Regional Transmission and Reassortment of 2.3.4.4b Highly Pathogenic Avian Influenza (HPAI) Viruses in Bulgarian Poultry 2017/18

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Epidemiology
2.2. Sequencing
2.3. Visualising Reassortment
2.4. Spatial Phylodynamic Analysis and Ancestral Host Reconstruction
3. Results
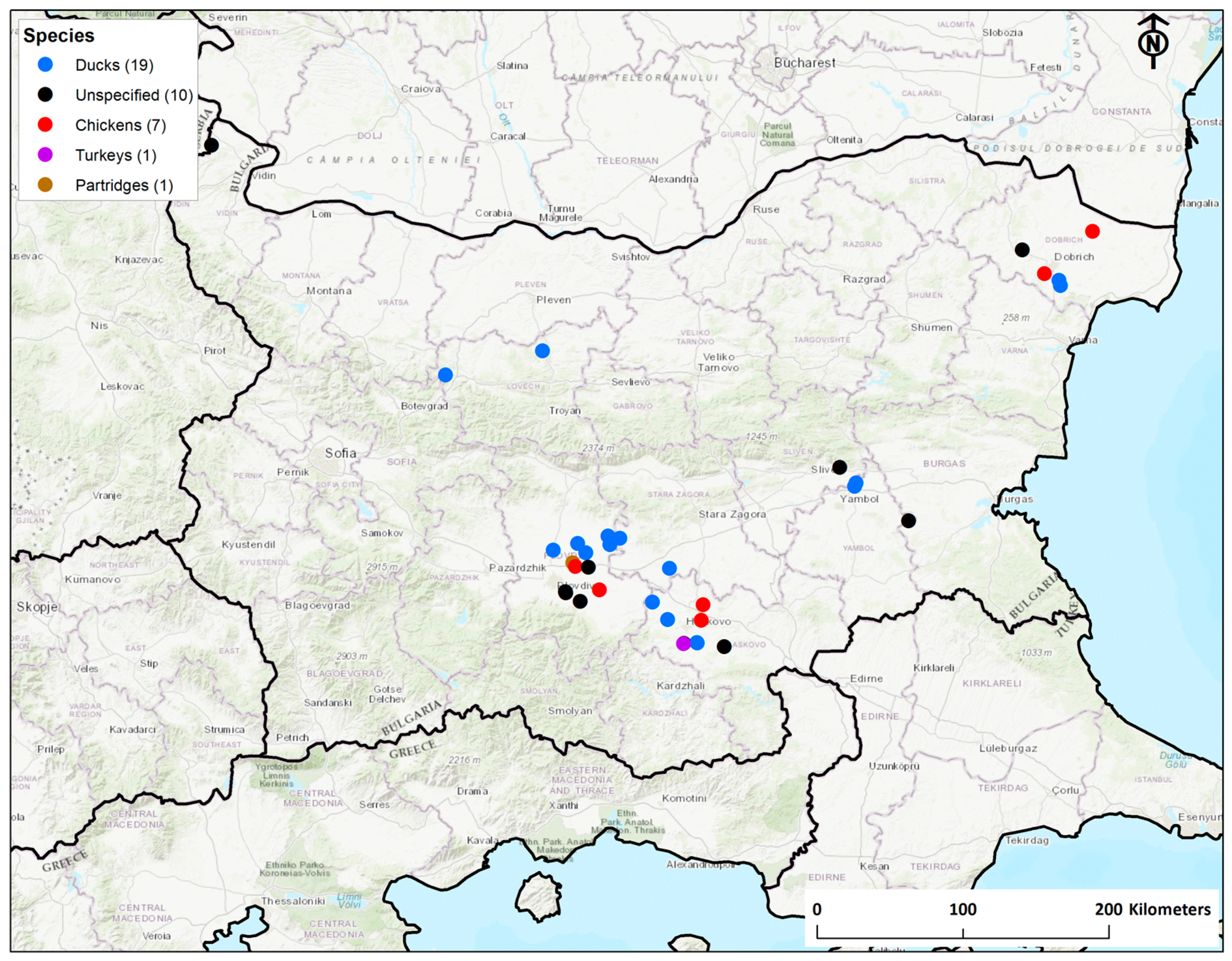
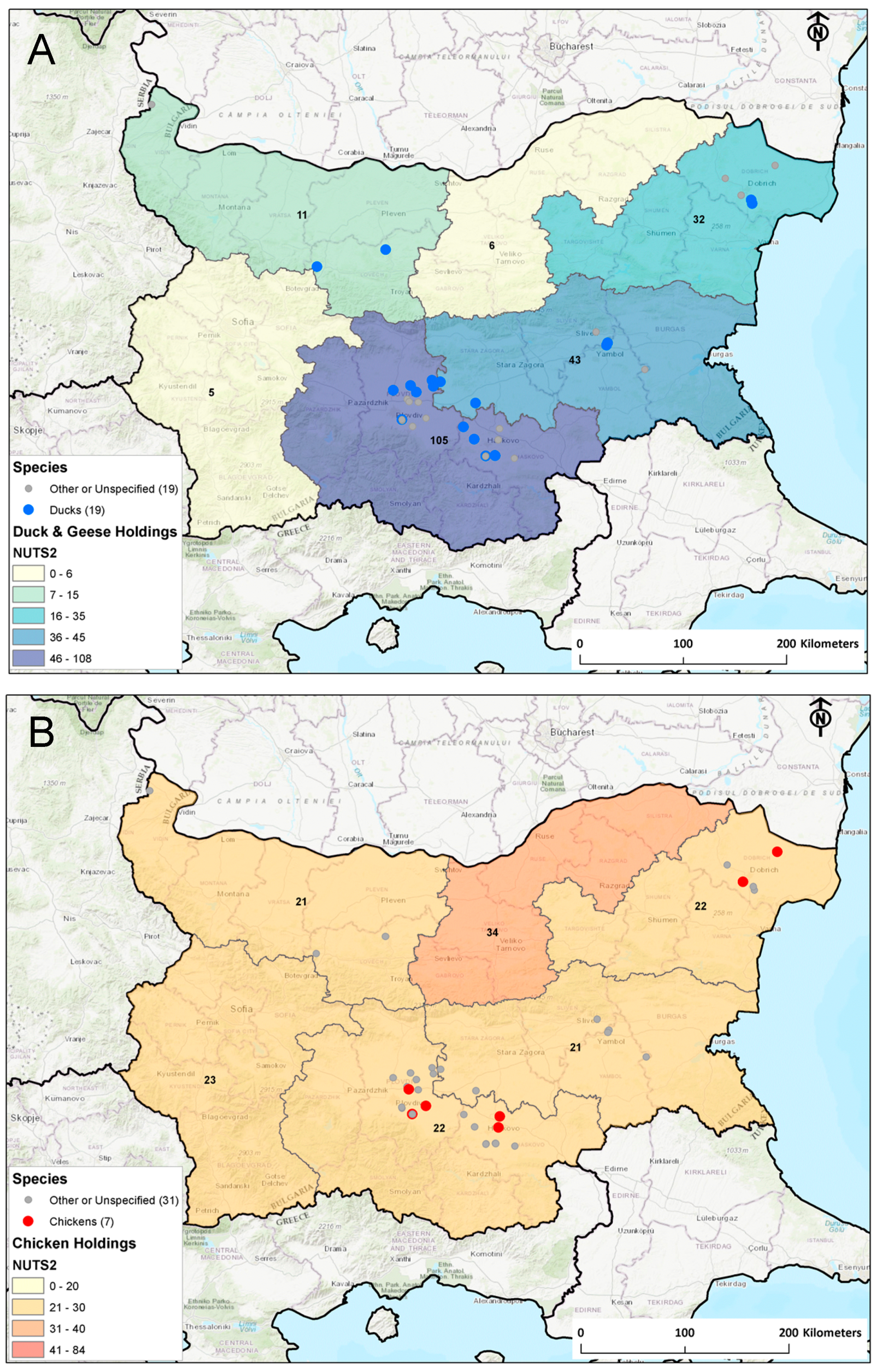
3.1. Epidemiology
3.2. Genetic Structure of Bulgarian H5 HA
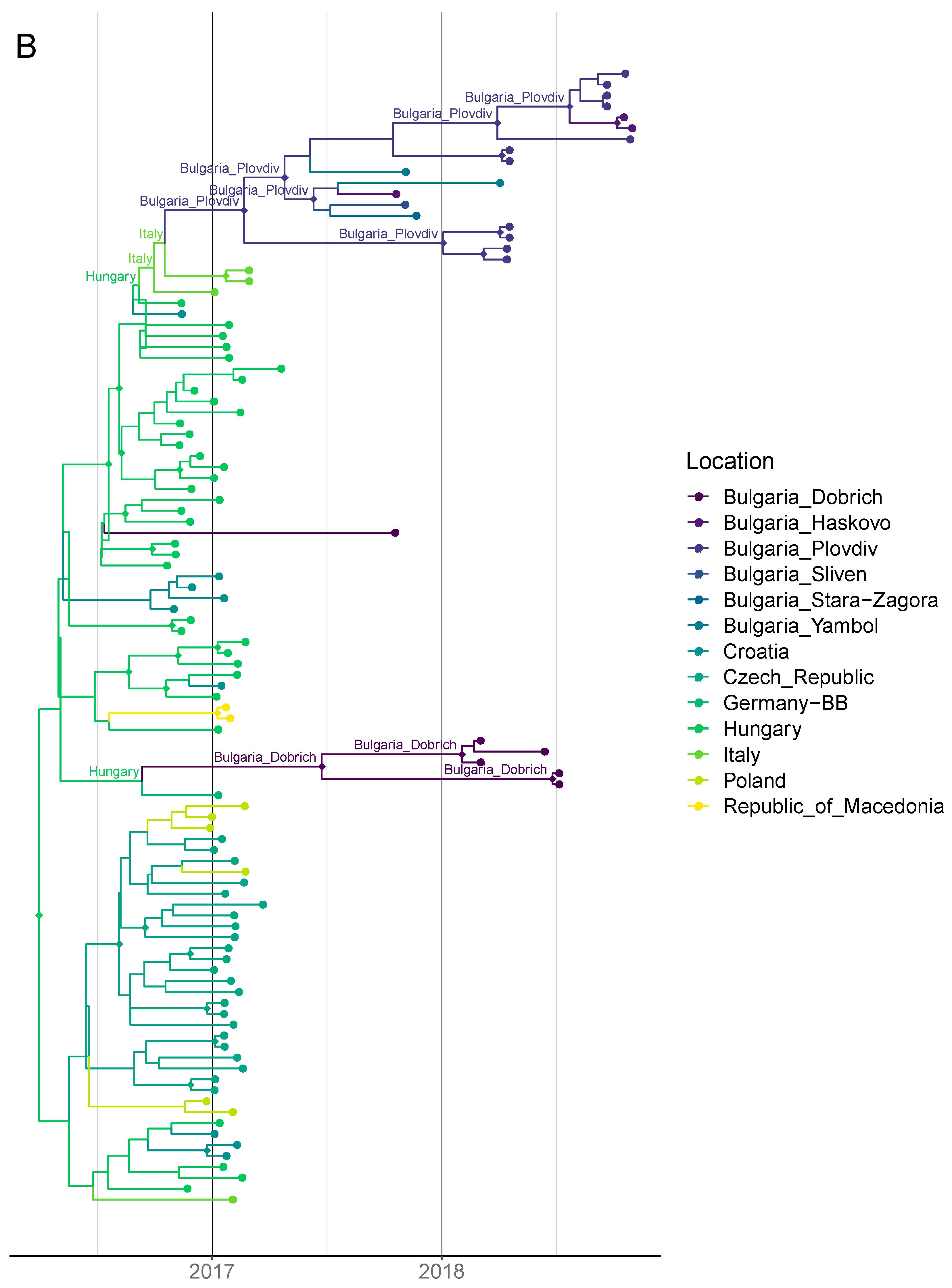
3.3. Timing of Introductions, Host Dynamics and Spatial Spread
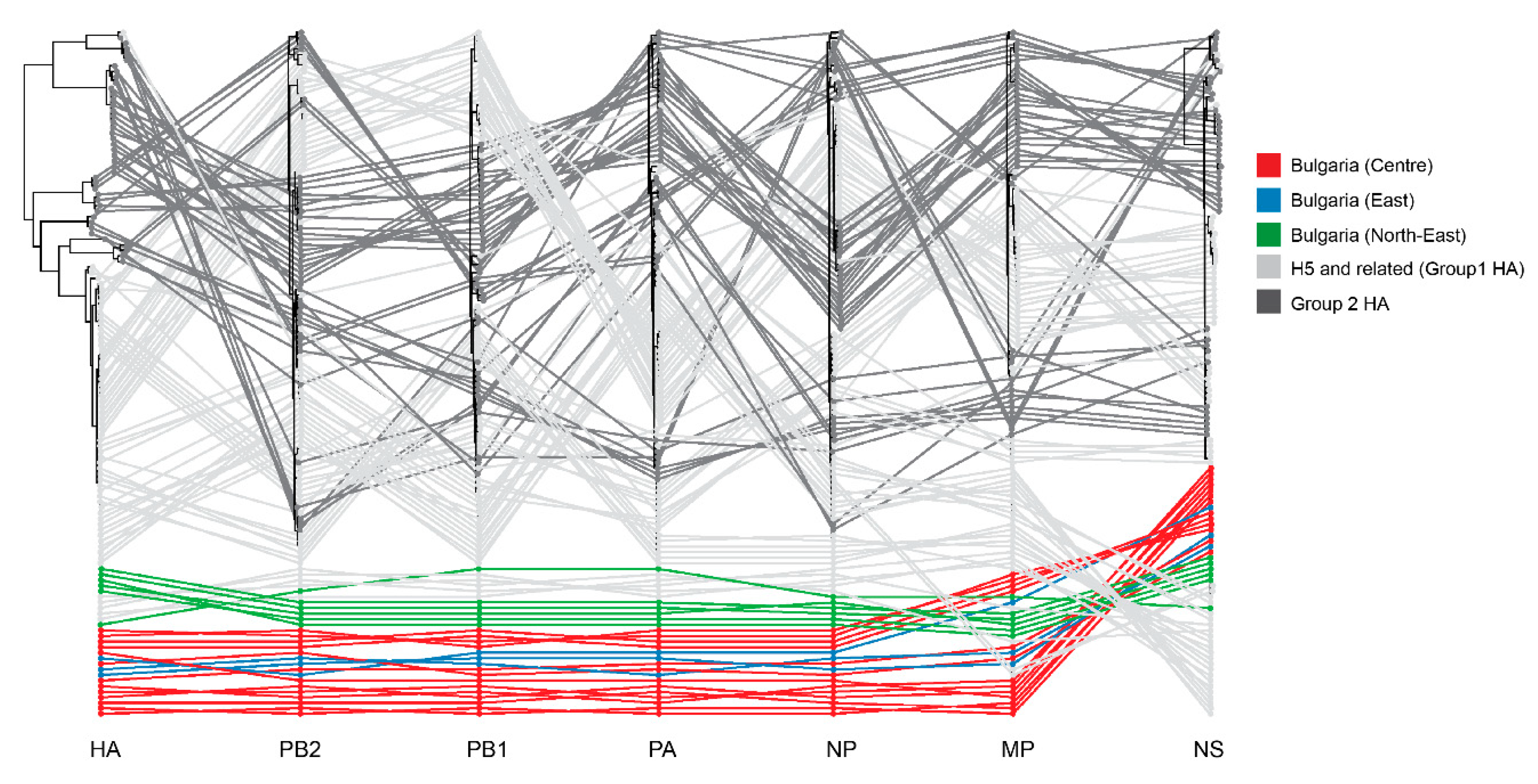
3.4. Whole-Genome Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bosch, F.X.; Orlich, M.; Klenk, H.-D.; Rott, R. The structure of the hemagglutinin, a determinant for the pathogenicity of influenza viruses. Virology 1979, 95, 197–207. [Google Scholar] [CrossRef]
- Röhm, C.; Horimoto, T.; Kawaoka, Y.; Süss, J.; Webster, R.G. Do Hemagglutinin Genes of Highly Pathogenic Avian influenza Viruses Constitute Unique Phylogenetic Lineages? Virology 1995, 209, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.J. A review of avian influenza in different bird species. Vet. Microbiol. 2000, 74, 3–13. [Google Scholar] [CrossRef]
- Alexander, D.J. An overview of the epidemiology of avian influenza. Vaccine 2007, 25, 5637–5644. [Google Scholar] [CrossRef] [PubMed]
