HIV-1 Latency and Latency Reversal: Does Subtype Matter?


Department of Microbiology, Immunology and Tropical Medicine, The George Washington University, Washington, DC 20052, USA
*
Author to whom correspondence should be addressed.
Viruses 2019, 11(12), 1104; https://doi.org/10.3390/v11121104
Submission received: 14 October 2019
/
Revised: 22 November 2019
/
Accepted: 27 November 2019
/
Published: 28 November 2019
(This article belongs to the Special Issue HIV-1 Latency: Regulation and Reversal)
Abstract
:Cells that are latently infected with HIV-1 preclude an HIV-1 cure, as antiretroviral therapy does not target this latent population. HIV-1 is highly genetically diverse, with over 10 subtypes and numerous recombinant forms circulating worldwide. In spite of this vast diversity, much of our understanding of latency and latency reversal is largely based on subtype B viruses. As such, most of the development of cure strategies targeting HIV-1 are solely based on subtype B. It is currently assumed that subtype does not influence the establishment or reactivation of latent viruses. However, this has not been conclusively proven one way or the other. A better understanding of the factors that influence HIV-1 latency in all viral subtypes will help develop therapeutic strategies that can be applied worldwide. Here, we review the latest literature on subtype-specific factors that affect viral replication, pathogenesis, and, most importantly, latency and its reversal.
1. Introduction
Human Immunodeficiency Virus (HIV-1) remains one of the highest causes of morbidity and mortality worldwide. Antiretroviral therapy (ART) has significantly improved the lives of those living with HIV-1, but a daily commitment to these medications is critical to managing the disease. If ART is interrupted, the virus can resurge, and the disease progresses to Acquired Immunodeficiency Syndrome (AIDS). This is because HIV-1 is able to establish latency [1,2,3,4]. Latently infected cells harbor an integrated but transcriptionally silent HIV-1 provirus that can be reactivated [5]. ART targets different steps of the viral life cycle of HIV-1, such as reverse transcription, viral entry, integration, and viral budding, but cannot target these latently infected cells [1,2,3,4,5,6,7]. Thus, these latently infected cells are a barrier to an HIV-1 cure. Resting memory CD4 T cells are thought to be one of the main latent reservoirs, though other cell types contribute to this reservoir [8,9,10,11,12,13]. Several strategies are being pursued towards eliminating this latent reservoir [14]. “Shock-and-kill” is a cure strategy currently in clinical trials (reviewed in [15,16]). This strategy involves revealing latently infected cells to the immune system by using latency reversing agents (LRAs). LRAs can promote viral protein expression through several distinct mechanisms, such as relieving repressive epigenetic modifications or supplying host transcription factors and other cellular factors necessary for viral gene expression, thus producing viral antigens (“Shock”) [17]. Reactivated cells are then revealed and are now sensitive to the cytotoxic arm of the immune system (“Kill”).
Significant progress has been made in developing novel LRAs and understanding the mechanisms underlying HIV-1 latency reversal, but these findings have been mostly conducted with subtype B viruses. The genetic diversity of HIV-1 is staggering, with over 10 subtypes worldwide and ever-increasing recombinant strains [18]. However, it is incompletely understood whether subtype has a biological impact on latency establishment and latency reversal. This complexity can become a challenge when developing a curative strategy to be implemented worldwide. For example, the vast genetic diversity of HIV-1 has long been a challenge to the development of a universal HIV-1 vaccine. The envelope protein, the main epitope utilized for vaccine development, can vary greatly from subtype to subtype, affecting vaccine efficacy [19]. Thus, the development of an HIV-1 vaccine that can provide cross-subtype protection is vital. This same reasoning should be applied to HIV-1 cure strategies, where the methods developed should elicit the most latency reversal across subtypes, not just subtype B. Currently, it is not well characterized as to whether subtype has an impact on latency or latency reversal. As such, understanding whether subtype may affect latency should be addressed early on in the development of curative strategies to be implemented worldwide.
Here, we provide a comprehensive literature review specifically focused on differences in viral pathogenesis, co-receptor usage, viral protein function, and viral long terminal repeat (LTR) sequence among subtypes and how this may influence the establishment and reactivation of the latent reservoir.
2. Role of Subtype in HIV-1 Pathogenesis
Within HIV-1 group M viruses, there are at least 10 subtypes and numerous circulating recombinant forms (CRFs) and unique recombinant forms (URFs). They are classified as A, B, C, D, F, G, H, J, K, and major CRFs such as AE and AG (CRF01_AE, CRF02_AG). Subtypes can be further divided into sub-subtypes, which are denoted with numbers, e.g., subtype A1 [20]. Subtype AE was previously described as subtype E (and is referred as such in older publications), but it has been shown to be a recombinant form of subtype A and was reclassified as subtype AE [20]. The distribution of subtype worldwide is not uniform, as shown in Figure 1. Geographically, subtype C has been shown to mainly circulate in the sub-Saharan African continent. Subtype G circulates in Western and Central African countries, where some CRFs also exist, such as AG. Western Europe is mostly subtype B, with some small populations of subtypes A, C, G, and AG. Eastern Europe is mostly subtype A, with a population of subtype B and smaller fractions of subtypes C, F, and AB. Northern American countries, Latin America, and the Caribbean are largely subtype B with some populations of subtypes C and F. South and Southeastern Asia is largely subtype AE, while Eastern Asia is comprised of subtypes B, AE, and subtype BC recombinants [18,21,22,23]. The locations of these various subtypes are not fixed in place, as there are reports showing an increased diversity of subtypes in traditionally subtype B-dominant regions [24,25,26]. Recent work by Yamaguchi et al. have helped establish the presence of a new subtype, L, through next-generation sequencing [27]. Using country-specific HIV-1 molecular epidemiology data from 2000 to 2007, Hemelaar et al. generated global estimates for the prevalence of the distinct subtypes. Subtype C comprises approximately 48% of infections, while subtype A comprises 12%, subtype B 11%, subtype G 5%, and subtype D 2% of worldwide infections. Subtypes F, H, J, and K are a relative global minority and comprise 0.45%, 0.26%, 0.12%, and 0.01%, respectively. Recombinant CRF and URF subtypes comprise a large portion of global infections, about 20%. The major CRFs, AE and AG, comprise approximately 5% and 8%, respectively [18,21,22,23].
Several studies have examined the association between viral subtype and disease progression. The results of these studies which examined pathogenesis in vivo are explained in detail below and are summarized in Table 1. Earlier studies found no difference in disease progression among viral subtypes [28,29,30], though most recent studies did find a significant difference [31,32,33,34,35,36,37]. These opposing conclusions may be due to differences in sample size, host or environmental factors, disease stage of participants, or subtype identification methods. Additionally, making direct comparisons between subtypes can be challenging, as any underlying differences in host or environmental factors can confound the results of the study [18]. Studies that examine pathogenesis where the subtypes co-circulate within the same population are the most appropriate, as it presumably eliminates some of these confounding variables. For example, in a prospective study involving Senegalese female sex workers, women who were infected with non-subtype A virus had a greater risk of developing AIDS than subtype-A-infected women in the same cohort [38]. Similarly, individuals infected with subtype D had a faster CD4 decline when compared to subtype A-infected individuals in a seroincident cohort in Uganda [31]. Baeten et al. also observed an association of subtype and faster CD4 decline in a cohort of Kenyan commercial sex workers, who were followed from the time of HIV-1 acquisition [32]. In this study, individuals infected with subtype D virus were also found to be at a higher risk of death compared to subtype A, and this risk persisted after correction for plasma HIV-1 viral loads. Similarly, Ssemwanga et al. found that subtype D-infected individuals had faster CD4 decline when compared to subtype A-infected individuals in rural Uganda [34]. In another study, subtype D infection was associated with the fastest CD4 decline, compared to subtype A or C, in a Tanzanian cohort of pregnant mothers [35]. Furthermore, subtype D infection was associated with the greatest CD4 cell decline in a cohort of women in Sub-Saharan Africa who were either subtype A, C, or D [36]. Kaleebu et al. found similar results within a cohort of >1000 subtype A or subtype D individuals, where subtype D individuals had faster disease progression compared to subtype A [33]. Finally, Amornkul et al. assessed disease progression in a cohort of subtypes A, C, and D individuals and found similar results where non-subtype A individuals had faster CD4 decline compared to subtype A individuals [37]. Taken together, the overall consensus is that there are differences in disease progression associated with viral subtype. This has obvious implications for the treatment of HIV-1, but it is less clear whether this difference in pathogenesis will impact latency establishment or latency-reversal.
3. HIV-1 Coreceptor Usage and Tropism Switch
As untreated HIV-1 infection progresses, the virus can switch from CCR5 to CXCR4 usage [39,40,41]. This switch to CXCR4 is correlated with disease progression [40], which is typical of subtype B viruses and can emerge late in disease in other subtypes as well [18,42]. The HIV-1 envelope, being the only protein that is exposed, is a target for antibody and cell-mediated immune responses and is obviously indispensable for entry into host cells (reviewed in [43]). As such, the sequence diversity within the viral env gene has been characterized extensively, mostly with subtype B and C. Between subtypes, the sequence identity of the env gene can vary by as much as 35% (for a review on Env diversity, see [44]). The sequence of the third variable loop (V3 loop) of the viral glycoprotein gp120 is critical for infection and is a determinant of coreceptor usage [45,46,47]. Interestingly, not all HIV-1 subtypes uniformly switch coreceptor usage, even in late stages of the disease. Subtype C and subtype A rarely undergo this switch; subtype C viruses favor CCR5 more extremely throughout infection than subtype A [20,48,49]. The V3 loop sequence length, amino acid charge, glycosylation site presence, and amino acid variations affect the development of CXCR4 usage [50]. Subtype C exhibits less sequence variation in the V3 loop compared to subtype B. Subtype A has been reported to be highly similar in its V3 loop to subtype C, though not identical [51]. These genetic features could explain the rarity of X4 variants in subtype C or subtype A infection. On the other hand, subtype D has been reported to be more X4-tropic, or exhibit dual (CXCR4/CCR5) usage in some cases [49,52,53,54]. The V3 loop of subtype D viruses is identical to R5-tropic viruses, suggesting other regions outside of the V3 loop affect CXCR4 usage for subtype D [52]. It has been shown that the majority of the latent reservoir in subtype B encompass viruses that use CCR5 as co-receptor [55]. However, it is unclear whether this is true for other subtypes. Furthermore, the tropism of the virus can influence the subset of CD4 T cells in which HIV can establish the latent reservoir. Different subsets of CD4 T cells respond differently to different LRAs [56,57]. Further research will be needed to characterize whether the differences in tropism influences the establishment of latency in different subsets and how this could influence cure strategies.
4. The Viral Long Terminal Repeat (LTR)
Upon integration, the HIV-1 LTR becomes the HIV-1 promoter and recruits host transcription factors for viral transcription [58]. There are numerous mechanisms that contribute to HIV-1 latency, one of which is lack of activated host transcription factors such as nuclear factor kappa light chain enhancer of activated B cells (NF-κB), nuclear factor of activated T cells (NFAT), or signal transducer and activator of transcription 5 (STAT5) in resting memory CD4 T cells [59,60,61,62,63]. One of the rationale for latency reversing strategies is to provide these activated transcription factors in order to promote viral transcription.
The presence and number of transcription factor binding sites vary depending on the subtype (Figure 2), which in some cases has been shown to alter their transcriptional activation in response to diverse stimuli in vitro [64,65,66,67,68,69,70,71,72]. These studies are described below and are also summarized in Table 2. Of note, the subtype C LTR harbors more NF-κB binding sites than other subtypes, with 3–4 NF-κB sites [70,73] compared to all other subtypes which typically harbor 2. Subtype AE is an exception to this, as it harbors only one NF-κB binding site [74]. Though these additional κB sites have not been shown to directly compete for NF-κB binding in subtype C [68], their presence has been associated with greater transcriptional activity in response to NF-κB activating stimuli [73]. This was shown in an LTR-reporter system in Jurkat cells, where viruses with 4 NF-κB binding sites produced higher amounts of viral transcripts, compared to viruses with only 3 [73]. This phenotype was further confirmed in a subtype-specific LTR-luciferase reporter system in Jurkat cells, where stimulation with NF-κB-activating stimuli induced more luciferase expression in subtype C LTRs which had 4 NF-κB sites versus those that had 3 [64]. If they are bonafide NF-κB binding sites, these additional NF-κB sites could potentially result in increased transcription by recruitment of p65/p50 heterodimers; however, it could potentially impute a more silent phenotype in latent cells, through binding of the p50 homodimer NF-κB and recruitment of inhibitory complexes such as histone deacetylases (HDAC) to the LTR [75].
