Mitovirus and Mitochondrial Coding Sequences from Basal Fungus Entomophthora muscae
by
 , , and
, , and
Max L. Nibert
1,*, 
Humberto J. Debat
2 ,
,
Austin R. Manny
1,
Igor V. Grigoriev
3,4 and
Henrik H. De Fine Licht
5 1
Department of Microbiology and Program in Virology, Harvard Medical School, Boston, MA 02115, USA
2
Instituto de Patología Vegetal, Centro de Investigaciones Agropecuarias, Instituto Nacional de Tecnología Agropecuaria (IPAVE-CIAP-INTA), Córdoba X5020ICA, Argentina
3
U.S. Department of Energy Joint Genome Institute, Walnut Creek, CA 94598, USA
4
Department of Plant and Microbial Biology, University of California Berkeley, Berkeley, CA 94720, USA
5
Department of Plant and Environmental Sciences, University of Copenhagen, DK-1871 Frederiksberg, Denmark
*
Author to whom correspondence should be addressed.
Viruses 2019, 11(4), 351; https://doi.org/10.3390/v11040351
Submission received: 29 March 2019
/
Revised: 11 April 2019
/
Accepted: 15 April 2019
/
Published: 17 April 2019
(This article belongs to the Special Issue Mycoviruses)
Abstract
:Fungi constituting the Entomophthora muscae species complex (members of subphylum Entomophthoromycotina, phylum Zoopagamycota) commonly kill their insect hosts and manipulate host behaviors in the process. In this study, we made use of public transcriptome data to identify and characterize eight new species of mitoviruses associated with several different E. muscae isolates. Mitoviruses are simple RNA viruses that replicate in host mitochondria and are frequently found in more phylogenetically apical fungi (members of subphylum Glomeromyoctina, phylum Mucoromycota, phylum Basidiomycota and phylum Ascomycota) as well as in plants. E. muscae is the first fungus from phylum Zoopagomycota, and thereby the most phylogenetically basal fungus, found to harbor mitoviruses to date. Multiple UGA (Trp) codons are found not only in each of the new mitovirus sequences from E. muscae but also in mitochondrial core-gene coding sequences newly assembled from E. muscae transcriptome data, suggesting that UGA (Trp) is not a rarely used codon in the mitochondria of this fungus. The presence of mitoviruses in these basal fungi has possible implications for the evolution of these viruses.
1. Introduction
The classification scheme for kingdom Fungi currently applied by the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) includes eight major phyla: Ascomycota, Basidiomycota, Mucoromycota (subphyla Glomeromycotina, Mortierellomycotina, and Mucoromycotina), Zoopagomycota (subphyla Entomophthoromycotina, Kickxellomycotina, and Zoopagomycotina), Blastocladiomycota, Chytridiomycota, Cryptomycota, and Microsporidia, in approximate order of increasing time since they emerged as divergent taxa [1,2]. Fungal mitoviruses have been reported to date only from the most recently (apically) diverging phyla, Ascomycota and Basidiomycota, as well as from subphylum Glomeromycotina in phylum Mucoromycota [3,4,5]. We remain interested to discover fungal mitoviruses from other, less recently (basally) diverging phyla or subphyla, as part of an effort to understand the deeper evolutionary history of these viruses.
Mitoviruses are currently classified in genus Mitovirus, family Narnaviridae. They have small plus-strand RNA genomes and replicate persistently in host cell mitochondria [3,6,7,8]. Though initially reported only from fungi [3,4,5], they have recently been found also in plants [9,10]. Several mitoviruses reported also from invertebrates [11,12] might instead have been derived from fungal symbionts of the sampled animals. Mitovirus genomes range between 2.0 and 4.5 kb in length, each encompassing a single long open reading frame that encodes a deduced protein with the conserved motifs of a viral RNA-dependent RNA polymerase (RdRp) [13]. Mitoviruses do not form virions and are thought to persist and replicate instead as ribonucleoprotein complexes inside infected mitochondria, which are transmitted to daughter cells during cell division, as well as horizontally during hyphal anastomosis in the case of fungal mitoviruses and vertically through spores or seeds in the case of fungal or plant mitoviruses, respectively [3,8,9,10,14]. Mitovirus infections in fungi have been linked to morphological abnormalities in mitochondria, defective in vitro growth, and hypovirulence in some phytopathogenic fungi [8,15,16,17,18]. In fact, a recent report based on exhaustive phylogenetic analyses of RNA-dependent polymerases suggests that mitoviruses represent one of the most phylogenetically basal groups of eukaryotic RNA viruses [19].