- Klenk, H.-D.; Rott, R. The Molecular Biology of Influenza Virus Pathogenicity. In Advances in Virus Research; Maramorosch, K., Murphy, F.A., Shatkin, A.J., Eds.; Academic Press: Cambridge, MA, USA, 1988; Volume 34, pp. 247–281. [Google Scholar]
- Rott, R. The pathogenic determinant of influenza virus. Vet. Microbiol. 1992, 33, 303–310. [Google Scholar] [CrossRef]
- Sonnberg, S.; Webby, R.J.; Webster, R.G. Natural history of highly pathogenic avian influenza H5N1. Virus Res. 2013, 178, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Lycett Samantha, J. Duchatel Florian; Digard Paul A brief history of bird flu. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180257. [Google Scholar] [CrossRef]
- Claes, F.; Kuznetsov, D.; Liechti, R.; Von Dobschuetz, S.; Dinh Truong, B.; Gleizes, A.; Conversa, D.; Colonna, A.; Demaio, E.; Ramazzotto, S.; et al. The EMPRES-i genetic module: A novel tool linking epidemiological outbreak information and genetic characteristics of influenza viruses. Database 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO); World Organisation for Animal Health (OIE); Food and Agriculture Organization (FAO). H5N1 Evolution Working Group Revised and updated nomenclature for highly pathogenic avian influenza A (H5N1) viruses. Influenza Other Respir. Viruses 2014, 8, 384–388. [Google Scholar] [CrossRef]
- Claes, F.; Morzaria, S.P.; Donis, R.O. Emergence and dissemination of clade 2.3.4.4 H5Nx influenza viruses—how is the Asian HPAI H5 lineage maintained. Curr. Opin. Virol. 2016, 16, 158–163. [Google Scholar] [CrossRef]
