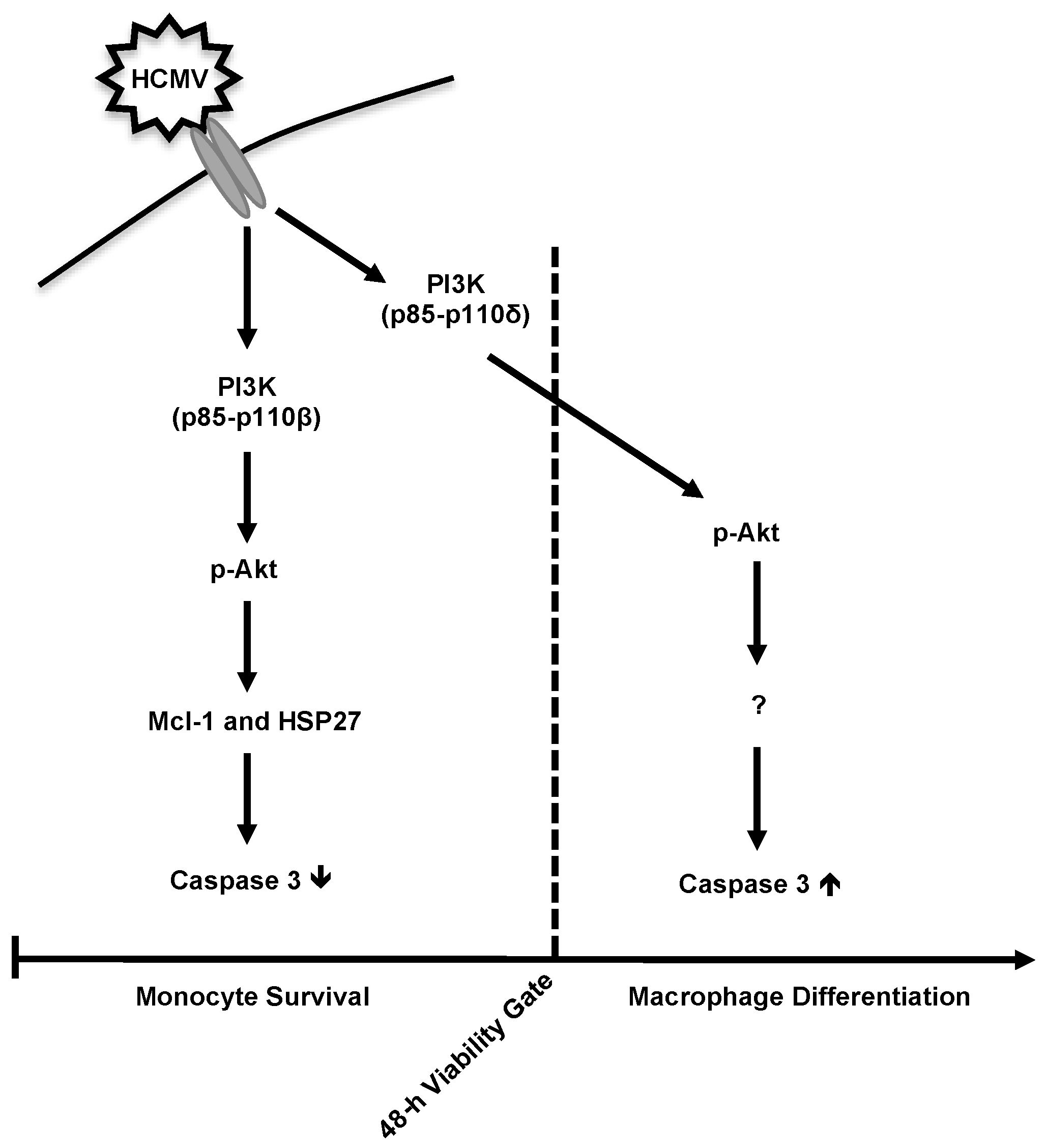
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ho, M. Virus infections after transplantation in man. Brief review. Arch. Virol. 1977, 55, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ho, M. Cytomegalovirus: Biology and Infection; Plenum Medical Book Company: New York, NY, USA, 1991. [Google Scholar]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masur, H.; Whitcup, S.M.; Cartwright, C.; Polis, M.; Nussenblatt, R. Advances in the management of AIDS-related cytomegalovirus retinitis. Ann. Intern. Med. 1996, 125, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Stagno, S.; Pass, R.F.; Dworsky, M.E.; Henderson, R.E.; Moore, E.G.; Walton, P.D.; Alford, C.A. Congenital cytomegalovirus infection: The relative importance of primary and recurrent maternal infection. N. Engl. J. Med. 1982, 306, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Emery, V. Investigation of CMV disease in immunocompromised patients. J. Clin. Pathol. 2001, 54, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humar, A.; St. Louis, P.; Mazzulli, T.; McGeer, A.; Lipton, J.; Messner, H.; MacDonald, K. Elevated serum cytokines are associated with cytomegalovirus infection and disease in bone marrow transplant recipients. J. Infect. Dis. 1999, 179, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Ljungman, P.; Engelhard, D.; Link, H.; Biron, P.; Brandt, L.; Brunet, S.; Cordonnier, C.; Debusscher, L.; De Laurenzi, A.; Kolb, H. Treatment of interstitial pneumonitis due to cytomegalovirus with ganciclovir and intravenous immune globulin: Experience of European Bone Marrow Transplant Group. Clin. Infect. Dis. 1992, 14, 831–835. [Google Scholar] [CrossRef]
- Lumbreras, C.; Manuel, O.; Len, O.; ten Berge, I.; Sgarabotto, D.; Hirsch, H. Cytomegalovirus infection in solid organ transplant recipients. Clin. Microbiol. Infect. 2014, 20, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J.; Sissons, P. Latent and persistent infections of monocytes and macrophages. Intervirology 1996, 39, 293–301. [Google Scholar] [CrossRef]
- Sinzger, C.; Jahn, G. Human cytomegalovirus cell tropism and pathogenesis. Intervirology 1996, 39, 302–319. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar] [CrossRef] [Green Version]
- Gnann, J.W.; Ahlmén, J.; Svalander, C.; Olding, L.; Oldstone, M.B.; Nelson, J.A. Inflammatory cells in transplanted kidneys are infected by human cytomegalovirus. Am. J. Pathol. 1988, 132, 239–248. [Google Scholar]
- Manez, R.; Kusne, S.; Rinaldo, C.; Aguado, J.M.; St George, K.; Grossi, P.; Frye, B.; Fung, J.J.; Ehrlich, G.D. Time to detection of cytomegalovirus (CMV) DNA in blood leukocytes is a predictor for the development of CMV disease in CMV-seronegative recipients of allografts from CMV-seropositive donors following liver transplantation. J. Infect. Dis. 1996, 173, 1072–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolovan-Fritts, C.A.; Mocarski, E.S.; Wiedeman, J.A. Peripheral blood CD14(+) cells from healthy subjects carry a circular conformation of latent cytomegalovirus genome. Blood 1999, 93, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Booss, J.; Dann, P.R.; Griffith, B.P.; Kim, J.H. Host defense response to cytomegalovirus in the central nervous system. Predominance of the monocyte. Am. J. Pathol. 1989, 134, 71–78. [Google Scholar] [PubMed]
- Larsson, S.; Soderberg-Naucler, C.; Wang, F.Z.; Moller, E. Cytomegalovirus DNA can be detected in peripheral blood mononuclear cells from all seropositive and most seronegative healthy blood donors over time. Transfusion 1998, 38, 271–278. [Google Scholar] [CrossRef]