Rodriguez et al. characterized the LTRs of five subtype A and seven subtype C circulating strains in India. Sequence analysis showed that subtype C isolates contained three NF-κB sites, and there were no significant differences in transcriptional activity between subtype A and subtype C [67]. Transcriptional activity was determined by transfecting LTR-luciferase reporter plasmids into Jurkat cells and measuring luciferase in the absence or presence of HIV-1 Tat. One limitation of this study is that subtype B Tat was used, instead of the subtype-specific Tat, which could explain why they observed no differences in transcriptional activity. In a study by Roof et al., subtypes B, C, or E LTRs and subtypes B, C, or E Tat were transiently transfected into Jurkat cells and then stimulated with tumor necrosis factor-α (TNF-α). Luciferase activity was measured 10 hours post-stimulation. In this system, differences in basal level transcription were observed among subtypes, and subtype E Tat was found to have the highest transactivation capacity regardless of LTR. [68]. Most major subtypes (A–G) harbor a signal transducers and activators of transcription (STAT) binding site, though their exact sequence and intensity of response to activated STAT5 stimulation vary [65]. The location and number of activator protein-1 (AP-1) and specificity protein-1 (Sp1) sites vary as well, depending on the subtype [66,69,71]. The variance in Sp1 and NF-κB sites in the LTR of the Chinese subtype B’ was associated with a greater capacity to drive gene expression in a reporter system [72]. Jeeninga et al. examined the contribution to transcription by each of the subtype-specific LTR sequences (A, B, C1, C2, D, E, F, G and G’’). This study found that although there was no difference in tat-transactivation among subtypes, the response to TNF-α was correlated with the number of NF-κB sites present in the various viral LTRs [66]. Subtype C, which harbored three NF-κB sites, exhibited the greatest response to TNF-α. Using subtype-specific LTR-luciferase constructs, they found that subtype E had higher basal transcriptional activity. The core of the subtype E promoter was subsequently cloned into a subtype B molecular clone (HIV-1 LAI) and was used to infect SupT1 cells. This subtype B/subtype E chimeric virus produced higher CA-p24 than subtype B LAI, suggesting that genetic differences in subtypes may partly drive differences in viral replication kinetics [66].
The impact of host cell environment and subtype on viral replication was evaluated by van Opijnen et al. By employing viruses with subtype-specific LTRs (subtypes A, B, C, D, E, F, G) in a subtype B LAI background, they examined replication rates in SupT1 and MT2 cell lines in the presence or absence of phytohemagglutinin (PHA) or TNF-α (mimicking a resting state or activated state in T cells). They tracked viral replication by measuring CA-p24 across seven days of infection. The authors also assessed viral fitness by performing competition experiments where two distinct subtypes of virus were used to infect SupT1 cells. Infected cells were sequenced in order to determine the frequencies of each of the subtypes. They found that subtype E was the most fit but that this fitness changed depending on the cellular environment [69]. These studies highlight the importance of the host’s cellular environment in concert with viral-intrinsic factors such as subtype-specific LTR and suggest that some subtypes may be more adaptable to different cellular environments than others. Whether this difference also extends to latency dynamics has not been fully characterized.
Thus far, there have been limited studies evaluating the ability of non-B subtypes to establish latency in T cell lines [76,77,78]. These studies have been described in detail below, as well as summarized in Table 2. Van der Sluis et al. characterized the latency profiles established by chimeric viruses containing subtype A, B, C1, C2, D, AE, F, G, or AG specific LTRs with a subtype B LAI backbone. SupT1 cells were infected with these clone variants and cultured in the presence of a fusion inhibitor to prevent further infection. Latency was measured 24 hours later after stimulation with TNF-α by assessing CA-p24 by flow cytometry and p24 release by an enzyme linked immunosorbent assay (ELISA) [76]. Using this acute HIV-1 model system, the authors found no differences in latency among subtypes with the exception of subtype AE, which had higher basal transcription and low responsiveness to TNF-α. This phenotype was proposed to be related to the presence of a GA-binding protein (GABP) site in the place of an NF-κB site within the subtype AE LTR [76].
Duverger et al. further explored how a subtype-specific LTR sequence may affect the establishment of latency. HIV-1 clones which had LTRs from subtypes A, B, C1, D, E, F, and G were cloned into HIV-1 LAI and used to infect the GFP reporter T cell line J2574. This cell line contains a subtype B NL4-3 LTR that controls the expression of GFP either in response to HIV-1 activating stimuli or HIV-1 tat. GFP was used as a surrogate of tat expression and transcriptional activity. Interestingly, they found that subtype A and subtype C LTR viruses generated higher levels of latent infections (assessed by phorbol myristate acetate or PMA stimulation of day 23 cultured cells), and the subtype E LTR virus had a reduced ability to establish latency [77]. Using transcription element search software, the authors found an AP-1 binding site that was extended by seven nucleotides in subtypes A and C, and absent in subtype E LTRs. They confirmed the relevance of this site by deleting the AP-1 site from NL4-3 and assessing latent infection in the J2574 cell line. Unexpectedly, the authors also found that viruses with a subtype B LTR or subtype A LTR responded similarly to TNF-α, hexamethylene bisacetamide (HMBA, a cell differentiating agent), suberoylanilide hydroxamic acid (SAHA, a histone deacetylase inhibitor), and trichostatin A (TSA, a histone deacetylase inhibitor). So, although subtype A generated a higher level of latent infections, the percentage of cells that could be reactivated was similar to subtype B even with strong stimuli such as TNF-α, suggesting that perhaps there are other mechanisms involved in maintaining latency in subtype A apart from epigenetic modification or NF-κB activation.
Recently, Dahabieh et al. used a doubly fluorescent virus system to distinguish between immediate silencing leading to latency (LTR-silent), or progressive latency due to epigenetic silencing [78]. The Red-Green-HIV-1 (RGH) virus encodes both enhanced green fluorescent protein (eGFP) and mCherry, a red fluorescent protein. The expression of mCherry is under a cytomegalovirus promoter and is constantly on, while eGFP is controlled by the HIV-1 LTR and can be packaged into virions during active infection. Thus, cells that are productively infected are either yellow (due to the combination of red and green) or green, cells that are latently infected are red, and uninfected cells will not express either fluorescent protein. Subtype-specific LTRs (subtypes B, A, C, D, AE, F, and G) were cloned into the RGH vector and used to infect Jurkat cells. The authors found that the ability to establish latency was different among subtypes, where subtype D and subtype F were more prone to LTR silencing than other subtypes, whereas subtype AE was less prone to latency.
Taken together, these studies highlight differences in establishment of latency that are attributable to the viral LTR. Since these studies only replace the LTR and keep all other subtype B viral proteins, this effect may be more pronounced in a system with complete subtype-specific genomes. The general consensus from these studies is that subtype AE is more transcriptionally active and less latent. This may be due in part to the GABP site present in the subtype AE LTR. Most of the studies to date have been done in cell lines. As such, further studies are warranted to evaluate whether these differences are also observed either in primary cell models or in cells isolated from HIV-infected participants ex vivo.
5. HIV-1 Viral Proteins and Latency
The role of HIV-1 viral proteins in HIV-1 pathogenesis and immune evasion has been extensively studied with mostly subtype B, and these studies have been extended to non-B subtypes and reviewed elsewhere. Here we review and discuss the potential role of HIV-1 proteins in the establishment or reactivation of latency.
Once HIV-1 is integrated into the host genome, the specific site of integration can have a dramatic impact on the persistence of latently infected cells. This can occur, for example, by integration into genes that are typically associated with cancer or cell cycle such as BACH2 or MKL2 [79,80]. These previous studies associating the integration site and persistence of the latent reservoir were likely with subtype B viruses due to the geographic location of the studies, and to our knowledge, this has not been extended to other subtypes. The genetic diversity of the HIV-1 integrase protein has obvious implications when it comes to integrase-inhibitor resistance development [81,82], but whether this leads to differential preference in the integration site has not been shown. The sequence of integrase is mostly (60%) conserved across subtypes in key catalytic domains to preserve general functionality, though there are several regions of the protein that are less conserved (extensively reviewed in [83]). In a study by Demeulemeester et al., naturally occurring HIV-1 integrase variants were found among subtypes A, B, C, D, F, and G. The study found that the variant INS119G re-targeted viral integration from gene-rich areas to gene-poor regions, characterized by heterochromatin and H3K9 bi and trimethylation. The abundance of these variants was disproportionate among subtypes, where the global re-targeting variants INS119G occurred most in subtype B, and INR231G in subtype C. Interestingly, these variants were associated with faster disease progression in a chronic infection subtype C cohort [84]. Since the prevalence of these integrase variants differs among subtypes in this study, this could conceivably affect integration site patterns from subtype to subtype, thus impacting latency through this mechanism. However, these findings have not been confirmed by others and represent rare variants of the integrase protein. Further work needs to be done to determine whether the integration site differs due to subtype, or if the findings with subtype B integration sites in cancer-associated genes can be extended to other subtypes.
During infection, subversion of the host’s immune system is vital to the virus’ survival. As such, HIV-1 has numerous viral proteins to achieve this immune evasion. HIV-1 viral infectivity factor (Vif) is a viral protein known to counteract host antiretroviral protein apolipoprotein B mRNA editing enzyme catalytic subunit 3G (APOBEC3G) [85]. APOBEC3G deaminates viral cDNA which causes premature stop codons or amino acid substitutions, rending the virus defective. Vif counteracts this host restriction factor by targeting it for degradation, thus preventing APOBEC3G antiviral functions [86]. Lisovsky et al. showed that subtype-specific Vif function affected viral replication in the presence of APOBEC3G in a Tzm-Bl luciferase reporter assay [87]. Subtypes B, C, and AG Vif sequences were used to complement subtype-specific ΔVif virus backbones and transfected into 293T cells in the presence or absence of APOBEC3G. The resulting virus generated was used to infect Tzm-Bl cells, and viral replication was measured using luciferase. Interestingly, subtype C was less functional and more sensitive to APOBEC3G-mediated viral inhibition, compared to subtype B or subtype AG in this system. Since subtype C Vif was not as effective at counteracting APOBEC3G, the viral genome was hypermutated by APOBEC3G, thus limiting the ability of the virus to infect and subsequently replicate. Because most of the latent reservoir found in patients contain defective and hypermutated proviruses [88], it is tempting to speculate that subtype-specific vif could influence the establishment of an intact and inducible latent reservoir.
The HIV-1 viral protein R (Vpr) is involved in the transport of the pre-integration complex to the nucleus, modulation of cell death, cell cycle arrest, and LTR-transctivation [85,89]. Vpr has been found highly conserved in subtype B latent proviruses isolated from ART-treated individuals, suggesting a selective advantage to preserve the vpr reading frame [90]. However, whether this plays a role in HIV-1 latency is not clear. Regarding its transactivation potential, subtype B has been shown to more potently transactivate the HIV-1 LTR than subtype C [91]. However, how this influences either the establishment of latency or reactivation has not been characterized.
HIV-1 viral protein U (Vpu) also plays a critical role in downmodulating host immune responses against HIV-1, such as counteracting Tethrin and downregulating CD4 [85,92]. Recently, it has been shown that Vpu potently inhibited NF-κB at the transcriptional level and prevented immune responses against HIV-1 [93]. In this study, subtype C and subtype B viruses, which were either wildtype for Vpu or lacked Vpu, were used to infect primary CD4 T cells. Infected cells were subjected to RNA-Seq. Transcription factor network analyses showed suppression of NF-κB target genes in the presence of Vpu, suggesting a role of Vpu in the establishment of latency.
Nef is one of the most studied HIV-1 accessory proteins, likely due to its prominent role in the host immune response to HIV-1 and pathogenesis [85]. An early study examined 41 viruses which represented subtypes A, B, C, E, F, G, and H and characterized their Nef sequence. Through phylogenetic analysis, they found that the distance between strains within the same subtype to be 9.6%, and the distance between subtypes to be 20.4% [94]. The impact of these sequence variations so far has been evaluated in vitro. Mann et al. examined over 300 single HIV plasma RNA Nef clones from ART-naïve chronically infected patients that were almost equal parts subtypes A, B, C, or D. They transfected cloned Nef sequences into an immortalized CD4 T cell line and measured CD4 and human leukocyte antigen (HLA) class I surface expression by flow cytometry. Interestingly, they found that with regard to CD4 and HLA I downregulation, subtype B was the most functional, followed by subtypes A or D, and lastly subtype C [95]. The authors also found substantial inter-subtype sequence diversity, which could potentially explain inter-subtype functional differences. In another publication, Mann et al. explored the underlying mechanisms of this functional difference between subtypes and examined genetic determinants of Nef function in subtypes B and C, the most functionally disparate subtypes [96]. Using the same experimental setup of transfecting Nef sequences into an immortalized T cell line and examining CD4 and HLA class I surface expression by flow cytometry, they mutated specific residues in NL4-3 (subtype B) Nef to examine the impact of subtype-specific mutations on Nef function. They found sequence variations at codons 20 and 88 significantly affected Nef’s ability to downregulate CD4 and HLA-I [96]. Mutations at codon 88, specifically S88G mutation, is significantly more frequent in subtype C viruses, which could partially explain the functional differences observed between subtypes. Nef has also been shown to reactivate latent HIV from latency in cell lines [97,98,99], but whether these subtype-specific differences directly impact latency is not well established.
