Hepatitis B Surface Antigen Activates Unfolded Protein Response in Forming Ground Glass Hepatocytes of Chronic Hepatitis B

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recombinant Adenoviruses Production
2.2. Cell Culture and AdVs Transduction
2.3. HBV Infection
2.4. RNA Preparation and Quantitative Real-Time PCRs
2.5. Western Blot Assay
2.6. Paraffin-Embedding of Cells and Liver Tissues
2.7. Immunofluorescent Staining for Cells and Liver Samples
2.8. Caspase-3/7 Assay and Cell Viability Assay
2.9. Statistical Analysis
3. Results
3.1. Activation of PERK Pathway by Overexpression of HBsAg
3.2. Induction of PERK Pathway Genes Correlates with Level of HBsAg Expression
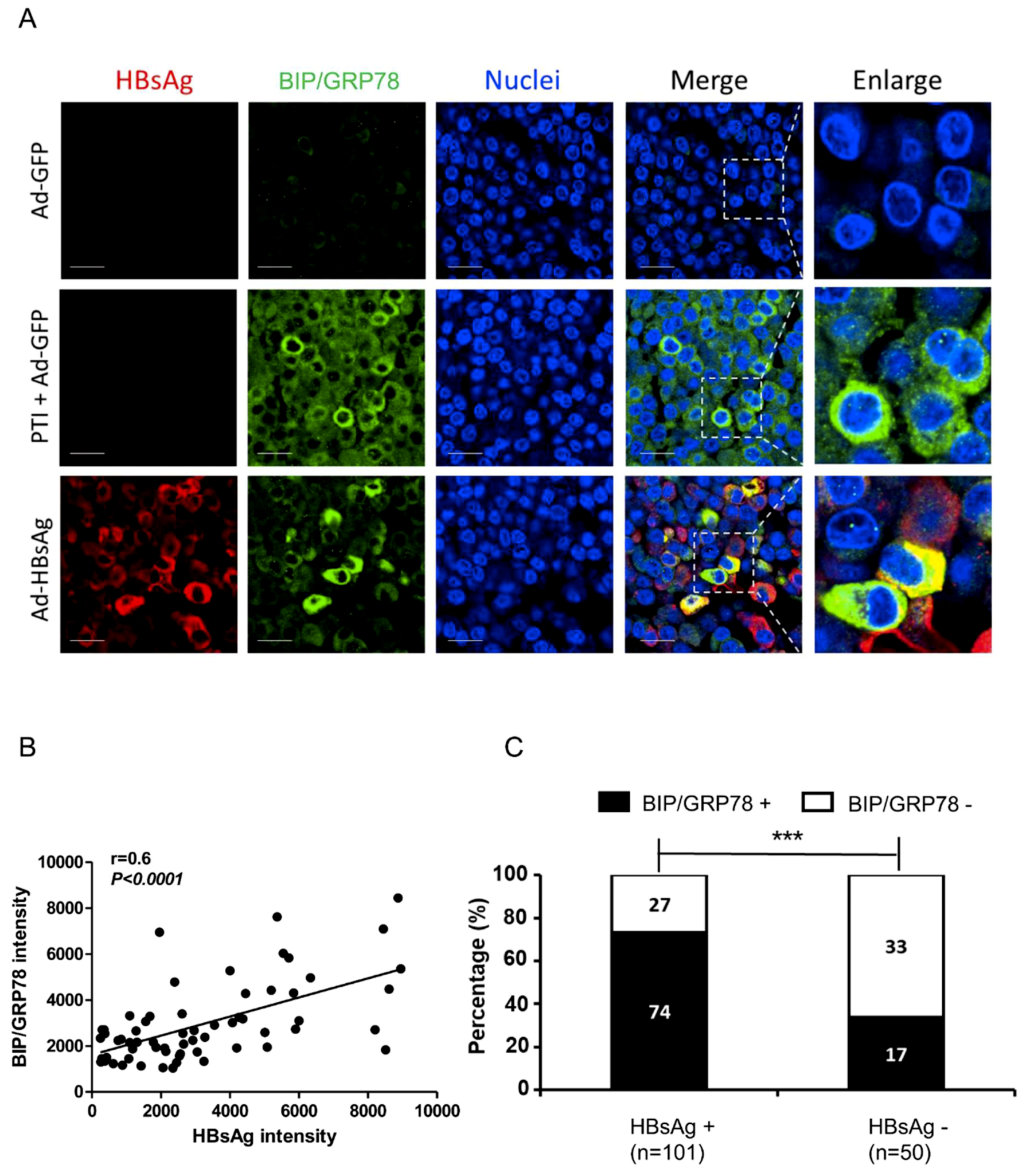
3.3. Activation of ER Stress Marker BIP/GRP78 by HBsAg at Single-cell Level
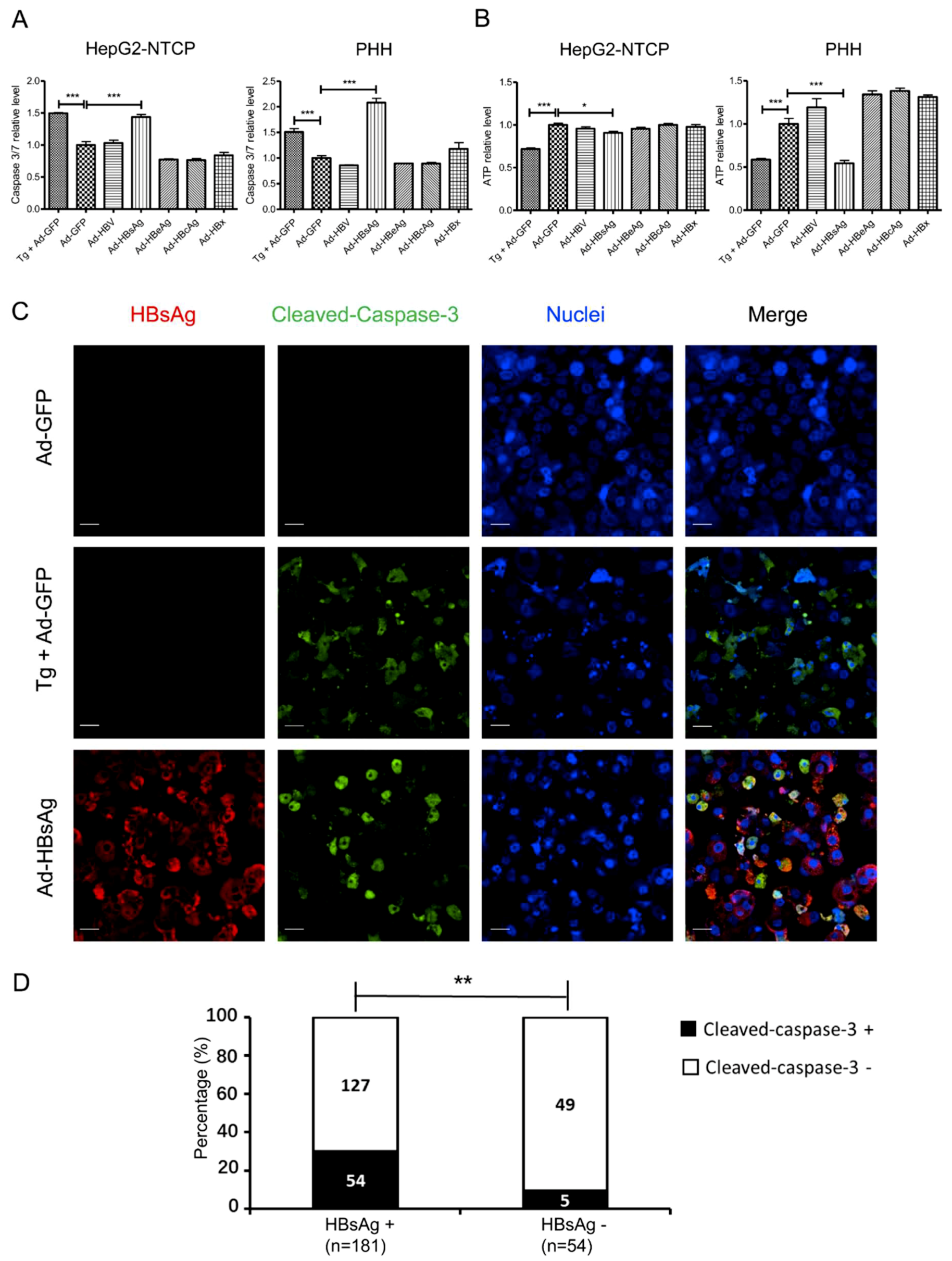
3.4. Induction of Apoptosis in HepG2-NTCP Cells and PHHs by HBsAg Overexpression and ER Stress
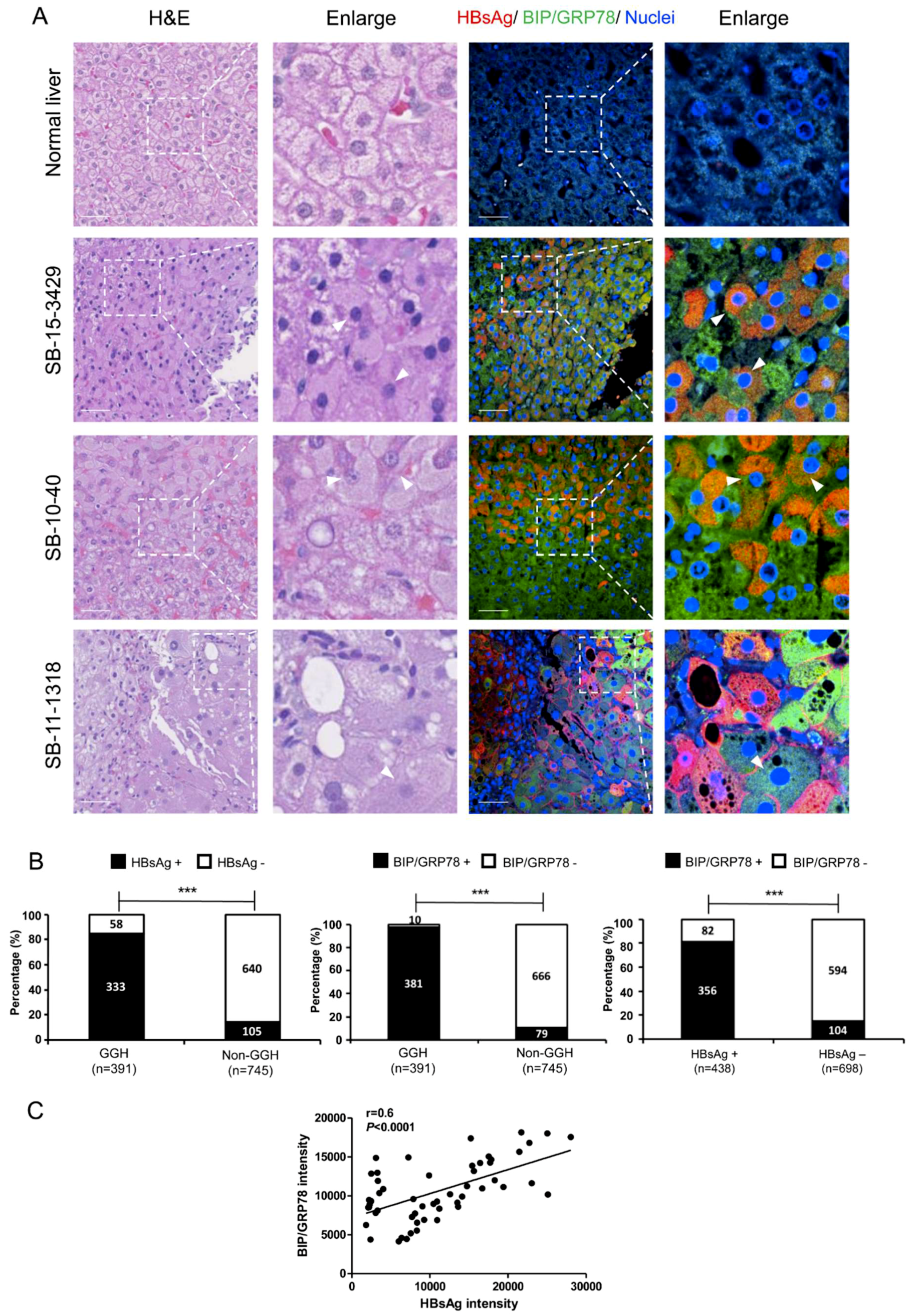
3.5. ER Stress and Ground Glass Hepatocytes in HBV-Infected Liver
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, C. Ground-glass hepatocytes. S. Afr. Med. J. 1975, 49, 1401–1403. [Google Scholar]
- Gerber, M.A.; Hadziyannis, S.; Vissoulis, C.; Schaffner, F.; Paronetto, F.; Popper, H. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am. J. Pathol. 1974, 75, 489–502. [Google Scholar]