- Sinzger, C.; Plachter, B.; Grefte, A.; The, T.H.; Jahn, G. Tissue macrophages are infected by human cytomegalovirus in vivo. J. Infect. Dis. 1996, 173, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Whitelaw, D.M. Observations on human monocyte kinetics after pulse labeling. Cell Tissue Kinet. 1972, 5, 311–317. [Google Scholar] [CrossRef]
- Ibanez, C.E.; Schrier, R.; Ghazal, P.; Wiley, C.; Nelson, J.A. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 1991, 65, 6581–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins-McMillen, D.; Kim, J.H.; Nogalski, M.T.; Stevenson, E.V.; Chan, G.C.; Caskey, J.R.; Cieply, S.J.; Yurochko, A.D. Human cytomegalovirus promotes survival of infected monocytes via a distinct temporal regulation of cellular Bcl-2 family proteins. J. Virol. 2015, 90, 2356–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Smith, P.M.; Yurochko, A.D. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J. Immunol. 2008, 181, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Yurochko, A.D. NF-kappaB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced atypical M1/M2 polarization of monocytes. Virus Res. 2009, 144, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.; Nogalski, M.T.; Stevenson, E.V.; Yurochko, A.D. Human cytomegalovirus induction of a unique signalsome during viral entry into monocytes mediates distinct functional changes: A strategy for viral dissemination. J. Leukoc. Biol. 2012, 92, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J. Virol. 2012, 86, 10714–10723. [Google Scholar] [CrossRef] [Green Version]
- Collins-McMillen, D.; Chesnokova, L.; Lee, B.J.; Fulkerson, H.L.; Brooks, R.; Mosher, B.S.; Yurochko, A.D. HCMV Infection and apoptosis: How do monocytes survive HCMV infection? Viruses 2018, 10, 533. [Google Scholar] [CrossRef] [Green Version]
- Collins-McMillen, D.; Stevenson, E.V.; Kim, J.H.; Lee, B.J.; Cieply, S.J.; Nogalski, M.T.; Chan, G.C.; Frost, R.W., 3rd; Spohn, C.R.; Yurochko, A.D. Human cytomegalovirus utilizes a nontraditional signal transducer and activator of transcription 1 activation cascade via signaling through epidermal growth factor receptor and integrins to efficiently promote the motility, differentiation, and polarization of infected monocytes. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Cojohari, O.; Peppenelli, M.A.; Chan, G.C. Human cytomegalovirus induces an atypical activation of Akt to stimulate the survival of short-lived monocytes. J. Virol. 2016, 90, 6443–6452. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Yurochko, A.D. Transcriptome analysis of NF-κB- and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J. Virol. 2008, 82, 1040–1046. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. USA 2009, 106, 22369–22374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.S.; Bentz, G.L.; Alexander, J.S.; Yurochko, A.D. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 2004, 78, 4444–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.S.; Bentz, G.L.; Smith, P.M.; Bivins, E.R.; Yurochko, A.D. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J. Leukoc. Biol. 2004, 76, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bivins-Smith, E.R.; Tilley, A.M.; Bentz, G.L.; Chan, G.; Minard, J.; Yurochko, A.D. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: Strategy for viral persistence. J. Virol. 2007, 81, 7683–7694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, E.V.; Collins-McMillen, D.; Kim, J.H.; Cieply, S.J.; Bentz, G.L.; Yurochko, A.D. HCMV reprogramming of infected monocyte survival and differentiation: A Goldilocks phenomenon. Viruses 2014, 6, 782–807. [Google Scholar] [CrossRef] [Green Version]