There are other experimental evidences suggesting that some accessory genes may play a role in HIV-1 latency, though these studies have been mostly with subtype B. Recently, the group of Susanna Valente has reported that mutations in both Nef and Vpr proteins that enhance their ability to activate NF-κB conferred resistance to the Tat inhibitor didehydro-cortistatin A (dCA) [100]. dCA is an alkaloid shown to repress HIV-1 reactivation from latency and promote long-term latency [101,102]. Furthermore, the presence of accessory genes may influence the establishment of latent infections in a primary cell model that uses fetal thymocytes [103]. The genetic sequence of accessory proteins can vary from 5% to 35% by subtype, potentially affecting function [104,105]. Thus, we speculate that perhaps non-B viral proteins may have varying degrees of impact on HIV latency. Further studies using non-subtype B viruses are needed to address this possibility.
6. The Size of the Latent Reservoir
Most of the studies evaluating the size, long-term dynamics, and composition of this latent reservoir in vivo have been done in subtype B individuals, though a few recent studies have examined the reservoir size in non-B participants. For a summary of these studies thus far, see Table 3. Two of these studies have examined the size of the replication-competent, inducible latent reservoir in ART-treated virally suppressed non-B individuals. In the first study, Prodger et al. examined the size of the latent viral reservoir in a Ugandan cohort and compared it to a previously published Baltimore cohort. Interestingly, they found that long-term virally suppressed individuals with non-B subtypes (subtypes A, D, and A/D recombinant) had a 3-fold lower frequency of inducible latently infected resting CD4 T cells as measured by the quantitative viral outgrowth assay (QVOA) [106]. The authors could not attribute this difference to subtype due to the Ugandan cohort having subtypes A, D, or A/D while the Baltimore cohort was exclusively subtype B. The majority (69%) of individuals in the Ugandan cohort were subtype D, which was shown to be more prone to latency than other subtypes in an in vitro model of latency [78]. This could be a potential explanation as to why the Ugandan cohort had a lower measured reservoir since the QVOA involves reactivating the cells in vitro. Alternatively, an unknown mechanism that may involve viral-intrinsic or host-intrinsic factors may be influencing the establishment of latency in this population.
In the second study, Omondi et al. assessed the viral reservoir in a smaller cohort of early-treated individuals (subtypes B, AE and G) and found a correlation between subtype-specific Nef function and reservoir size [107]. This study measured the size of the latent HIV-1 reservoir using QVOA from acute/early treated individuals and also cloned viral Nef sequences to assess Nef function using an in vitro flow cytometric assay. They found that subtype B Nef was most effective at downregulating CD4 and HLA. These results are in line with the previous observation that subtype B Nef is most functional compared to other subtypes [95,96]. The authors reasoned that Nef function could be allowing infected cells to evade HLA-mediated immune responses, thus explaining the larger reservoir size of subtype B individuals in this cohort. However, the complete mechanism underlying this phenotype of a reduced size of the latent reservoir could not be solely attributed to Nef function [107]. The results from these studies can be interpreted in one of two ways. First, individuals with non-B subtype viruses potentially establish latency at a reduced frequency than subtype B, due to differential immune responses or functionality of viral proteins. Perhaps making the argument that subtype B is more evolutionarily fit to persist through viral latency in unsuitable cellular conditions. Second, it could be that non-B subtypes achieve a deeper state of latency, perhaps due to changes in integration sites, methylation of viral promoter, or LTR sequence, making them less inducible than subtype B. Further work needs to be done to understand the mechanisms behind this reduction of inducible reservoir observed in these two studies.
In the third study, the long-term dynamics of the viral reservoir were measured in non-B individuals. In a large cohort study of 1057 ART-suppressed individuals, Bachmann et al. evaluated host and viral characteristics and whether any of these variables were associated with reservoir size [108]. The viral reservoir was determined by total HIV-1 DNA after 1.5 years of ART initiation. The initiation of ART within a year of infection was associated with the size of the reservoir. This finding confirms other studies where this was also observed [109,110]. They found that decay of the latent reservoir was significantly faster in non-B individuals, compared to subtype B individuals in a multivariable model. HIV-1 subtype was only associated with a smaller reservoir in univariable analysis, and this significant difference did not persist in multivariable analysis, though there was a trend. Interestingly, this study also found that ethnicity affected reservoir size. Non-white individuals were associated with a lower reservoir size. As distinct subtypes circulate in separate regions of the world, ethnicity is an important variable to consider. Using a Wilcoxon rank sum test, the authors found that subtype C individuals had lower total levels of HIV-1 DNA than subtype B, and that this difference was still observed after controlling for ethnicity.
To our knowledge, these are the only studies comparing the size of the reservoir of non-subtype B to subtype B. Though the studies are quite dissimilar in cohort sizes, subtypes included, and cohort type (long term vs. early treated), two of the three report the same finding: non-B individuals have a reduced frequency of latently infected cells that harbor replication-competent virus. The third study found a trend towards this phenotype in their cohort of individuals who were on ART for 1.5 years, suggesting that subtype was not associated with reservoir size. One potential explanation for this opposing conclusion could be that the polymerase chain reaction (PCR)-based assays and the QVOA used to measure the reservoir do not correlate very well [111]. Total HIV-1 DNA may over-estimate the size of the latent reservoir compared to the QVOA [111]. The large majority of integrated proviruses are defective [112,113], which are detected in PCR assays but not in the QVOA. The QVOA assay has drawbacks as well, as it can underestimate the true size of the reservoir, as a subset of these non-induced viruses are intact and indeed replication-competent [112]. Clearly, the development of better methods to measure the latent reservoir, in addition to expanding our understanding of the dynamics affecting the latent reservoir in non-B individuals, is greatly needed. With that said, if non-B subtype is indeed associated with smaller reservoir size, this can have dramatic implications for cure strategies, as a cure strategy may be more feasible with individuals who have smaller latent reservoirs. Additionally, understanding the mechanisms that are responsible for this reduction can help with efforts to reduce or preclude the establishment of latency.
The complete mechanisms that affect the size of the latent reservoir in both subtype B and non-B infections are incompletely understood but there are several known factors that can contribute, such as the lack of host transcription factors, the lack of viral tat, and epigenetic silencing (reviewed in [5]). Furthermore, it is well accepted that the timing of initiation of ART can also limit the establishment of the latent reservoir [109,110,114,115]. The host immune response has also been associated with the size of the latent reservoir. It has been shown that there is an association between early immune responses and the size of the latent reservoir in a small cohort of early treated individuals [114]. Yue et al. found that granzyme B T cell responses against Tat/Rev, Env, Gag, and Vif negatively correlated with HIV-1 proviral loads and size of the latent reservoir (measured by QVOA) at both the beginning of the study and 48 weeks [114]. These findings suggest that initial immune responses to HIV-1 may prevent the establishment of the latent reservoir. Though the subtype of the individuals was not included, study participants were recruited in Toronto, Ontario, Canada and are likely to be subtype B. To our knowledge, these observations on immune responses and reservoir size have not been extended to other subtypes; however, there is some limited data on the quality of CD8 T cell responses in non-B subtypes. In a study by Baker et al., CD8 T cells from 56 HIV-1-infected participants (which were either subtypes A or D) were stimulated with consensus subtype A or D Gag peptides ex vivo. The subsequent CD8 T cell response, defined in this study by Interferon-γ (IFN-γ) production and CD107a/b (a degranulation marker), was measured by flow cytometry. The authors found that IFN-γ production was not significantly different between the two subtypes but observed a significant reduction in CD107 expression in individuals infected with subtype D compared to subtype A. These data suggest that distinct subtypes can elicit varying CD8 T cell responses. The molecular mechanisms underlying this difference in immune response have been somewhat evaluated in vitro.
There is also some limited in vitro evidence that the nucleotide composition of HIV-1 affects type-I Interferon responses, as HIV-1 has a different average composition of amino acids than human sequences. Vabret et al. found that nucleotide divergence was correlated with type-I Interferon response. Among subtypes A, B, C, D, F, and G, subtype D had the highest nucleotide divergence, which was correlated with subtype D’s increased pathogenicity [116]. Thus, the general composition of nucleotides in HIV-1’s genome may play a part in immune responses. In addition to nucleotide composition, viral protein function can also vary by subtype [47,87,91,95,117,118,119]. Because of these two factors, one can speculate that the immune responses generated against different subtypes may vary, potentially impacting the generation of the latent reservoir through this mechanism. The relationship between non-B subtypes, immune responses elicited against them, whether or not they differ in quality or quantity, and the size of the latent reservoir has yet to be proven. This area of research requires further investigation to understand HIV-1 latency in the non-B subtype, which is extremely relevant as the majority of the worldwide HIV-1 infections are with non-B subtypes [18,21,22,23].
7. Latency Reversal—The “Shock”
“Shock and kill” strategies are based on eliciting viral gene expression from latently infected cells using an LRA, thus allowing for the elimination of latently infected cells by virus-mediated cytopathicity or immune-mediated removal by NK or CD8 T cells. LRAs are small molecules that can induce viral gene expression in latently infected cells (for reviews in LRAs see [120,121,122]). Thus far, the pre-clinical screening and development of LRAs have used in vitro systems with subtype B HIV-1 viruses. There is an assumption that pre-clinical LRAs that are effective with subtype B will be just as effective for all other subtypes. However, given that there are substantial sequence differences in LTR [66,67,68,69,76,123,124], and over 80% of the world is not subtype B [23], this is an area of research that requires further attention. HIV-1 cure strategies may require subtype-specific approaches if they are not universally applicable. Below, we summarize several different classes of LRAs that have been developed thus far, and in what contexts they have been tested relevant to different HIV-1 subtypes. These studies are also summarized in Table 4.
7.1. Protein Kinase C (PKC) Agonists
PKC agonists mainly activate the transcription factor NF-κB. There are several isoforms of PKC, and their regulation can be independent or dependent on calcium and diacylglycerol [125]. PKC agonists can activate distinct isoforms of PKC, ultimately activating NF-κB but through slightly different ways. Under basal conditions, NF-κB is complexed with IκBα in the cytoplasm and this prevents NF-κB’s transcriptional activity (reviewed in [126,127]). Activation of PKC induces signaling that allows release from this inhibitory complex, allowing NF-κB to be activated and translocate to the nucleus. In the setting of HIV-1 infection, NF-κB can be recruited to NF-κB binding sites present in the viral LTR, thus appropriating NF-κB to promote its own transcription. There are several PKC agonists developed thus far, some of which have advanced to clinical trials (extensively reviewed in [15,121]).
The success of using NF-κB activation as a strategy to reactivate latent HIV-1 is impacted by whether the NF-κB binding sites exist in the LTR. Subtypes A, B, and D harbor two, subtype AE harbors one, and subtype C typically harbors three to four NF-κB binding sites [66,73]. The effect that these additional NF-κB sites has on latency reversal has been somewhat evaluated in cell lines, with a first study finding no difference in latency reversal with the exception of recombinant subtype AE [76] and a second study finding differences with several subtypes [71].
Prostratin is a non-tumor promoting phorbol ester that reactivates in both cell lines and ex vivo patient cells [128,129]. Prostratin, alone or in combination with the HDAC inhibitor vorinostat (also known as SAHA) was examined in one study across several subtypes [71]. Using both a lentiviral latency system and a primary cell latency model, Burnett et al. explored the latency profiles of subtypes A, A2, B, C, C’, D, F, H and recombinant subtypes A/G, B/C and B/F [71]. Using the LTR-GFP-IRES-Tat (LGIT) virus system in Jurkat cells, the study evaluated single and combinatorial latency reversal strategies involving PKC agonists and epigenetic drugs. Interestingly, subtype C exhibited the weakest response, while subtypes A and A/G, B/F, and F exhibited strong responses. These studies were extended to a more biologically relevant cell type, primary CD4 T cells, using the same LGIT virus system, and similar patterns were observed. These data suggest differences in the magnitude of responses that are solely attributable to subtype-specific LTR differences as the LGIT virus does not differ in other viral genome elements, and that some subtypes may be more amenable to these single or combinatorial agents for latency reversal.