Entomophthoromycotina is one of three subphyla that currently constitute fungal phylum Zoopagomycota [1,2]. A few entomophthoroid fungi that are normally soil saprobes can also cause human infections, including Conidiobolus coronatus (order Entomophthorales) and Basidiobolus ranarum (order Basidiobolales) [20,21]. Entomophthoroid fungi are probably best known, however, as insect parasites, which exhibit generally narrow host ranges and can cause epizootic events in which large numbers of susceptible insects are killed within local geographic regions [22]. The Entomophthora muscae species complex (order Entomophthorales) [23] is one such group of entomopathogenic fungi, which infect and kill their dipteran, commonly muscoid fly, hosts. E. muscae is also known to manipulate fly behaviors, in particular inducing the behavior known as “summiting”, in which the fly climbs to a high surface where it becomes affixed via fungal outgrowths and then strikes a characteristic pose considered to aid in dispersal of spores from the subsequent carcass [24].
De Fine Licht et al. [25] have reported on the transcriptomics of E. muscae isolates, in a broad effort to understand their host specificity and pathogenicity. In that report, the authors mention the presence of several apparent viral transcripts in their transcriptome shotgun assemblies but provide no further descriptions of those viruses. As described in detail below, we subsequently discovered a number of apparent mitovirus sequences from that transcriptome study by searching for mitovirus-like sequences within the Transcriptome Shotgun Assembly (TSA) database at NCBI. Recognizing several interesting features of these apparent new mitoviruses from E. muscae, we have gone on to characterize their sequences in detail, as we report here. As a part of this work, we have also identified additional strains of these mitoviruses by assembling sequence reads from two other transcriptome studies of E. muscae, as obtained from the Sequence Read Archive (SRA) database at NCBI. Included among our findings is identification of numerous UGA (Trp) codons in the mitovirus sequences from E. muscae, as well as in mitochondrial core-gene coding sequences from E. muscae, leading us to conclude that UGA (Trp) is not a rarely used codon in E. muscae, unlike the case in many other basal fungi [4]. E. muscae is hereby the first fungus from phylum Zoopagomycota, and also the most phylogenetically basal fungus [1,2], that has been reported to harbor mitoviruses to date, with possible implications for mitovirus evolution.
2. Materials and Methods
2.1. Assembly and Analysis of Mitovirus Sequences from E. muscae
The main steps in this process are detailed in Results. All searches of the TSA, SRA, and NR databases were performed using the BLAST web server at NCBI. The 15 TSA hits from the initial TBLASTN searches were: GEMZ01003603.1, GEMZ01006022.1, GEMZ01006112.1, GEMZ01008256.1, GEMZ01008924.1, GEMZ01011847.1, GEMZ01008924.1, and GEMZ01021189.1; GENA01003603.1, GENA01006022.1, GENA01006112.1, GENA01008256.1, GENA01008924.1, GENA01011847.1, GENA01008924.1, and GENA01021189.1; and GEND01018947.1 [25]. The mitovirus queries for those searches were: BAJ23143.2, BAN85985.1, AVA17449.1–AVA17452.1, and AXY40441.1–AXY40444.1 [5,26,27,28]. To identify mitovirus-matching reads in SRA accessions prior to contig assembly, searches were performed using Discontiguous MegaBLAST or BLASTN. Sequence reads were then assembled into contigs using CAP3 [29] as implemented at http://biosrv.cab.unina.it/webcap3/ or galaxy.pasteur.fr/. Terminal residues in each assembled contig were trimmed back to those that agree between at least two mitovirus strains of the same species. Following contig assembly, mapped reads were re-identified using MegaBLAST, and coverage depths were determined using the “Map Reads to Reference” tool in CLC Genomics Workbench v7. RPKM and coverage depth values are shown in Table S1.