- Su, I.J.; Lai, M.Y.; Hsu, H.C.; Chen, D.S.; Yang, P.M.; Chuang, S.M.; Sung, J.L. Diverse virological, histopathological and prognostic implications of seroconversion from hepatitis B e antigen to anti-HBe in chronic hepatitis B virus infection. J. Hepatol. 1986, 3, 182–189. [Google Scholar] [CrossRef]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- Hsu, H.C.; Lai, M.Y.; Su, I.J.; Chen, D.S.; Chang, M.H.; Yang, P.M.; Wu, C.Y.; Hsieh, H.C. Correlation of hepatocyte HBsAg expression with virus replication and liver pathology. Hepatology 1988, 8, 749–754. [Google Scholar] [CrossRef]
- Caligiuri, P.; Cerruti, R.; Icardi, G.; Bruzzone, B. Overview of hepatitis B virus mutations and their implications in the management of infection. World J. Gastroenterol. 2016, 22, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Xu, Z.; Jensen, G.; Yen, T.S. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J. Virol. 1997, 71, 7387–7392. [Google Scholar] [PubMed]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Chang, W.W.; Lei, H.Y.; Lai, M.D.; Chang, W.T.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.C.; Chang, W.T.; Chang, W.W.; Wu, H.C.; Huang, W.; Lei, H.Y.; Lai, M.D.; Fausto, N.; Su, I.J. Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 2005, 41, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.C.; Neo, J.C.; Wu, J.C.; Chen, Y.F.; Kao, C.H.; Tsai, T.F. Expression of a hepatitis B virus pre-S2 deletion mutant in the liver results in hepatomegaly and hepatocellular carcinoma in mice. J. Pathol. 2017, 241, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Protzer, U.; Hu, Z.; Jacob, J.; Liang, T.J. Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J. Virol. 2004, 78, 4566–4572. [Google Scholar] [CrossRef]