In the last decade, other PKC agonists have been described, such as bryostatin-1 [130], ingenol dibenzoate [131,132], and ingenol mebutate (also known as Ingenol 3-angelate) [133,134]. Other semi-synthetic ingenols have been developed [64]. These agonists have been shown to be potent reactivators of latent HIV-1 with subtype B but have not been evaluated against non-subtype B viruses, with the exception of a study with ingenol B. Jurkat cells were transfected with subtype B and subtype C LTRs (some of which had 3 NF-κB or 4 NF-κB sites) and stimulated with various PKC-activating compounds such as TNF-α, PMA, or ingenol B. They observed that subtype C LTRs with 4 NF-κB sites had the greatest response. However, this study was limited to cell lines and transient transfection of LTR constructs. Further work needs to be done in other systems that better recapitulate latency in order to confirm these findings. With that said, this study highlights how the presence of an additional NF-κB site can enhance the response to NF-κB-activating stimuli.
All these PKC agonists activate the general PKC-NF-κB signaling axis. So, it can be speculated that some subtypes may be more amenable to latency reversal through the PKC-NF-κB signaling axis. Further studies are necessary to confirm these results using primary cell models and ideally latently infected cells from well-suppressed HIV-1-infected participants.
7.2. Toll-Like Receptor Agonists
Toll-like receptors (TLRs) are pattern recognition receptors that play critical roles in detecting microbial pathogens [135]. They are localized at either the cell membrane (TLR-1, -2, -6, -4, -5) or in endosomes (TLR-3, -7, -8, -9) (for a review, see [16,136]). Our lab has shown that TLR-1/2 agonist Pam3CSK4, TLR-2/6 agonist Pam2CSK4, and dual TLR-2/7 agonists reactivate latency in a primary cell model of latency and ex vivo patient cells in a NF-κB-dependent manner [137,138]. MGN-1703 is another TLR agonist that reactivates latent HIV-1 through TLR-9 [139]. There have been several exciting LRAs under this class that have moved onto clinical trials, one of which being GS-9620 (vesatolomid). GS-9620 is a TLR-7 agonist that has been shown to reactivate latent HIV-1 from aviremic participants and improve effector cell functions [140]. Currently, clinical trials are underway to determine the safety of GS-9620, in addition to other secondary measures such as time-to-rebound following an analytical treatment interruption (NCT03060447, NCT02858401). Due to the geographical location, the clinical trial participants will likely be subtype B. To the best of our knowledge, TLR ligands have not been evaluated against non-B subtypes although, making the same speculation based on NF-κB sites, there may be varying degrees of efficacy using this class of LRAs.
7.3. Non-Canonical NF-κB Agonists
NF-κB is part of a family of transcription factors that can form hetero or homo dimers composed of RelA/p65, c-Rel, RelB, p50 or p52 subunits. The composition of NF-κB has differential effects on transcription [126]. Homodimers of p50 or p52 can act as transcriptional repressors since they do not contain a transactivating domain, which is known to be critical for transcriptional activity. Second mitochondrial-derived activator of caspases (SMAC) mimetics mimic the tetrapeptide region of SMAC, which binds to the baculoviral inhibitor of apoptosis protein repeat (BIR) domain. These BIR domains are present in inhibitor of apoptosis (IAP) family member proteins, including the baculoviral IAP repeat containing-2 (BIRC2) protein. This binding to BIRC2 prevents the degradation of the NF-κB inducing kinase (NIK), allowing for the accumulation of NIK and induction of p100 processing into p52 [126]. Using an RNA interference screen in HEK293T cells using vesicular stomatitis virus-pseudotyped NL4-3 (a subtype B virus), it was found that BIRC2/cIAP1 negatively regulates HIV-1 transcription and its pharmacological inhibition reactivated latent HIV-1 [141]. They found that this reactivation occurred through the non-canonical NF-κB pathway. Sampey et al. found that AZD5582, a SMAC mimetic, also reactivated latent HIV-1 through the non-canonical NF-κB pathway. AZD5582 has been tested in ex vivo resting CD4 T cells from aviremic participants [142] and in vivo mouse models and non-human primates [143]. To the best of our knowledge, this has not been tested with other HIV-1 subtypes apart from subtype B, and SIVmac239. These compounds may also have varying effects on viral reactivation, likely dependent on the subtypes evaluated.
7.4. STAT Modulators
The study of STAT binding sites across subtypes has been somewhat limited. Crotti et al. examined the LTR of subtypes A, B, C, D, F, G, AE, AG, and found differences in putative STAT binding sites. Further, they found that in a U937 reporter system, the subtype G LTR was most responsive to phosphorylated STAT5, the activated form of the transcription factor [65]. This could be pertinent to the development of HIV-1 cure strategies which employ STAT activation as a method of reversing latency [63], as this study suggests some subtypes would be more responsive to STAT5-mediated reactivation. Thus far, STAT modulators have been tested in subtype B contexts, in vitro. Further studies are needed to confirm whether different subtypes possess the same sensitivity to latency reversal through the STAT5 signaling pathway.
7.5. Epigenetic Modifiers and PTEF-b Release Agents
Epigenetic modification is another critical factor of viral latency (reviewed extensively in [5,144]). There are several types of epigenetic modifiers, including bromodomain and extra terminal (BET) inhibitors, DNA and histone methyltransferase (DMT, HMT, respectively) inhibitors, and HDAC inhibitors. Many of these epigenetic modifiers have anti-cancer properties [145,146,147] in addition to their HIV-1 latency-reversing activity [148,149,150,151,152,153,154,155,156].
Repressive epigenetic marks such as methylation can be alleviated with HMTs, and lack of chromatin accessibility can be remedied with HDAC inhibitors. These strategies have been shown to be effective for reversing latency in both in vitro latency models and ex vivo HIV-1-positive participant cells [150,151,152,153]. Within HDAC inhibitors, some drugs can affect all HDACs (pan-HDAC inhibitors such as SAHA), as well as selective class 1 HDAC inhibitors like MS-275 (entinostat). Entinostat has been evaluated as a latency-reversing agent (either as a single agent or in combination with other LRAs) in latent cell models [153].
Thus far, the HDAC inhibitor vorinostat (SAHA) and valproic acid have advanced the farthest in clinical trials, but studies have shown it has not had an impact on the size of the latent reservoir in clinical trials as a single agent [152,157,158]. Importantly, these studies were conducted in Europe/United States, meaning that the study population was likely subtype B. DNA methyltransferas inhibitors such as 5’Azacytidine have been developed as LRA [150], though the role of DNA methylation in viral latency remains controversial [159,160]. Thus far, studies that have examined the efficacy of these epigenetic modifiers have been limited to subtype B, with the exception of Vorinostat [71]. Vorinostat did not meaningfully reactivate latent HIV-1 in both cell lines and primary cells alone, but in combination with Prostratin showed varying efficacy across subtypes A, A2, B, C, C’, D, F, H and recombinant subtypes A/G, B/C and B/F [71] (Table 4). Apart from this study, it has not been examined whether some subtypes are more or less sensitive to epigenetic modifiers either alone or in combination with other LRAs.
For HIV-1 latency reversal, repressive chromatin environments (methylated, lack of acetylation) can limit transcription initiation, but transcription initiation is not the only block for efficient viral reactivation [161]. Transcription elongation also has to occur for a successful reactivation. P-TEFb is normally sequestered in a transcriptionally inactive complex, the 7SK small ribonucleoprotein, and P-TEFb is critical for promoting elongation of transcripts through RNA polymerase II phosphorylation (reviewed in [162,163]). Release from this complex by small molecules or drugs allows P-TEFb to bind to HIV-1 tat to promote HIV-1 transcript elongation. However, there is competition for this binding as P-TEFb can also be bound by host BET proteins [164]. It has been shown that BET inhibitors such as JQ-1, can reactivate latency by releasing P-TEFb or interfering with BRD4 binding to P-TEFb, thus allowing for Tat binding [148,155,156]. To our knowledge, this class of LRA has not been tested in systems that use non-B subtype viruses. As Tat transactivation potential differs by subtype [68], it is possible that bromodomain inhibitors may be more effective in some subtypes than others. Further work is necessary to confirm whether this is true.
7.6. Enhancing Viral Reactivation with Combination Treatments
LRAs that have been developed thus far are not exceptionally potent as single agents, compared to in vitro maximal activating stimuli such as T cell receptor engagement [165]. This issue can be potentially solved by combining distinct classes of LRAs to hopefully synergize and achieve the maximal viral reactivation without overt toxicity. Recent work by Laird et al. have identified a few combinations of LRAs that are effective at reversing latency in ex vivo HIV-1-infected participant cells, namely PKC agonists combined with bromodomain inhibitors or HDAC inhibitors [166]. There is limited data on whether the combination of LRAs could also be effective in non-B subtypes. A study by Burnett et al. included prostratin, a PKC agonist, and several epigenetic modifiers such as SAHA and HMBA. They found that the greatest responses were produced when the LRAs were used in combination, though not all subtypes were equally responsive to the combination [71]. Even so, the viral reactivation obtained from a combination was greater than single-agent treatment for the majority of subtypes.
Interestingly, checkpoint inhibitors have also been combined with LRAs to increased viral reactivation. It has been shown that checkpoint inhibitors alone have limited effects ex vivo on HIV-1 reactivation in HIV-1-infected participant cells [167]. However, the checkpoint molecule programmed cell death-1 (PD-1) plays a role in suppressing viral reactivation [168]. Fromentin et al. showed that PD-1 engagement inhibited viral production and blocked T cell receptor engagement-mediated viral reactivation in ex vivo HIV-1-infected participant cells. They further showed that combination treatment with an anti-PD-1 antibody and potent PKC agonist bryostatin-1, latency reversal is enhanced without deleterious effects of T cell activation [168]. In this study, the subtype of the participants is not denoted but are from countries in North America, so it is likely that the majority of participants were subtype B. These studies have not been extended to other subtypes, but it is a novel and interesting path to potentially enhancing latency reversal with the traditional LRAs.
8. Future Perspectives
Despite the progress in our general knowledge of HIV-1 infection and HIV-1 latency, there still remains much to be explored with regard to the study of non-subtype B viruses in both model systems and in vivo studies. Thus far, it is clear that there are differences in viral pathogenesis, viral protein function, and some early studies in vivo showing that the size of the latent reservoir differs in subtype B from non-subtype B infected individuals. The mechanisms underlying the reduced reservoir in non-subtype B infected individuals in vivo are likely multifaceted. As the field makes advances towards developing more effective LRAs or combinations of LRAs, the matter of subtype should not be forgotten. Availability of infectious molecular clones of non-B subtypes can facilitate their study in in vitro systems for pre-clinical development of LRAs. Thus far, work in cell lines has given some credence to the idea that viral subtype may impact the establishment of latency or the ability to reactivate from latency. These studies should be extended to primary cells with viruses that encode all viral accessory genes and are replication-competent. For pre-clinical LRA development, primary cell models with non-B viruses can also be used to test efficacy, and ideally non-B ART-suppressed HIV-1-infected participant cells. Primary in vitro systems can also be used to investigate the mechanisms responsible for the establishment of latent infections. Further work needs to be done to show whether subtype is a factor in viral latency in order to develop a global and relevant cure strategy.
Author Contributions
Conceptualization, I.S. and A.B.; writing—original draft preparation, I.S. and A.B.; writing—review and editing, I.S. and A.B..; supervision, A.B..; funding acquisition, I.S. and A.B.
Funding
I.S. is supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number F31AI147814. The work in A.B.’s lab is currently supported by the National Institute of Allergy and Infectious Diseases and National Institute of Health grants R01-AI124722, R21/R33-AI116212, R01-AI147845, UM1-AI126617 and P30-AI117970.