For generating transcriptome shotgun assemblies from SRA libraries, we used Trinity v2.2.0 [30] with default parameters as implemented through https://usegalaxy.org/ and rnaSPAdes v3.13.0 [31] with default parameters as implemented locally. The contigs from each Trinity assembly were subjected to searches with TBLASTN using mitovirus RdRp sequences as queries. Identified contigs were then extended by iterative mapping of reads from the corresponding BioProject using the “Map to Reference” tool in Geneious v8.1.9 with low-sensitivity parameters. The contigs from each rnaSPAdes assembly were subjected to taxonomic classification using locally implemented DIAMOND v0.9.22.123 [32] in BLASTX mode against the NCBI NR database, with an E-value threshold of 1e−3 and a “top” parameter of 1 to identify the best species-level hits for each contig.
Codon usage was analyzed using the Codon Usage tool in Sequence Manipulation Suite at http://www.bioinformatics.org/sms2/. Pairwise and multiple sequence alignments were performed using MAFFT v7 [33] as implemented at https://mafft.cbrc.jp/alignment/server/. For determining % sequence identity, pairwise alignments were performed using EMBOSS Needleall as implemented at http://www.bioinformatics.nl/cgi-bin/emboss/needleall. Maximum-likelihood phylogenetic analyses were performed using the program IQ-Tree [34], including the “Find best and apply” option (ModelFinder) [35] and the “Bootstrap ultrafast” option (UFboot) [36], as implemented at https://www.hiv.lanl.gov/content/sequence/HIV/HIVTools.html.
2.2. Assembly and Analysis of Mitochondrial Gene Coding Sequences from E. muscae
Programs were used as described above for mitovirus sequences. The following TSA hits arose from the initial TBLASTN searches for mitochondrial gene coding sequences from E. muscae and were then used as queries of the SRA transcriptome libraries from E. muscae isolate Berkeley: atp6, GENA01019820.1; atp9, GENA01019993.1; cob, GEND01033240.1; cox1, GEND01036136.1 and GEND01034538.1; cox2, GEND01003800.1; cox3, GEND01033140.1; nad1, GENA01026764.1 and GENA01028096.1; nad2, GENA01003576.1; nad3, GENA01020377.1; nad4, GEND01006792.1 and GEND01006791.1; nad5, GENA01001061.1; and nad6, GEND01032932.1 and GENA01021432.1 [25]. RPKM and coverage depth values are shown in Table S2.
2.3. SRA Accessions Used for Assembling Contigs
SRA accessions that we used in this study for assembling mitovirus or mitochondrial transcript contigs from each E. muscae isolate are listed in Table S3. For E. muscae isolate KVL-14-117, the SRA accessions only from media-grown fungus (not fly-grown fungus) were used for assembling mitovirus contigs, in an effort to ensure that the identified viruses were derived from E. muscae, and not the fly hosts or a different fly symbiont. For E. muscae isolate Berkeley, the SRA accessions from only (i) flies infected with E. muscae (not uninfected flies) and (ii) later times post-infection (72, 96, and 120 h, not 24 and 48 h) were used for assembling mitovirus or mitochondrial transcript contigs, in an effort to increase the proportion of reads that were derived from E. muscae, not the fly hosts.
2.4. Newly Reported Sequences
Coding-complete mitovirus sequences from E. muscae isolates KVL-14-117, KVL-14-118, HHdFL130914-1, HHdFL050913-1, and Berkeley have been deposited in GenBank as accessions MK682513.1–MK682534.1 and BK010729.1–BK010736.1. Mitochondrial core-gene coding sequences from E. muscae isolate Berkeley have been deposited in GenBank as accessions BK010748–BK010759. In addition, the mitovirus and mitochondrial core-gene sequences from all five of these E. muscae isolates have been included in the Supplementary Materials for this report as Data S1 and Data S2, respectively.
2.5. Sequencing at the Joint Genome Institute, 1000 Fungal Genomes Project
The sequence reads from E. muscae isolate HHdFL130914-1 (accession SRX2782457) have not been reported in a peer-reviewed article to date, and the methods are therefore described here. A stranded cDNA library was generated using an Illumina TruSeq Stranded RNA LT kit (San Diego, CA, USA). mRNA was purified from 1 µg of total RNA using magnetic beads containing poly-T oligos. mRNA was fragmented and reverse-transcribed using random hexamers and Superscript II (Invitrogen, Carlsbad, CA, USA) followed by second strand synthesis. The fragmented cDNA was treated with end repair, A-tailing, adapter ligation, and eight cycles of PCR. The prepared library was quantified using KAPA Biosystem’s next-generation sequencing library qPCR kit (Wilmington MA, USA) and run on a Roche LightCycler 480 real-time PCR instrument (Pleasanton CA, USA). The quantified libraries were then prepared for sequencing on the Illumina HiSeq platform using an Illumina TruSeq Rapid paired-end cluster kit. Sequencing of the flow cell was performed on the Illumina HiSeq2500 sequencer using Illumina HiSeq TruSeq SBS sequencing kits, following a 2 × 150-nt indexed run recipe.