- Sprinzl, M.F.; Oberwinkler, H.; Schaller, H.; Protzer, U. Transfer of hepatitis B virus genome by adenovirus vectors into cultured cells and mice: crossing the species barrier. J. Virol. 2001, 75, 5108–5118. [Google Scholar] [CrossRef]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.-T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-γ and tumor necrosis factor-α produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Cheng, X.; Li, Y.; Valdez, K.; Chen, W.; Liang, T.J. Hepatitis B virus deregulates cell cycle to promote viral replication and a premalignant phenotype. J. Virol. 2018, 92, e00722. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, I.J.; Wang, H.C.; Wu, H.C.; Huang, W.Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef] [Green Version]
- Chisari, F.V.; Filippi, P.; Buras, J.; McLachlan, A.; Popper, H.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc. Natl. Acad. Sci. USA 1987, 84, 6909–6913. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. ER stress: Can the liver cope? J. Hepatol. 2006, 45, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Tsai, H.; Teng, C.; Wang, L.H.; Huang, W. Ground glass hepatocytes provide targets for therapy or prevention of hepatitis B virus-related hepatocellular carcinoma. AIMS Med. Sci. 2018, 5, 90–101. [Google Scholar] [CrossRef]
- Liu, N.; Jiao, T.; Huang, Y.; Liu, W.; Li, Z.; Ye, X. Hepatitis B virus regulates apoptosis and tumorigenesis through the microRNA-15a-Smad7-transforming growth factor beta pathway. J. Virol. 2015, 89, 2739–2749. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Diao, J.; Khine, A.A.; Sarangi, F.; Hsu, E.; Iorio, C.; Tibbles, L.A.; Woodgett, J.R.; Penninger, J.; Richardson, C.D. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J. Biol. Chem. 2001, 276, 8328–8340. [Google Scholar] [CrossRef]