Acknowledgments
We would like to thank Ryan Durga for his careful proofreading of the manuscript. We apologize to those scientists whose outstanding work was not cited or was not directly cited in the text but only referred through reviews.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on HIghly Active Antiretroviral Therapy. Science 1997, 278. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Sierra, S.; Kupfer, B.; Kaiser, R. Basics of the virology of HIV-1 and its replication. J Clin Virol 2005, 34, 233–244. [Google Scholar] [CrossRef]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Hill, B.J.; Ambrozak, D.R.; Price, D.A.; Guenaga, F.J.; Casazza, J.P.; Kuruppu, J.; Yazdani, J.; Migueles, S.A.; Connors, M.; et al. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: Implications for HIV pathogenesis. J. Virol. 2004, 78, 1160–1168. [Google Scholar] [CrossRef]
- McNamara, L.A.; Ganesh, J.A.; Collins, K.L. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-kappaB activation. J. Virol. 2012, 86, 9337–9350. [Google Scholar] [CrossRef] [PubMed]
- Venanzi Rullo, E.; Cannon, L.; Pinzone, M.R.; Ceccarelli, M.; Nunnari, G.; O’Doherty, U. Genetic evidence that Naive T cells can contribute significantly to the HIV intact reservoir: Time to re-evaluate their role. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Deeks, S.G. Immunologic strategies for HIV-1 remission and eradication. Science 2014, 345, 169–174. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting Cellular and Tissue HIV Reservoirs With Toll-Like Receptor Agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef]
- Hemelaar, J. Implications of HIV diversity for the HIV-1 pandemic. J. Infect. 2013, 66, 391–400. [Google Scholar] [CrossRef]
- Hsu, D.C.; O’Connell, R.J. Progress in HIV vaccine development. Hum. Vaccin. Immunother. 2017, 13, 1018–1030. [Google Scholar] [CrossRef]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The Challenge of HIV-1 Subtype Diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S.; WHO-UNAIDS Network for HIV Isolation and Characterisation. Global trends in molecular epidemiology of HIV-1 during 2000-2007. AIDS 2011, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006, 20, W13–W23. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.T.; Hackett, J., Jr.; Holzmayer, V.; Hillyard, D.R. Large-scale analysis of the prevalence and geographic distribution of HIV-1 non-B variants in the United States. J. Clin. Microbiol. 2013, 51, 2662–2669. [Google Scholar] [CrossRef]
- Oster, A.M.; Switzer, W.M.; Hernandez, A.L.; Saduvala, N.; Wertheim, J.O.; Nwangwu-Ike, N.; Ocfemia, M.C.; Campbell, E.; Hall, H.I. Increasing HIV-1 subtype diversity in seven states, United States, 2006-2013. Ann. Epidemiol. 2017, 27, 244–251 e241. [Google Scholar] [CrossRef] [PubMed]
- Delgado, E.; Benito, S.; Montero, V.; Cuevas, M.T.; Fernandez-Garcia, A.; Sanchez-Martinez, M.; Garcia-Bodas, E.; Diez-Fuertes, F.; Gil, H.; Canada, J.; et al. Diverse Large HIV-1 Non-subtype B Clusters Are Spreading Among Men Who Have Sex With Men in Spain. Front. Microbiol. 2019, 10, 655. [Google Scholar] [CrossRef]
- Yamaguchi, J.; McArthur, C.; Vallari, A.; Sthreshley, L.; Cloherty, G.A.; Berg, M.G.; Rodgers, M.A. Complete genome sequence of CG-0018a-01 establishes HIV-1 subtype L. J. Acquir. Immune Defic. Syndr. 2019. [Google Scholar] [CrossRef]
- Alaeus, A.; Lidman, K.; Bjorkman, A.; Giesecke, J.; Albert, J. Similar rate of disease progression among individuals infected with HIV-1 genetic subtypes A-D. AIDS 1999, 13, 901–907. [Google Scholar] [CrossRef]
- Amornkul, P.N.; Tansuphasawadikul, S.; Limpakarnjanarat, K.; Likanonsakul, S.; Young, N.; Eampokalap, B.; Kaewkungwal, J.; Naiwatanakul, T.; Von Bargen, J.; Hu, D.J.; et al. Clinical disease associated with HIV-1 subtype B’ and E infection among 2104 patients in Thailand. AIDS 1999, 13, 1963–1969. [Google Scholar] [CrossRef]
- Galai, N.; Kalinkovich, A.; Burstein, R.; Vlahov, D.; Bentwich, Z. African HIV-1 subtype C and rate of progression among Ethiopian immigrants in Israel. Lancet 1997, 349, 180–181. [Google Scholar] [CrossRef]
- Kiwanuka, N.; Robb, M.; Laeyendecker, O.; Kigozi, G.; Wabwire-Mangen, F.; Makumbi, F.E.; Nalugoda, F.; Kagaayi, J.; Eller, M.; Eller, L.A.; et al. HIV-1 viral subtype differences in the rate of CD4+ T-cell decline among HIV seroincident antiretroviral naive persons in Rakai district, Uganda. J. Acquir. Immune. Defic. Syndr. 2010, 54, 180–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeten, J.M.; Chohan, B.; Lavreys, L.; Chohan, V.; McClelland, R.S.; Certain, L.; Mandaliya, K.; Jaoko, W.; Overbaugh, J. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J. Infect. Dis. 2007, 195, 1177–1180. [Google Scholar] [CrossRef] [Green Version]
- Kaleebu, P.; French, N.; Mahe, C.; Yirrell, D.; Watera, C.; Lyagoba, F.; Nakiyingi, J.; Rutebemberwa, A.; Morgan, D.; Weber, J.; et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J. Infect. Dis. 2002, 185, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Ssemwanga, D.; Nsubuga, R.N.; Mayanja, B.N.; Lyagoba, F.; Magambo, B.; Yirrell, D.; Van der Paal, L.; Grosskurth, H.; Kaleebu, P. Effect of HIV-1 subtypes on disease progression in rural Uganda: A prospective clinical cohort study. PLoS ONE 2013, 8, e71768. [Google Scholar] [CrossRef] [PubMed]
- Vasan, A.; Renjifo, B.; Hertzmark, E.; Chaplin, B.; Msamanga, G.; Essex, M.; Fawzi, W.; Hunter, D. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin. Infect. Dis. 2006, 42, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venner, C.M.; Nankya, I.; Kyeyune, F.; Demers, K.; Kwok, C.; Chen, P.L.; Rwambuya, S.; Munjoma, M.; Chipato, T.; Byamugisha, J.; et al. Infecting HIV-1 Subtype Predicts Disease Progression in Women of Sub-Saharan Africa. EBioMedicine 2016, 13, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Amornkul, P.N.; Karita, E.; Kamali, A.; Rida, W.N.; Sanders, E.J.; Lakhi, S.; Price, M.A.; Kilembe, W.; Cormier, E.; Anzala, O.; et al. Disease progression by infecting HIV-1 subtype in a seroconverter cohort in sub-Saharan Africa. AIDS 2013, 27, 2775–2786. [Google Scholar] [CrossRef] [Green Version]
- Kanki, P.J.; Hamel, D.J.; Sankale, J.L.; Hsieh, C.; Thior, I.; Barin, F.; Woodcock, S.A.; Gueye-Ndiaye, A.; Zhang, E.; Montano, M.; et al. Human immunodeficiency virus type 1 subtypes differ in disease progression. J. Infect. Dis. 1999, 179, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 Coreceptors— Central to Understanding the Transmission and Pathogenesis of Human Immunodeficiency VIrus Type 1 Infection. AIDS Res. Hum. Retroviruses 2004, 20, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Esbjornsson, J.; Mansson, F.; Martinez-Arias, W.; Vincic, E.; Biague, A.J.; da Silva, Z.J.; Fenyo, E.M.; Norrgren, H.; Medstrand, P. Frequent CXCR4 tropism of HIV-1 subtype A and CRF02_AG during late-stage disease--indication of an evolving epidemic in West Africa. Retrovirology 2010, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitari, S.; Wang, Y.; Liu, S.L.; Liang, C. HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion. Viruses 2019, 11, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, R.M.; Shen, T.; Gnanakaran, S.; Derdeyn, C.A. Appreciating HIV type 1 diversity: Subtype differences in Env. AIDS Res. Hum. Retroviruses 2009, 25, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Tang, M.; Zhang, M.Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; et al. Structure of a V3-containing HIV-1 gp120 core. Science 2005, 310, 1025–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.S.; Boyle, T.J.; Lyerly, H.K.; Cullen, B.R. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 1991, 253, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.G.; Seillier-Moiseiwitsch, F.; Ahn, J.; Walker, J.M.; Swanstrom, R. Variability in the human immunodeficiency virus type 1 gp120 Env protein linked to phenotype-associated changes in the V3 loop. J. Virol. 2002, 76, 3852–3864. [Google Scholar] [CrossRef] [Green Version]
- Alaeus, A. Significance of HIV-1 genetic subtypes. Scand. J. Infect. Dis. 2000, 32, 455–463. [Google Scholar] [CrossRef]
- Tscherning, C.; Alaeus, A.; Fredriksson, R.; Bjorndal, A.; Deng, H.; Littman, D.R.; Fenyo, E.M.; Albert, J. Differences in chemokine coreceptor usage between genetic subtypes of HIV-1. Virology 1998, 241, 181–188. [Google Scholar] [CrossRef]
- Coetzer, M.; Cilliers, T.; Ping, L.H.; Swanstrom, R.; Morris, L. Genetic characteristics of the V3 region associated with CXCR4 usage in HIV-1 subtype C isolates. Virology 2006, 356, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Felsovalyi, K.; Nadas, A.; Zolla-Pazner, S.; Cardozo, T. Distinct sequence patterns characterize the V3 region of HIV type 1 gp120 from subtypes A and C. AIDS Res. Hum. Retroviruses 2006, 22, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Eshleman, S.H.; Toma, J.; Fransen, S.; Stawiski, E.; Paxinos, E.E.; Whitcomb, J.M.; Young, A.M.; Donnell, D.; Mmiro, F.; et al. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: High prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J. Virol. 2007, 81, 7885–7893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ataher, Q.; Portsmouth, S.; Napolitano, L.A.; Eng, S.; Greenacre, A.; Kambugu, A.; Wood, R.; Badal-Faesen, S.; Tressler, R. The epidemiology and clinical correlates of HIV-1 co-receptor tropism in non-subtype B infections from India, Uganda and South Africa. J. Int. AIDS Soc. 2012, 15, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Huang, Y.; He, T.; Cao, Y.; Ho, D.D. HIV-1 subtype and second-receptor use. Nature 1996, 383, 768. [Google Scholar] [CrossRef]
- Bednar, M.M.; Hauser, B.M.; Zhou, S.; Jacobson, J.M.; Eron, J.J., Jr.; Frank, I.; Swanstrom, R. Diversity and Tropism of HIV-1 Rebound Virus Populations in Plasma Level After Treatment Discontinuation. J. Infect. Dis. 2016, 214, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef] [PubMed]