- Yurochko, A.D.; Huang, E.S. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J. Immunol. 1999, 162, 4806–4816. [Google Scholar]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppenelli, M.A.; Arend, K.C.; Cojohari, O.; Moorman, N.J.; Chan, G.C. Human cytomegalovirus stimulates the synthesis of select Akt-dependent antiapoptotic proteins during viral entry to promote survival of infected monocytes. J. Virol. 2016, 90, 3138–3147. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.A.V.; Megeney, L.A. Evolution of caspase-mediated cell death and differentiation: Twins separated at birth. Cell Death Differ. 2017, 24, 1359–1368. [Google Scholar] [CrossRef]
- Cathelin, S.; Rébé, C.; Haddaoui, L.; Simioni, N.; Verdier, F.; Fontenay, M.; Launay, S.; Mayeux, P.; Solary, E. Identification of proteins cleaved downstream of caspase activation in monocytes undergoing macrophage differentiation. J. Biol. Chem. 2006, 281, 17779–17788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droin, N.; Cathelin, S.; Jacquel, A.; Guery, L.; Garrido, C.; Fontenay, M.; Hermine, O.; Solary, E. A role for caspases in the differentiation of erythroid cells and macrophages. Biochimie 2008, 90, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Jacquel, A.; Benikhlef, N.; Paggetti, J.; Lalaoui, N.; Guery, L.; Dufour, E.K.; Ciudad, M.; Racoeur, C.; Micheau, O.; Delva, L.; et al. Colony-stimulating factor-1-induced oscillations in phosphatidylinositol-3 kinase/AKT are required for caspase activation in monocytes undergoing differentiation into macrophages. Blood 2009, 114, 3633–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solier, S.; Fontenay, M.; Vainchenker, W.; Droin, N.; Solary, E. Non-apoptotic functions of caspases in myeloid cell differentiation. Cell Death Differ. 2017, 24, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Rebe, C.; Plenchette, S.; Zermati, Y.; Hermine, O.; Vainchenker, W.; Garrido, C.; Solary, E.; Dubrez-Daloz, L. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 2002, 100, 4446–4453. [Google Scholar] [CrossRef] [Green Version]
- Rebe, C.; Cathelin, S.; Launay, S.; Filomenko, R.; Prevotat, L.; L’Ollivier, C.; Gyan, E.; Micheau, O.; Grant, S.; Dubart-Kupperschmitt, A.; et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood 2007, 109, 1442–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cojohari, O.; Burrer, C.M.; Peppenelli, M.A.; Abulwerdi, F.A.; Nikolovska-Coleska, Z.; Chan, G.C. BH3 profiling reveals selectivity by herpesviruses for specific Bcl-2 proteins to mediate survival of latently infected cells. J. Virol. 2015, 89, 5739–5746. [Google Scholar] [CrossRef] [Green Version]
- Nogalski, M.T.; Chan, G.C.T.; Stevenson, E.V.; Collins-McMillen, D.K.; Yurochko, A.D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 2013, 9, e1003463. [Google Scholar] [CrossRef]
- Sinzger, C.; Schmidt, K.; Knapp, J.; Kahl, M.; Beck, R.; Waldman, J.; Hebart, H.; Einsele, H.; Jahn, G. Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J. Gen. Virol. 1999, 80, 2867–2877. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Murphy, E.A. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef] [Green Version]