- Dhingra, M.S.; Artois, J.; Robinson, T.P.; Linard, C.; Chaiban, C.; Xenarios, I.; Engler, R.; Liechti, R.; Kuznetsov, D.; Xiao, X.; et al. Global mapping of highly pathogenic avian influenza H5N1 and H5Nx clade 2.3.4.4 viruses with spatial cross-validation. eLife 2016, 5, e19571. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Bertran, K.; Kwon, J.-H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Highly pathogenic avian influenza A subtype H5N8. EFSA J. 2014, 12, 3941. [Google Scholar] [CrossRef]
- Beerens, N.; Heutink, R.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Multiple Reassorted Viruses as Cause of Highly Pathogenic Avian Influenza A(H5N8) Virus Epidemic, the Netherlands, 2016. Emerg. Infect. Dis. 2017, 23, 1974–1981. [Google Scholar] [CrossRef] [PubMed]
- Kleyheeg, E.; Slaterus, R.; Bodewes, R.; Rijks, J.M.; Spierenburg, M.A.H.; Beerens, N.; Kelder, L.; Poen, M.J.; Stegeman, J.A.; Fouchier, R.A.M.; et al. Deaths among Wild Birds during Highly Pathogenic Avian Influenza A(H5N8) Virus Outbreak, the Netherlands. Emerg. Infect. Dis. 2017, 23, 2050–2054. [Google Scholar] [CrossRef]
- Pohlmann, A.; Starick, E.; Harder, T.; Grund, C.; Höper, D.; Globig, A.; Staubach, C.; Dietze, K.; Strebelow, G.; Ulrich, R.G.; et al. Outbreaks among Wild Birds and Domestic Poultry Caused by Reassorted Influenza A(H5N8) Clade 2.3.4.4 Viruses, Germany, 2016. Emerg. Infect. Dis. 2017, 23, 633–636. [Google Scholar] [CrossRef]