- Grau-Exposito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badia, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.A.; Kwon, H.; Chen, L.F.; Greene, W.C. Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 2007, 81, 6043–6056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Pandelo Jose, D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Crotti, A.; Chiara, G.D.; Ghezzi, S.; Lupo, R.; Jeeninga, R.E.; Liboi, E.; Lievens, P.M.; Vicenzi, E.; Bovolenta, C.; Berkhout, B.; et al. Heterogeneity of signal transducer and activator of transcription binding sites in the long-terminal repeats of distinct HIV-1 subtypes. Open Virol. J. 2007, 1, 26–32. [Google Scholar] [CrossRef]
- Jeeninga, R.E.; Hoogenkamp, M.; Armand-Ugon, M.; de Baar, M.; Verhoef, K.; Berkhout, B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 2000, 74, 3740–3751. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.A.; Shen, C.; Ratner, D.; Paranjape, R.S.; Kulkarni, S.S.; Chatterjee, R.; Gupta, P. Genetic and functional characterization of the LTR of HIV-1 subtypes A and C circulating in India. AIDS Res. Hum. Retroviruses 2007, 23, 1428–1433. [Google Scholar] [CrossRef]
- Roof, P.; Ricci, M.; Genin, P.; Montano, M.A.; Essex, M.; Wainberg, M.A.; Gatignol, A.; Hiscott, J. Differential regulation of HIV-1 clade-specific B, C, and E long terminal repeats by NF-kappaB and the Tat transactivator. Virology 2002, 296, 77–83. [Google Scholar] [CrossRef] [Green Version]
- van Opijnen, T.; Jeeninga, R.E.; Boerlijst, M.C.; Pollakis, G.P.; Zetterberg, V.; Salminen, M.; Berkhout, B. Human immunodeficiency virus type 1 subtypes have a distinct long terminal repeat that determines the replication rate in a host-cell-specific manner. J. Virol. 2004, 78, 3675–3683. [Google Scholar] [CrossRef] [Green Version]
- Montano, M.A.; Novitsky, V.A.; Blackard, J.T.; Cho, N.L.; Katzenstein, D.A.; Essex, M. Divergent transcriptional regulation among expanding human immunodeficiency virus type 1 subtypes. J. Virol. 1997, 71, 8657–8665. [Google Scholar]
- Burnett, J.C.; Lim, K.I.; Calafi, A.; Rossi, J.J.; Schaffer, D.V.; Arkin, A.P. Combinatorial latency reactivation for HIV-1 subtypes and variants. J. Virol. 2010, 84, 5958–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, D.; Li, C.; Sang, F.; Li, Q.; Jiang, Z.Q.; Xu, L.R.; Guo, H.J.; Zhang, C.; Wang, J.H. The variances of Sp1 and NF-kappaB elements correlate with the greater capacity of Chinese HIV-1 B′-LTR for driving gene expression. Sci. Rep. 2016, 6, 34532. [Google Scholar] [CrossRef] [PubMed]
- Bachu, M.; Yalla, S.; Asokan, M.; Verma, A.; Neogi, U.; Sharma, S.; Murali, R.V.; Mukthey, A.B.; Bhatt, R.; Chatterjee, S.; et al. Multiple NF-kappaB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J. Biol. Chem. 2012, 287, 44714–44735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoef, K.; Sanders, R.W.; Fontaine, V.; Kitajima, S.; Berkhout, B. Evolution of the human immunodeficiency virus type 1 long terminal repeat promoter by conversion of an NF-kappaB enhancer element into a GABP binding site. J. Virol. 1999, 73, 1331–1340. [Google Scholar] [PubMed]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional intiation. EMBO J. 2006, 24, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Sluis, R.M.; Pollakis, G.; van Gerven, M.L.; Berkhout, B.; Jeeninga, R.E. Latency profiles of full length HIV-1 molecular clone variants with a subtype specific promoter. Retrovirology 2011, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Duverger, A.; Wolschendorf, F.; Zhang, M.; Wagner, F.; Hatcher, B.; Jones, J.; Cron, R.Q.; van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B.; et al. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 2013, 87, 2264–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahabieh, M.S.; Ooms, M.; Simon, V.; Sadowski, I. A doubly fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. J. Virol. 2013, 87, 4716–4727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Geretti, A.M.; Zahonero, N.; Booth, C.; Strang, A.; Soriano, V.; De Mendoza, C. Integrase variability and susceptibility to HIV integrase inhibitors: Impact of subtypes, antiretroviral experience and duration of HIV infection. J Antimicrob. Chemother. 2010, 65, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spira, S. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J. Antimicrob. Chemother. 2003, 51, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccherini-Silberstein, F.; Malet, I.; D’Arrigo, R.; Antinori, A.; Marcelin, A.G.; Perno, C.F. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev. 2009, 11, 17–29. [Google Scholar] [PubMed]
- Demeulemeester, J.; Vets, S.; Schrijvers, R.; Madlala, P.; De Maeyer, M.; De Rijck, J.; Ndung’u, T.; Debyser, Z.; Gijsbers, R. HIV-1 integrase variants retarget viral integration and are associated with disease progression in a chronic infection cohort. Cell Host. Microbe 2014, 16, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [Green Version]
- Lisovsky, I.; Schader, S.M.; Sloan, R.D.; Oliveira, M.; Coutsinos, D.; Bernard, N.F.; Wainberg, M.A. HIV-1 subtype variability in Vif derived from molecular clones affects APOBEC3G-mediated host restriction. Intervirology 2013, 56, 258–264. [Google Scholar] [CrossRef]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host. Microbe 2017, 21, 494–506 e494. [Google Scholar] [CrossRef] [Green Version]
- Romani, B.; Kamali Jamil, R.; Hamidi-Fard, M.; Rahimi, P.; Momen, S.B.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr reactivates latent HIV-1 provirus by inducing depletion of class I HDACs on chromatin. Sci. Rep. 2016, 6, 31924. [Google Scholar] [CrossRef]
- Shen, A.; Siliciano, J.D.; Pierson, T.C.; Buck, C.B.; Siliciano, R.F. Establishment of latent HIV-1 infection of resting CD4(+) T lymphocytes does not require inactivation of Vpr. Virology 2000, 278, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Bano, A.S.; Gupta, N.; Sood, V.; Banerjea, A.C. Vpr from HIV-1 subtypes B and C exhibit significant differences in their ability to transactivate LTR-mediated gene expression and also in their ability to promote apoptotic DNA ladder formation. Aids 2007, 21, 1832–1834. [Google Scholar] [CrossRef] [PubMed]
- Lindwasser, O.W.; Chaudhuri, R.; Bonifacino, J.S. Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr. Mol. Med. 2007, 7, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; De Jesus, P.D.; Herbert, K.M.; Pache, L.; Smith, N.; van der Merwe, J.A.; et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-kappaB-elicited antiviral immune responses. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Jubier-Maurin, V.; Saragosti, S.; Perret, J.L.; Mpoudi, E.; Esu-Williams, E.; Mulanga, C.; Liegeois, F.; Ekwalanga, M.; Delaporte, E.; Peeters, M. Genetic characterization of the nef gene from human immunodeficiency virus type 1 group M strains representing genetic subtypes A, B, C, E, F, G, and H. AIDS Res. Hum. Retroviruses 1999, 15, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.K.; Byakwaga, H.; Kuang, X.T.; Le, A.Q.; Brumme, C.J.; Mwimanzi, P.; Omarjee, S.; Martin, E.; Lee, G.Q.; Baraki, B.; et al. Ability fo HIV-1 Nef to downregulate CD4 and HLA class I differs among viral subtypes. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.K.; Omarjee, S.; Khumalo, P.; Ndung’u, T. Genetic determinants of Nef-mediated CD4 and HLA class I down-regulation differences between HIV-1 subtypes B and C. Virol. J. 2015, 12, 200. [Google Scholar] [CrossRef]
- Fujinaga, K.; Zhong, Q.; Nakaya, T.; Kameoka, M.; Meguro, T.; Yamada, K.; Ikuta, K. Extracellular Nef protein regulates productive HIV-1 infection from latency. J. Immunol. 1995, 155, 5289–5298. [Google Scholar]
- Wolf, D.; Witte, V.; Clark, P.; Blume, K.; Lichtenheld, M.G.; Baur, A.S. HIV Nef enhances Tat-mediated viral transcription through a hnRNP-K-nucleated signaling complex. Cell Host. Microbe 2008, 4, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.N.; Refaeli, Y.; Weiner, D.B. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J. Virol. 1995, 69, 1243–1252. [Google Scholar]
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host. Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsden, M.D.; Burke, B.P.; Zack, J.A. HIV latency is influenced by regions of the viral genome outside of the long terminal repeats and regulatory genes. Virology 2011, 417, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.T.; Allen, E.E.; Farmer, A.D.; Myers, G.L. Heterogeneity of HIV-1 and HIV-2. AIDS 1995, 9 (Suppl. A), S5–S18. [Google Scholar]
- Iordanskly, S.; Waltke, M.; Feng, Y.; Wood, C. Subtype-associated differences in HIV-1 reverse transcription affect the viral replication. Retrovirology 2010, 7, 85. [Google Scholar]
- Prodger, J.L.; Lai, J.; Reynolds, S.J.; Keruly, J.C.; Moore, R.D.; Kasule, J.; Kityamuweesi, T.; Buule, P.; Serwadda, D.; Nason, M.; et al. Reduced Frequency of Cells Latently Infected With Replication-Competent Human Immunodeficiency Virus-1 in Virally Suppressed Individuals Living in Rakai, Uganda. Clin. Infect. Dis. 2017, 65, 1308–1315. [Google Scholar] [CrossRef] [Green Version]
- Omondi, F.H.; Chandrarathna, S.; Mujib, S.; Brumme, C.J.; Jin, S.W.; Sudderuddin, H.; Miller, R.L.; Rahimi, A.; Laeyendecker, O.; Bonner, P.; et al. HIV Subtype and Nef-Mediated Immune Evasion Function Correlate with Viral Reservoir Size in Early-Treated Individuals. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, N.; von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; Kok, Y.L.; et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat. Commun. 2019, 10, 3193. [Google Scholar] [CrossRef] [Green Version]
- Ananworanich, J.; Dube, K.; Chomont, N. How does the timing of antiretroviral therapy initiation in acute infection affect HIV reservoirs? Curr. Opin. HIV AIDS 2015, 10, 18–28. [Google Scholar] [CrossRef]