3. Results
3.1. Mitovirus-like Sequences in Transcriptome Data from E. muscae
We used TBLASTN to search entries from basal fungi (not phylum Ascomycota, phylum Basidiomycota, or subphylum Glomeromycotina) in the TSA database at NCBI, using RdRp sequences of previously reported fungal mitoviruses as queries. The searches yielded 15 strong hits (E-values, 5e−52 to 2e−10 with different queries; see Materials and Methods for further details), all derived from the same transcriptomics study of muscoid-fly pathogens from the E. muscae species complex (BioProject PRJEB10825) [25], including E. muscae sensu stricto isolates from house flies (Musca domestica) and E. muscae sensu lato isolates from cabbage flies (Delia radicum). The lengths of these 15 hits range from 2321 to 2813 nt, within the expected interval for mitovirus genomes [3,4].
Before analyzing these apparent new mitovirus sequences, we examined the TSA metadata to learn about the samples and to identify the SRA accessions from which the TSA hits had been assembled. After reviewing the metadata and the sequences themselves, we chose to reassemble the mitovirus-like contigs for each E. muscae isolate, using the SRA-derived sequence reads that mapped to each respective TSA hit and including any additional reads that lengthened or shortened each contig. We were thereby able to reassemble seven distinct mitovirus-like sequences from media-grown E. muscae house fly isolate KVL-14-117, seven distinct mitovirus-like sequences from fly-grown E. muscae house fly isolate KVL-14-118 (one of the seven sequences includes two small gaps in sequencing coverage, as inferred by alignment with related sequences from other isolates), and one mitovirus-like sequence from fly-grown E. muscae cabbage fly isolate HHdFL050913-1. A small number of reads (22 total) matching the mitovirus from isolate HHdFL050913-1 were also identified from fly-grown E. muscae cabbage fly isolate HHdFL040913-2 [25] but were sufficient in coverage to assemble only a few short contigs.
By reviewing the SRA database for other transcriptome-associated accessions that are annotated as relating to E. muscae or other Entomophthoromycotina members, we found such accessions from five other BioProjects: PRJNA372837 for E. muscae house fly isolate HHdFL130914-1 (also known as KVL-14-115 [37]), PRJNA435715 for E. muscae fruit fly isolate Berkeley (originally obtained from Drosophila hydei) [24], PRJNA259024 for Conidiobolus thromboides isolate FSU 785, PRJNA67455 for Conidiobolus coronatus isolate NRRL 28638, and PRJNA501640 for Zoophthora radicans isolate ATCC 208865/ARSEF 4784. We therefore used the newly assembled mitovirus-like sequences described above as queries to search the SRA transcriptome libraries from these five other BioProjects and obtained numerous strong hits from two of them, those for E. muscae house fly isolate HHdFL130914-1 and E. muscae fruit fly isolate Berkeley. Assembling these SRA reads from isolates HHdFL130914-1 and Berkeley then gave rise to seven distinct mitovirus-like sequences from each of these additional isolates, for a total of 29 mitovirus-like sequences assembled from five different E. muscae isolates (Figure 1).
Given the large number of mitovirus-like sequences associated with E. muscae, we reasoned that there might be sequences of yet other, divergent mitoviruses remaining to be identified in the SRA transcriptome libraries. To address this possibility, we generated our own shotgun assemblies from the libraries analyzed in the preceding paragraph and then searched these new assemblies for other mitoviruses. In this manner, we were able to identify one additional mitovirus-like sequence from E. muscae isolate Berkeley (sequence length, 2479 nt) (Figure 1), bringing the overall total to 30 such sequences from the five E. muscae isolates.