- Poen, M.J.; Venkatesh, D.; Bestebroer, T.M.; Vuong, O.; Scheuer, R.D.; Oude Munnink, B.B.; de Meulder, D.; Richard, M.; Kuiken, T.; Koopmans, M.P.G.; et al. Co-circulation of genetically distinct highly pathogenic avian influenza A clade 2.3.4.4 (H5N6) viruses in wild waterfowl and poultry in Europe and East Asia, 2017–2018. Virus Evol. 2019, 5. [Google Scholar] [CrossRef]
- Alarcon, P.; Brouwer, A.; Venkatesh, D.; Duncan, D.; Dovas, C.I.; Georgiades, G.; Monne, I.; Fusaro, A.; Dan, A.; Śmietanka, K.; et al. Comparison of 2016–17 and Previous Epizootics of Highly Pathogenic Avian Influenza H5 Guangdong Lineage in Europe. Emerg. Infect. Dis. 2018, 24, 2270–2283. [Google Scholar] [CrossRef]
- OIE. OIE Situation Report for Avian Influenza 2017; World Organisation for Animal Health: Paris, France, 2017. [Google Scholar]
- Kwon, J.-H.; Jeong, S.; Lee, D.-H.; Swayne, D.E.; Kim, Y.; Lee, S.; Noh, J.-Y.; Erdene-Ochir, T.-O.; Jeong, J.-H.; Song, C.-S. New Reassortant Clade 2.3.4.4b Avian Influenza A(H5N6) Virus in Wild Birds, South Korea, 2017–2018. Emerg. Infect. Dis. 2018, 24, 1953–1955. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.; Mulatti, P.; Smietanka, K.; Staubach, C.; Willeberg, P.; Adlhoch, C.; Candiani, D.; Fabris, C.; Zancanaro, G.; Morgado, J.; et al. Avian influenza overview October 2016–August 2017. EFSA J. 2017, 15, e05018. [Google Scholar] [CrossRef]