- Al Faraj, A.; Sultana Shaik, A.; Pureza, M.A.; Alnafea, M.; Halwani, R. Preferential macrophage recruitment and polarization in LPS-induced animal model for COPD: Noninvasive tracking using MRI. PLoS ONE 2014, 9, e90829. [Google Scholar] [CrossRef] [PubMed]
- Shono, J.; Sakaguchi, S.; Suzuki, T.; Do, M.K.; Mizunoya, W.; Nakamura, M.; Sato, Y.; Furuse, M.; Yamada, K.; Ikeuchi, Y.; et al. Preliminary time-course study of antiinflammatory macrophage infiltration in crush-injured skeletal muscle. Anim. Sci. J. 2013, 84, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Magatti, M.; Vertua, E.; De Munari, S.; Caro, M.; Caruso, M.; Silini, A.; Delgado, M.; Parolini, O. Human amnion favours tissue repair by inducing the M1-to-M2 switch and enhancing M2 macrophage features. J. Tissue Eng. Regen. Med. 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Krausz, S.; van Eijk, M.; Hamann, J.; Radstake, T.R.; Reedquist, K.A.; Tak, P.P.; Baeten, D.L. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J. Immunol. Methods 2012, 375, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Leidi, M.; Gotti, E.; Bologna, L.; Miranda, E.; Rimoldi, M.; Sica, A.; Roncalli, M.; Palumbo, G.A.; Introna, M.; Golay, J. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. J. Immunol. 2009, 182, 4415–4422. [Google Scholar] [CrossRef] [Green Version]
- Bayer, C.; Varani, S.; Wang, L.; Walther, P.; Zhou, S.; Straschewski, S.; Bachem, M.; Soderberg-Naucler, C.; Mertens, T.; Frascaroli, G. Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J. Virol. 2013, 87, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Perlman, H.; Pagliari, L.J.; Pope, R.M. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-kappaB, Bad, or caspase activation. J. Exp. Med. 2001, 194, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Kelley, T.W.; Graham, M.M.; Doseff, A.I.; Pomerantz, R.W.; Lau, S.M.; Ostrowski, M.C.; Franke, T.F.; Marsh, C.B. Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J. Biol. Chem. 1999, 274, 26393–26398. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.T.; Craggs, G.; Wilson, L.; Kellie, S. Mechanism of phosphatidylinositol 3-kinase-dependent increases in BAC1.2F5 macrophage-like cell density in response to M-CSF: Phosphatidylinositol 3-kinase inhibitors increase the rate of apoptosis rather than inhibit DNA synthesis. Inflam. Res. 2000, 49, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Hamilton, J.A.; Scholz, G.M.; Masendycz, P.; Macaulay, S.L.; Elsegood, C.L. Phosphatidylinostitol-3 kinase and phospholipase C enhance CSF-1-dependent macrophage survival by controlling glucose uptake. Cell. Signal. 2009, 21, 1361–1369. [Google Scholar] [CrossRef]
- Busca, A.; Saxena, M.; Iqbal, S.; Angel, J.; Kumar, A. PI3K/Akt regulates survival during differentiation of human macrophages by maintaining NF-kappaB-dependent expression of antiapoptotic Bcl-xL. J. Leukoc. Biol. 2014, 96, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Xie, L.; Liu, C.; Zhang, Q.; Sun, S. PTEN/PI3k/AKT regulates macrophage polarization in emphysematous mice. Scand. J. Immunol. 2017, 85, 395–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocher, C.; Singla, D.K. SMAD-PI3K-Akt-mTOR pathway mediates BMP-7 polarization of monocytes into M2 macrophages. PLoS ONE 2013, 8, e84009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beharka, A.A.; Crowther, J.E.; McCormack, F.X.; Denning, G.M.; Lees, J.; Tibesar, E.; Schlesinger, L.S. Pulmonary surfactant protein A activates a phosphatidylinositol 3-kinase/calcium signal transduction pathway in human macrophages: Participation in the up-regulation of mannose receptor activity. J. Immunol. 2005, 175, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Weisser, S.B.; McLarren, K.W.; Voglmaier, N.; van Netten-Thomas, C.J.; Antov, A.; Flavell, R.A.; Sly, L.M. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur. J. Immunol. 2011, 41, 1742–1753. [Google Scholar] [CrossRef]