- Terradillos, O.; de La Coste, A.; Pollicino, T.; Neuveut, C.; Sitterlin, D.; Lecoeur, H.; Gougeon, M.-L.; Kahn, A.; Buendia, M.-A. The hepatitis B virus X protein abrogates Bcl-2-mediated protection against Fas apoptosis in the liver. Oncogene 2002, 21, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Lee, H.; Yun, Y. X-gene product of hepatitis B virus induces apoptosis in liver cells. J. Biol. Chem. 1998, 273, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, S.Y.; Kim, J.; Lee, H.; Choi, M.; Kim, J.K.; Ahn, J.K. Hepatitis B virus X protein induces apoptosis by enhancing translocation of Bax to mitochondria. IUBMB Life 2008, 60, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, B.P. An overview of unfolded protein response signaling and its role in cancer. Cancer Biother. Radiopharm. 2017, 32, 275–281. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum wtress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef]
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeben, A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Sahin, F.; Kannanga, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef]
- Roy, H.K. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.-M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef]
- Wu, H.C.; Tsai, H.W.; Teng, C.F.; Hsieh, W.C.; Lin, Y.J.; Wang, L.H.C.; Yuan, Q.; Su, I.J. Ground-glass hepatocytes co-expressing hepatitis B virus X protein and surface antigens exhibit enhanced oncogenic effects and tumorigenesis. Hum. Pathol. 2014, 45, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Mathai, A.M.; Alexander, J.; Kuo, F.Y.; Torbenson, M.; Swanson, P.E.; Yeh, M.M. Type II ground-glass hepatocytes as a marker of hepatocellular carcinoma in chronic hepatitis B. Hum. Pathol. 2013, 44, 1665–1671. [Google Scholar] [CrossRef]
- Tsai, H.W.; Lin, Y.J.; Wu, H.C.; Chang, T.T.; Wu, I.C.; Cheng, P.N.; Yen, C.J.; Chan, S.H.; Huang, W.; Su, I.J.; et al. Resistance of ground glass hepatocytes to oral antivirals in chronic hepatitis B patients and implication for the development of hepatocellular carcinoma. Oncotarget 2016, 7, 27724–27734. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Xia, Y.; Cheng, X.; Kleiner, D.E.; Hewitt, S.M.; Sproch, J.; Li, T.; Zhuang, H.; Liang, T.J. Hepatitis B Surface Antigen Activates Unfolded Protein Response in Forming Ground Glass Hepatocytes of Chronic Hepatitis B. Viruses 2019, 11, 386. https://doi.org/10.3390/v11040386
Li Y, Xia Y, Cheng X, Kleiner DE, Hewitt SM, Sproch J, Li T, Zhuang H, Liang TJ. Hepatitis B Surface Antigen Activates Unfolded Protein Response in Forming Ground Glass Hepatocytes of Chronic Hepatitis B. Viruses. 2019; 11(4):386. https://doi.org/10.3390/v11040386
Chicago/Turabian StyleLi, Yao, Yuchen Xia, Xiaoming Cheng, David E. Kleiner, Stephen M. Hewitt, Julia Sproch, Tong Li, Hui Zhuang, and T. Jake Liang. 2019. "Hepatitis B Surface Antigen Activates Unfolded Protein Response in Forming Ground Glass Hepatocytes of Chronic Hepatitis B" Viruses 11, no. 4: 386. https://doi.org/10.3390/v11040386