- Brown, I.; Kuiken, T.; Mulatti, P.; Smietanka, K.; Staubach, C.; Stroud, D.; Therkildsen, O.R.; Willeberg, P.; Baldinelli, F.; Verdonck, F.; et al. Avian influenza overview September–November 2017. EFSA J. 2017, 15, e05141. [Google Scholar] [CrossRef]
- Adlhoch, C.; Brouwer, A.; Kuiken, T.; Mulatti, P.; Smietanka, K.; Staubach, C.; Guajardo, I.M.; Verdonck, F.; Amato, L.; Baldinelli, F. Avian influenza overview February–May 2018. EFSA J. 2018, 16, e05358. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Kuiken, T.; Monne, I.; Smietanka, K.; Staubach, C.; Guajardo, I.M.; Baldinelli, F. Avian influenza overview February–August 2019. EFSA J. 2019, 17, e05843. [Google Scholar] [CrossRef]
- Environmental Systems Research Institute. ArcGIS Desktop 10.2.2 2014; Environmental Systems Research Institute: Redlands, CA, USA, 2014. [Google Scholar]
- PAFF Avian. PAFF Avian Influenza in Bulgaria; PAFF Avian: Bulgaria, Bulgaria, 2018. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Bedford, T. Modern-day SIV viral diversity generated by extensive recombination and cross-species transmission. PLoS Pathog. 2017, 13, e1006466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Brouwer, A.; Gonzales, J.; Huneau, A.; Mulatti, P.; Kuiken, T.; Staubach, C.; Stegeman, A.; Antoniou, S.