- Goyal, A.; Wang, Y.; Graham, M.M.; Doseff, A.I.; Bhatt, N.Y.; Marsh, C.B. Monocyte survival factors induce Akt activation and suppress caspase-3. Am. J. Respir. Cell Mol. Biol. 2002, 26, 224–230. [Google Scholar] [CrossRef]
- Hunter, M.; Wang, Y.; Eubank, T.; Baran, C.; Nana-Sinkam, P.; Marsh, C. Survival of monocytes and macrophages and their role in health and disease. Front. Biosci. 2009, 14, 4079–4102. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544. [Google Scholar] [CrossRef]
- Brooks, R.; Fuhler, G.M.; Iyer, S.; Smith, M.J.; Park, M.Y.; Paraiso, K.H.; Engelman, R.W.; Kerr, W.G. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 2010, 184, 3582–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, S.; Iyer, S.; Kerr, W.G. Role of SHIP1 in cancer and mucosal inflammation. Ann. N. Y. Acad. Sci. 2013, 1280, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.G. Inhibitor and activator: Dual functions for SHIP in immunity and cancer. Ann. N. Y. Acad. Sci. 2011, 1217, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppenelli, M.A.; Miller, M.J.; Altman, A.M.; Cojohari, O.; Chan, G.C. Aberrant regulation of the Akt signaling network by human cytomegalovirus allows for targeting of infected monocytes. Antivir. Res. 2018, 158, 13–24. [Google Scholar] [CrossRef]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Liu, R.; Fan, T.; Geng, W.; Chen, Y.H.; Ruan, Q.; Zhang, C. Negative immune regulator TIPE2 promotes M2 macrophage differentiation through the activation of PI3K-AKT signaling pathway. PLoS ONE 2017, 12, e0170666. [Google Scholar] [CrossRef]
- Arranz, A.; Doxaki, C.; Vergadi, E.; Martinez de la Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef] [Green Version]
- Papakonstanti, E.A.; Zwaenepoel, O.; Bilancio, A.; Burns, E.; Nock, G.E.; Houseman, B.; Shokat, K.; Ridley, A.J.; Vanhaesebroeck, B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J. Cell Sci. 2008, 121, 4124–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sly, L.M.; Ho, V.; Antignano, F.; Ruschmann, J.; Hamilton, M.; Lam, V.; Rauh, M.J.; Krystal, G. The role of SHIP in macrophages. Front. Biosci. 2007, 12, 2836–2848. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W.; States, D.J. Colony-stimulating factor-1 requires PI3-kinase-mediated metabolism for proliferation and survival in myeloid cells. Cell Death Differ. 2006, 13, 1900–1914. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cojohari, O.; Mahmud, J.; Altman, A.M.; Peppenelli, M.A.; Miller, M.J.; Chan, G.C. Human Cytomegalovirus Mediates Unique Monocyte-to-Macrophage Differentiation through the PI3K/SHIP1/Akt Signaling Network. Viruses 2020, 12, 652. https://doi.org/10.3390/v12060652
Cojohari O, Mahmud J, Altman AM, Peppenelli MA, Miller MJ, Chan GC. Human Cytomegalovirus Mediates Unique Monocyte-to-Macrophage Differentiation through the PI3K/SHIP1/Akt Signaling Network. Viruses. 2020; 12(6):652. https://doi.org/10.3390/v12060652
Chicago/Turabian StyleCojohari, Olesea, Jamil Mahmud, Aaron M. Altman, Megan A. Peppenelli, Michael J. Miller, and Gary C. Chan. 2020. "Human Cytomegalovirus Mediates Unique Monocyte-to-Macrophage Differentiation through the PI3K/SHIP1/Akt Signaling Network" Viruses 12, no. 6: 652. https://doi.org/10.3390/v12060652