- Archin, N.M.; Vaidya, N.K.; Kuruc, J.D.; Liberty, A.L.; Wiegand, A.; Kearney, M.F.; Cohen, M.S.; Coffin, J.M.; Bosch, R.J.; Gay, C.L.; et al. Immediate antiviral therapy appears to restrict resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection. Proc. Natl. Acad. Sci. USA 2012, 109, 9523–9528. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, F.Y.; Cohen, J.C.; Ho, M.; Rahman, A.; Liu, J.; Mujib, S.; Saiyed, A.; Hundal, S.; Khozin, A.; Bonner, P.; et al. HIV-Specific Granzyme B-Secreting but Not Gamma Interferon-Secreting T Cells Are Associated with Reduced Viral Reservoirs in Early HIV Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzón, M.J.; Yang, Y.; Ouyang, Z.; Sun, H.; Seiss, K.; Rogich, J.; Le Gall, S.; Pereyra, F.; Rosenberg, E.S.; Yu, X.G.; et al. Susceptibility to CD8 T-cell-mediated killing influences the reservoir of latently HIV-1-infected CD4 T cells. J. Acquir. Immune. Defic. Syndr. 2014, 65, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vabret, N.; Bailly-Bechet, M.; Najburg, V.; Muller-Trutwin, M.; Verrier, B.; Tangy, F. The biased nucleotide composition of HIV-1 triggers type I interferon response and correlates with subtype D increased pathogenicity. PLoS ONE 2012, 7, e33502. [Google Scholar] [CrossRef]
- Kiguoya, M.W.; Mann, J.K.; Chopera, D.; Gounder, K.; Lee, G.Q.; Hunt, P.W.; Martin, J.N.; Ball, T.B.; Kimani, J.; Brumme, Z.L.; et al. Subtype-Specific Differences in Gag-Protease-Driven Replication Capacity Are Consistent with Intersubtype Differences in HIV-1 Disease Progression. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, L.; Mzobe, Z.; Jin, S.W.; Rajkoomar, E.; Reddy, T.; Brockman, M.A.; Brumme, Z.L.; Ndung’u, T.; Mann, J.K. Nef-mediated inhibition of NFAT following TCR stimulation differs between HIV-1 subtypes. Virology 2019, 531, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Ojwach, D.B.A.; MacMillan, D.; Reddy, T.; Novitsky, V.; Brumme, Z.L.; Brockman, M.A.; Ndung’u, T.; Mann, J.K. Pol-Driven Replicative Capacity Impacts Disease Progression in HIV-1 Subtype C Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host. Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.M.; Archin, N.M. Proviral Latency, Persistent Human Immunodeficiency Virus Infection, and the Development of Latency Reversing Agents. J. Infect. Dis. 2017, 215, S111–S118. [Google Scholar] [CrossRef] [Green Version]
- van der Sluis, R.M.; Derking, R.; Breidel, S.; Speijer, D.; Berkhout, B.; Jeeninga, R.E. Interplay between viral Tat protein and c-Jun transcription factor in controlling LTR promoter activity in different human immunodeficiency virus type I subtypes. J. Gen. Virol. 2014, 95, 968–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Rajagopalan, P.; Lotke, R.; Varghese, R.; Selvam, D.; Kundu, T.K.; Ranga, U. Functional Incompatibility between the Generic NF-kappaB Motif and a Subtype-Specific Sp1III Element Drives the Formation of the HIV-1 Subtype C Viral Promoter. J. Virol. 2016, 90, 7046–7065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Dandekar, S. Targeting NF-kappaB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retroviruses 2015, 31, 4–12. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiscott, J.; Kwon, H.; Génin, P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: Activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Fenard, D.; Bisgrove, D.; Verdin, E.; Greene, W.C. Prostratin antagonizes HIV latency by activating NF-kappaB. J. Biol. Chem. 2004, 279, 42008–42017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS ONE 2010, 5, e11160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Bosque, A.; Balch, A.H.; Smyth, D.; Martins, L.; Planelles, V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4(+) T Cells from Aviremic Patients. Antimicrob. Agents Chemother. 2015, 59, 5984–5991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, L.J.; Bonczkowski, P.; Spivak, A.M.; De Spiegelaere, W.; Novis, C.L.; DePaula-Silva, A.B.; Malatinkova, E.; Trypsteen, W.; Bosque, A.; Vanderkerckhove, L.; et al. Modeling HIV-1 Latency in Primary T Cells Using a Replication-Competent Virus. AIDS Res. Hum. Retroviruses 2016, 32, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogdon, J.; Ziani, W.; Wang, X.; Veazey, R.S.; Xu, H. In vitro effects of the small-molecule protein kinase C agonists on HIV latency reactivation. Sci. Rep. 2016, 6, 39032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Maverakis, E.; Cheng, M.Y.; Elsheikh, M.M.; Deleage, C.; Mendez-Lagares, G.; Shimoda, M.; Yukl, S.A.; Hartigan-O’Connor, D.J.; Thompson, G.R., 3rd; et al. Disruption of latent HIV in vivo during the clearance of actinic keratosis by ingenol mebutate. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Mifsud, E.J.; Tan, A.C.; Jackson, D.C. TLR Agonists as Modulators of the Innate Immune Response and Their Potential as Agents Against Infectious Disease. Front. Immunol. 2014, 5, 79. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; De Assis, C.M.; Sorensen, E.S.; Moszczynski, P.; Huang, S.-h.; Ren, Y.; Spivak, A.M.; Jones, R.B.; Planelles, V.; et al. Dual TLR2 and TLR7 agonists as HIV latency-reversing agents. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Novis, C.L.; Archin, N.M.; Buzon, M.J.; Verdin, E.; Round, J.L.; Lichterfeld, M.; Margolis, D.M.; Planelles, V.; Bosque, A. Reactivation of latent HIV-1 in central memory CD4(+) T cells through TLR-1/2 stimulation. Retrovirology 2013, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Offersen, R.; Nissen, S.K.; Rasmussen, T.A.; Ostergaard, L.; Denton, P.W.; Sogaard, O.S.; Tolstrup, M. A Novel Toll-Like Receptor 9 Agonist, MGN1703, Enhances HIV-1 Transcription and NK Cell-Mediated Inhibition of HIV-1-Infected Autologous CD4+ T Cells. J. Virol. 2016, 90, 4441–4453. [Google Scholar] [CrossRef] [Green Version]
- Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D.D.; Murry, J.P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host. Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampey, G.C.; Irlbeck, D.M.; Browne, E.P.; Kanke, M.; McAllister, A.B.; Ferris, R.G.; Brehm, J.H.; Favre, D.; Routy, J.P.; Jones, C.D.; et al. The SMAC Mimetic AZD5582 is a Potent HIV Latency Reversing Agent. BioRxic Prepr. 2018. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Lawrence, D.; McDonald, D.; Refsland, E.; Bridges, S.; Smiley, S.; Tressler, R.L.; Beaubien, C.; Salzwedel, K. Highlights from the Fourth Biennial Strategies for an HIV Cure Meeting, 10-12 October 2018, Bethesda, MD, USA. J. Virus. Erad. 2019, 5, 50–59. [Google Scholar] [PubMed]
- Mbonye, U.; Karn, J. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr. HIV Res. 2011, 9, 554–567. [Google Scholar]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Zhang, C.; Su, Z.Y.; Wang, L.; Shu, L.; Yang, Y.; Guo, Y.; Pung, D.; Bountra, C.; Kong, A.N. Epigenetic blockade of neoplastic transformation by bromodomain and extra-terminal (BET) domain protein inhibitor JQ-1. Biochem. Pharmacol. 2016, 117, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef] [Green Version]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaikos, T.D.; Painter, M.M.; Sebastian Kettinger, N.T.; Terry, V.H.; Collins, K.L. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef] [Green Version]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [Green Version]
- Sagot-Lerolle, N.; Lamine, A.; Chaix, M.L.; Boufassa, F.; Costagliola, D.; Goujard, C.; Paller, C.; Delfraissy, J.F.; Lambotte, O. Prolonged valproic acid treatment does not reduce the size of the latent HIV reservoir. AIDS 2008, 22, 1125–1129. [Google Scholar] [CrossRef]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.W.; Fauci, A.S. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J. Virol. 2012, 86, 5390–5392. [Google Scholar] [CrossRef] [Green Version]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef] [Green Version]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4(+) T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef] [Green Version]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Invest. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Bui, J.K.; Cyktor, J.C.; Fyne, E.; Campellone, S.; Mason, S.W.; Mellors, J.W. Blockade of the PD-1 axis alone is not sufficient to activate HIV-1 virion production from CD4+ T cells of individuals on suppressive ART. PLoS ONE 2019, 14, e0211112. [Google Scholar] [CrossRef]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R.; et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4(+) T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 814. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Global distribution of HIV-1 subtypes. The major subtype that circulates in distinct geographical locations are listed first and are in bold, followed by other reported subtypes in the region of interest. This depiction is based on estimates of circulating subtypes from 2004 to 2007 [18,21,22,23]. CRF: circulating recombinant form; this describes other CRFs which are not AE, AB, or AG. URFs: unique recombinant forms.
Figure 1.
Global distribution of HIV-1 subtypes. The major subtype that circulates in distinct geographical locations are listed first and are in bold, followed by other reported subtypes in the region of interest. This depiction is based on estimates of circulating subtypes from 2004 to 2007 [18,21,22,23]. CRF: circulating recombinant form; this describes other CRFs which are not AE, AB, or AG. URFs: unique recombinant forms.

Figure 2.
Long terminal repeat (LTR) from diverse subtypes. Main transcription factor binding sites in each LTR. Activated protein-1 (AP-1); specificity protein-1 (Sp1); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); GA-binding protein (GABP); TATA box promoter sequence for transcription initiation. Several other transcription factor binding sites have been identified and are not denoted in the graph for simplicity. This schematic is based on binding sites reported elsewhere [66,69,71].
Figure 2.
Long terminal repeat (LTR) from diverse subtypes. Main transcription factor binding sites in each LTR. Activated protein-1 (AP-1); specificity protein-1 (Sp1); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); GA-binding protein (GABP); TATA box promoter sequence for transcription initiation. Several other transcription factor binding sites have been identified and are not denoted in the graph for simplicity. This schematic is based on binding sites reported elsewhere [66,69,71].


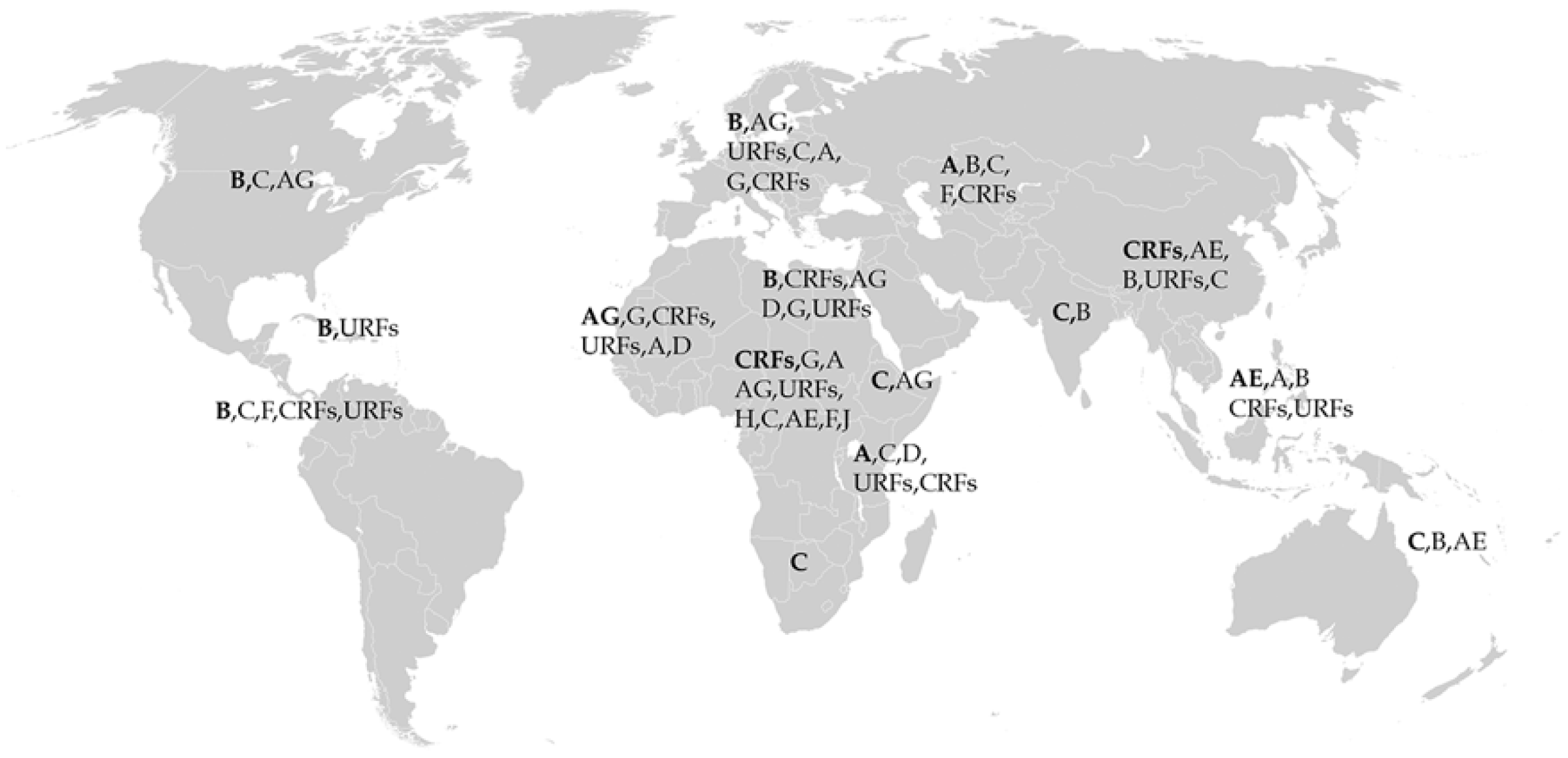{kind=link}
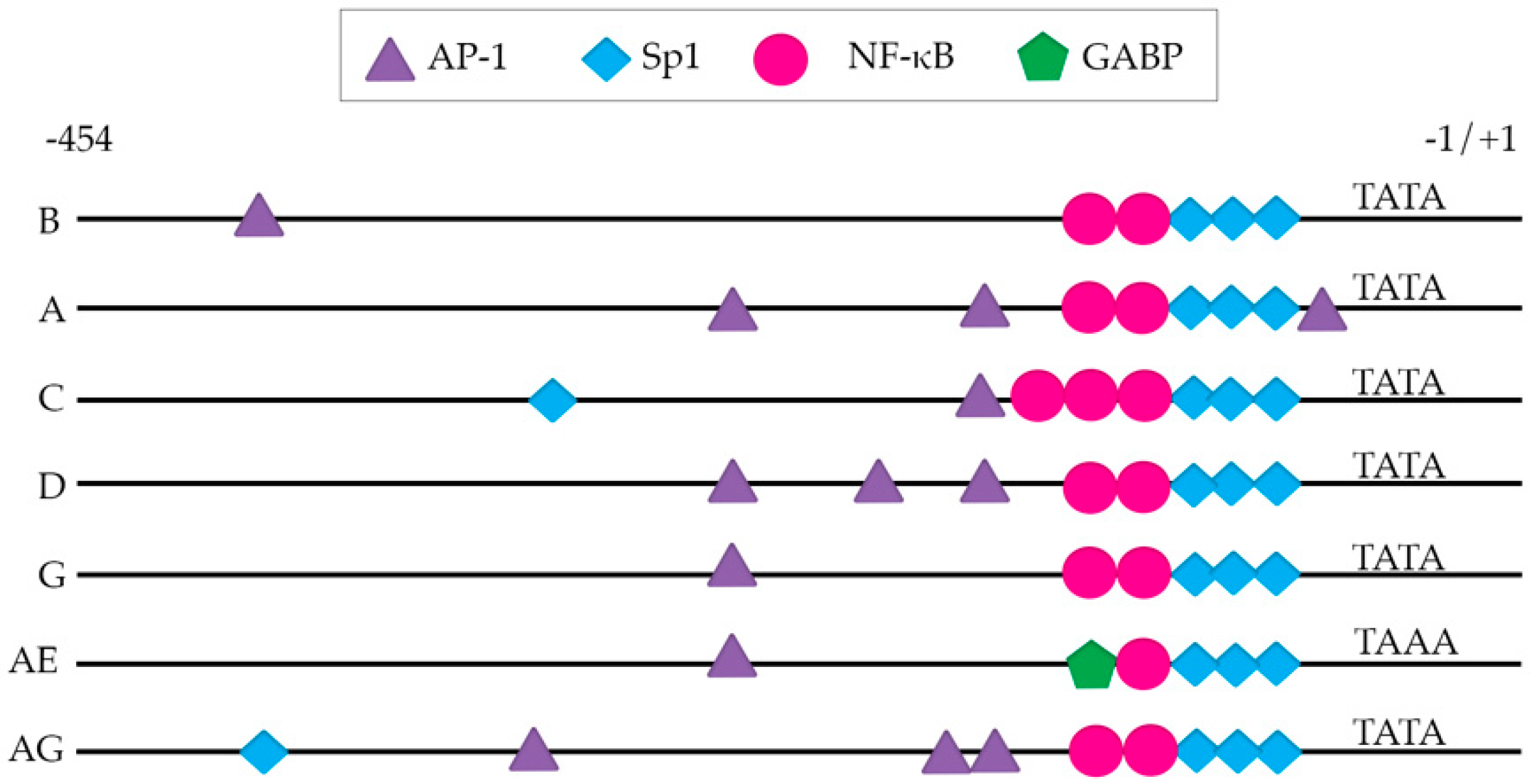{kind=link}
Table 1.