-E.; Baldinelli, F.; der Stede, Y.V.; et al. Annual Report on surveillance for avian influenza in poultry and wild birds in Member States of the European Union in 2018. EFSA J. 2019, 17, e05945. [Google Scholar] [CrossRef]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem. J. Virol. 2018, 92, e00433-18. [Google Scholar] [CrossRef] [Green Version]
- Marinova-Petkova, A.; Georgiev, G.; Petkov, T.; Darnell, D.; Franks, J.; Kayali, G.; Walker, D.; Seiler, P.; Danner, A.; Graham, A.; et al. Influenza surveillance on ‘foie gras’ duck farms in Bulgaria, 2008–2012. Influenza Other Respir Viruses 2016, 10, 98–108. [Google Scholar] [CrossRef]
- Guinat, C.; Durand, B.; Vergne, T.; Corre, T.; Rautureau, S.; Scoizec, A.; Lebouquin-Leneveu, S.; Guérin, J.-L.; Paul, M.C. Role of Live-Duck Movement Networks in Transmission of Avian Influenza, France, 2016–2017. Emerg. Infect. Dis. 2020, 26, 472–480. [Google Scholar] [CrossRef] [Green Version]
- EC Commission Decision 2010/367/EU of 25 June 2010 on the implementation by Member States of surveillance programmes for avian influenza in poultry and wild birds. Off. J. Eur. Union 2010, 166, 22.
- OFFLU. OFFLU Animal Influenza Report: September 2019 to February 2020 2020; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Guinat, C.; Artois, J.; Bronner, A.; Guérin, J.L.; Gilbert, M.; Paul, M.C. Duck production systems and highly pathogenic avian influenza H5N8 in France, 2016–2017. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dent, J.E.; Kiss, I.Z.; Kao, R.R.; Arnold, M. The potential spread of highly pathogenic avian influenza virus via dynamic contacts between poultry premises in Great Britain. BMC Vet. Res. 2011, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, J.; Gauger, P.; Harmon, K.; Zhang, J.; Connor, J.; Yeske, P.; Loula, T.; Levis, I.; Dufresne, L.; Main, R. Role of Transportation in Spread of Porcine Epidemic Diarrhea Virus Infection, United States. Emerg. Infect. Dis. 2014, 20, 872–874. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatesh, D.; Brouwer, A.; Goujgoulova, G.; Ellis, R.; Seekings, J.; Brown, I.H.; Lewis, N.S. Regional Transmission and Reassortment of 2.3.4.4b Highly Pathogenic Avian Influenza (HPAI) Viruses in Bulgarian Poultry 2017/18. Viruses 2020, 12, 605. https://doi.org/10.3390/v12060605
Venkatesh D, Brouwer A, Goujgoulova G, Ellis R, Seekings J, Brown IH, Lewis NS. Regional Transmission and Reassortment of 2.3.4.4b Highly Pathogenic Avian Influenza (HPAI) Viruses in Bulgarian Poultry 2017/18. Viruses. 2020; 12(6):605. https://doi.org/10.3390/v12060605
Chicago/Turabian StyleVenkatesh, Divya, Adam Brouwer, Gabriela Goujgoulova, Richard Ellis, James Seekings, Ian H. Brown, and Nicola S. Lewis. 2020. "Regional Transmission and Reassortment of 2.3.4.4b Highly Pathogenic Avian Influenza (HPAI) Viruses in Bulgarian Poultry 2017/18" Viruses 12, no. 6: 605. https://doi.org/10.3390/v12060605