Summary of in vivo studies assessing the relationship between subtype and pathogenesis.
| Geographical Location | Study Population | Study Size | Subtypes Included | Subtyping Method | End Points | Results | Reference |
|---|---|---|---|---|---|---|---|
| Israel | Ethiopian immigrants, non-Ethiopian Israeli men | 168 (77 subtype C Ethiopian immigrants, 91 subtype B non-Ethiopian Israeli men) | C, B | V3 env peptide immunoassay, direct sequencing of V3 env region PCR | CD4 and CD8 counts | No difference in rates of CD4 decline between both groups | [30] |
| Senegal | Seronegative registered female sex workers | 1683 seronegative enrolled, 81 seroconverted, 54 samples were subtyped | A, C, D, G | C2-V3 env region | AIDS-free survival, defined by <200 CD4 cells/mm3 | Non-A subtypes were 8 times more likely to develop AIDS than A subtypes | [38] |
| Thailand | HIV-1 positive inpatients | 2104 subtyped individuals | B’, E | V3 loop env-based peptide EIA | Total CD4 counts | No difference in immuno-suppression between subtypes | [29] |
| Sweden | HIV-1 infected outpatients | 98 individuals (49 ethnic Swedes, 39 ethnic Africans) | A, B, C, D | V3 env sequencing | CD4 count, CD4 decline, | No association in disease progression or CD4 decline and subtype | [28] |
| Uganda | HIV-1 infected adults | 1045 either A or D subtype individuals | A, D | Peptide serology, HMA | Progression to death, CD4 cell count | Subtype D associated with faster progression to death than subtype A | [33] |
| Tanzania | HIV-1 seropositive pregnant mothers | 428 samples where subtype was determined | A, C, D, Recombinants | C2-C5 env region and 3’ p24/5’-p7 region of gag | Progression to death, WHO stage 4 clinical disease, CD4 cell count | Subtype D associated with the fastest progression to death, WHO stage 4 of illness, CD4 <200 cells/mm3 than subtype A or C | [35] |
| Kenya | HIV-1 seronegative commercial female sex workers | 145 women | A, C, D | V1-V3 loops of env, HMA, sequencing and phylogenetic analysis | Mortality, CD4 counts | Subtype D associated with higher mortality and faster CD4 decline | [32] |
| Uganda | HIV-1 seroconverters | 312 individuals | A, D, Recombinants, multiple | Multiregion hybridization assay | CD4 decline | Subtype D associated with faster CD4 decline than subtype A | [31] |
| Uganda | HIV-1 incident ART-naïve individuals | 292 individuals | A, D, A/D, C, other recombinants | Partial gag, env, pol sequencing | CD4 ≤250 cells/mm3, WHO clinical stage 4 AIDS, death before and after ART introduction | Subtype D associated with faster disease progression than subtype A | [34] |
| Kenya, Rwanda, South Africa, Uganda, Zambia | Adult and youths with documented HIV-1 infection | 579 individuals were subtyped | A, C, D | pol sequencing | CD4 count <350 cells/µL, viral load of 1x105 copies/mL, clinical AIDS | Subtype C progressed faster than subtype A, subtype D progressed faster than subtype A | [37] |
| Sub-Saharan Africa (Uganda, Zimbabwe) | Newly infected HIV-1 women | 303 women | A, C, D | PR, RT, and C2-V3 env region | CD4 decline | Subtype D was associated with faster CD4 decline, followed by subtype A, then subtype C | [36] |
WHO: World Health Organization; PCR: polymerase chain reaction; HIV-1: human immunodeficiency virus-1; AIDS: acquired immunodeficiency; EIA: enzyme immunoassay; HMA: heteroduplex mobility assay; ART: antiretroviral therapy; PR: HIV-1 protease; RT: HIV-1 reverse transcriptase.
Table 2.
Summary of in vitro studies that have assessed non-B subtypes and the establishment of latency.
Table 2.
Summary of in vitro studies that have assessed non-B subtypes and the establishment of latency.
| Model System | Viruses Used | Subtype Assessed | Experimental Methods | Conclusions | Reference |
|---|---|---|---|---|---|
| C33A, HeLa, COS, U87, U373, SupT1 | Subtype-specific LTR in a subtype B LAI background, subtype-specific LTR-luciferase reporter plasmid | A, C1, C2, D, E, F, G, G’’ | Transfection of subtype-specific LTR- luciferase constructs to measure basal LTR activity and LTR activity in response to subtype B tat protein and TNF-α stimulation | Correlation between number of NF-κB sites and TNF-α response, subtype C had the greatest response Basal LTR activity in non-B subtypes significantly higher than subtype B, no difference in response to subtype B Tat transactivation Subtype-E LTR -LAI virus replicated better than subtype B-LAI | [66] |
| U937, Jurkat | LTR-luciferase reporter | B, C, E | Transfection of subtype-specific LTR-luciferase construct and addition of subtype B, C, E Tat to measure Tat transactivation, with or without TSA | Clade E Tat has the most transactivation activity Duplicated NF-κB sites in subtype C do not compete for NF-κB binding Basal activity of LTRs varied by subtype | [68] |
| SupT1, MT2 | Subtype-specific LTR in a subtype B LAI background | A, B, C, D, E, F, G | Used CA-p24 ELISA to measure viral fitness/replication | Subtype-specific LTR impacts viral replication, viral fitness influenced by cellular environment | [69] |
| Jurkat, Jurkat Tat-T | Subtype-specific LTR luciferase reporter plasmid | A, C | Used transfection of LTR-luciferase in presence or absence of subtype B tat, to measure transcriptional activity | No difference in transcriptional activity between subtypes A and C | [67] |
| 293T/U937 | Subtype-specific LTR-luciferase construct | A, B, C, D, E, F, G, G’’ | LTR transactivation studied by Co-transfection of subtype-specific LTR-luciferase construct and active pSTAT5 | Potency of LTR transactivation by active STAT5 differs between subtype | [65] |
| Jurkat, Primary CD4 T cells | LTR-GFP-IRES-Tat (LGIT) virus with subtype-specific LTRs | A, B, A2, A/G, B/C, C’, C, B/F, D, F, H | Flow cytometry to determine LGIT-virus infected cells and latency reactivation with LRAs | Varying degrees of sensitivity to reactivation by single agent or combination of LRAs across subtypes | [71] |
| Jurkat, SUPT1 | Subtype-specific LTR in a subtype B LAI background | A, B, C1, C2, D, AE, F, G, AG | Flow cytometry and ELISA for CAp24 for assessing reactivation | No differences except subtype AE and G were less prone to become latent, subtype AE had a significantly different response to LRA Vorinostat | [76] |
| Jurkat | Double-labeled (Red mCherry protein, Green eGFP protein) with subtype-specific LTR in a subtype B LAI background | A, B, C, D, F, G, AE | Flow cytometry to measure mCherry(red) expression and eGFP(green) expression. mCherry+eGFP- cells are latently infected | Difference in degree of silent infections across subtypes, as well as sensitivity to PMA/ionomycin-mediated reactivation. | [77] |
| J2574 | Subtype-specific LTR in a subtype B LAI background | A, B, C1, D, E, F, G | Flow cytometry: difference in %GFP between untreated infected population and PMA-treated infected population is latent population | AP-1 binding site in LTR is important for establishment of latency. Subtype E promoter lacks this site and has reduced ability to establish latency, Subtype A and subtype C exhibited greater latency establishment. LRAs reactivated similarly in subtype A compared to subtype B. | [78] |
| HEK293T, Primary CD4 T cells | Subtype-specific LTR in a HIV-eGFP/VSV-G single cycle infectious virus, LTR driven luciferase reporter and LTR-driven dual luciferase/renilla reporter | AE, B, B’, C, BC | measurement of luciferase/renilla as HIV-1 gene expression, measurement of GFP positive cells as a measure of viral reactivation | Blockade of Sp1 or NF-κB sites using dead Cas9 suppresses viral reactivation. HIV-1 B’ LTR had the greatest transcriptional activity when stimulated with TNF-α | [72] |
Subtype E has been now re-classified as subtype AE. LTR: long terminal repeat. ELISA: enzyme-linked immunosorbent assay. AP-1: activated protein-1. GFP: Green fluorescent protein. pSTAT5: phosphorylated signal transducers and activators of transcription-5. TSA: trichostatin A. IRES: internal ribosomal entry site. VSV-G: vesicular stomatitis virus-G protein. Sp-1: specificity protein 1. NF-κB: nuclear factor kappa-light chain enhancer of activated B cells.
Table 3.
Summary of in vivo studies of the latent viral reservoir that include non-B cohorts.
| Cohort Size | Cohort Type | Subtype | Measure of Viral Reservoir | Conclusions | Reference |
|---|---|---|---|---|---|
| Ugandan cohort: 70 Baltimore cohort: 51 | ART-treated, virally suppressed adults | A, D, AD, B | QVOA | Non-B individuals had a reduced frequency of latently infected cells, the mechanism underlying this observation could not be determined | [106] |
| 30 | ART-naïve men with acute/early HIV-1 infection | B, G, AE | QVOA | Subtype-specific Nef-mediated HLA downregulation correlates with reservoir size; HIV subtype is a statistically significant multivariable correlate of reservoir size. | [107] |
| 1057 | ART-treated long term suppressed individuals | B, AE, AG, A, C, D, F, G, Numerous recombinant forms | Total HIV-1 DNA | HIV-1 non-B subtype was associated with faster decay of reservoir in multivariable analysis; HIV-1 non-B subtype was only significantly associated with a smaller reservoir in a univariable model, trend observed in multivariable analysis | [108] |
ART: antiretroviral therapy. QVOA: quantitative viral outgrowth assay. HIV-1: human immunodeficiency virus-1.
Table 4.
Summary of in vitro testing of LRAs with non-B subtypes.
| Model System | LRAs Assessed | Subtypes Tested | Reference |
|---|---|---|---|
| LTR-GFP-IRES-Tat (LGIT) virus-infected Jurkat cells and primary CD4 T cells | SAHA Prostratin HMBA, TSA, Valproic acid | A, A2, B, C, C, D, F, AG, BC, BF, H | [71] |
| Subtype-specific LTR in a subtype B LAI virus background infected SUPT1 or Jurkat | Vorinostat | A, B, C1, C2, D, AE, F, G, AG | [76] |
| LTR-luciferase reporter construct and tat transfection into U937, Jurkat cells | TSA | B, C, E | [68] |
| Subtype specific LTR in a subtype B LAI virus-infected J2574 cells | TSA SAHA HMBA | A | [78] |
LTR: long terminal repeat. GFP: green fluorescent protein. IRES: internal ribosomal entry site. HMBA: hexamethylene bisacetamide. TSA: trichostatin A. SAHA: suberoylanilide hydroxamic acid.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sarabia, I.; Bosque, A. HIV-1 Latency and Latency Reversal: Does Subtype Matter? Viruses 2019, 11, 1104. https://doi.org/10.3390/v11121104
AMA Style
Sarabia I, Bosque A. HIV-1 Latency and Latency Reversal: Does Subtype Matter? Viruses. 2019; 11(12):1104. https://doi.org/10.3390/v11121104
Chicago/Turabian StyleSarabia, Indra, and Alberto Bosque. 2019. "HIV-1 Latency and Latency Reversal: Does Subtype Matter?" Viruses 11, no. 12: 1104. https://doi.org/10.3390/v11121104
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.