Especially in plants, fragments of mitovirus genome sequences have been commonly endogenized in host DNA [9,38,39,40]. To address this possibility for the mitovirus sequences reported here, we noted that there are six SRA libraries at NCBI from PacBio SMRT runs on genomic DNA from E. muscae isolate HHdFL130914-1 (BioProject PRJNA346904), an isolate that we showed above to harbor sequences from seven mitovirus strains in its RNA transcriptome data. These six SRA libraries from E. muscae DNA were found to register no MegaBLAST hits to the seven mitovirus sequences, providing evidence against these virus sequences being derived from endogenized elements. A small number of mitovirus-matching reads (28 total) found in three SRA libraries arising from Illumina runs on genomic DNA from the same BioProject as the PacBio SMRT runs seem unlikely to be significant, though they may be reminiscent of evidence for non-genomic DNA fragments detected for Gigaspora margarita mitoviruses [28]. Contigs for E. muscae mitoviruses found as hits from the Whole Genome Shotgun (WGS) database at NCBI are in fact derived from the transcriptome study BioProject PRJEB10825 [25], and thus from RNA not DNA (H.H.D.F.L.).
3.2. Basic Features of the Apparent Mitovirus Sequences from E. muscae
The lengths of our 30 newly assembled mitovirus-like contigs range from 2300 to 2806 nt (Figure 1), within the expected interval for mitovirus genomes [3,4]. Using genetic code 4 in which UGA encodes Trp not “stop” (as expected for translation in the mitochondria of most animals and fungi including members of phylum Zoopagomycota [41,42,43]), a single long open reading frame (ORF) is found in each contig, flanked by one or more stop codon at the plus-strand 5´ end in 27 of the 30 sequences and one or more stop codon at the plus-strand 3´ end in all 30 sequences, suggesting to us that all of the contigs are coding complete. The contigs encode proteins of nine different lengths (assuming that the first in-frame AUG is the start codon in each): four encode a 757-aa protein, five encode a 709-aa protein, four encode a 708-aa protein, four encode a 701-aa protein, three encode a 689-aa protein, one encodes a 684-aa protein, four encode a 681-aa protein, four encode a 680-aa protein, and one encodes a 636-aa protein, suggesting that there may be multiple mitovirus species represented by these sequences. When used in BLASTP searches of the Nonredundant Protein Sequences (NR) database for Viruses at NCBI, each of these deduced protein sequences showed strongest similarities to mitovirus RdRps, and the approximate position of the RdRp catalytic motif GDD is shown and centered for each in Figure 1 (also shown in the multiple sequence alignment of the eight sequences from E. muscae isolate Berkeley in Figure S1). For nine contigs, the top hit was to the Erysiphe necator mitovirus 1 (ATS94398; E-values, 1e−91 to 8e−82; identity scores, 37–40%); for nine other contigs, the top hit was to Erysiphe necator mitovirus 2 (ATS94399; E-values, 1e−94 to 5e−70; identity scores, 34–38%); for eight other contigs, the top hit was to Erysiphe necator mitovirus 3 (ATS94400; E-values, 0.0 to 5e−119; identity scores, 36–45%); and for the remaining four contigs, the top hit was to Hubei narna-like virus 25 (APG77157; E-values, 0.0; identity scores, 90%), which is also an apparent mitovirus [10]. These BLASTP findings suggest again that there may be multiple mitovirus species represented by these sequences. The mitovirus-like contigs may be missing some residues at one or both ends relative to the full-length viral genomes, but even so, there is a generally long nontranslated region (NTR) at the plus-strand 5´ end of each contig (105–477 nt, median 317 nt; again assuming that the first in-frame AUG is the start codon in each) and a generally shorter NTR at the plus-strand 3´ end of each contig (1–175 nt, median 106 nt) (Figure 1).
Most fungal mitoviruses contain a number of in-frame UGA codons in the RdRp open reading frame, encoding Trp per genetic code 4; however, some do not, and almost all of those that do not derive from fungal hosts from phylum Basidiomycota or subphylum Glomeromycotina in which UGA(Trp) is a rarely used mitochondrial codon [4,28]. The 30 mitovirus-like sequences from E. muscae described above fit the more typical pattern for fungal mitoviruses, in that a substantial fraction of their Trp codons are UGA (vs. UGG): 38–77%, numbering between 5 and 13 UGA(Trp) codons in the different apparent mitovirus sequences (Figure 1).
3.3. Pairwise and Phylogenetic Comparisons of the Mitovirus Sequences from E. muscae
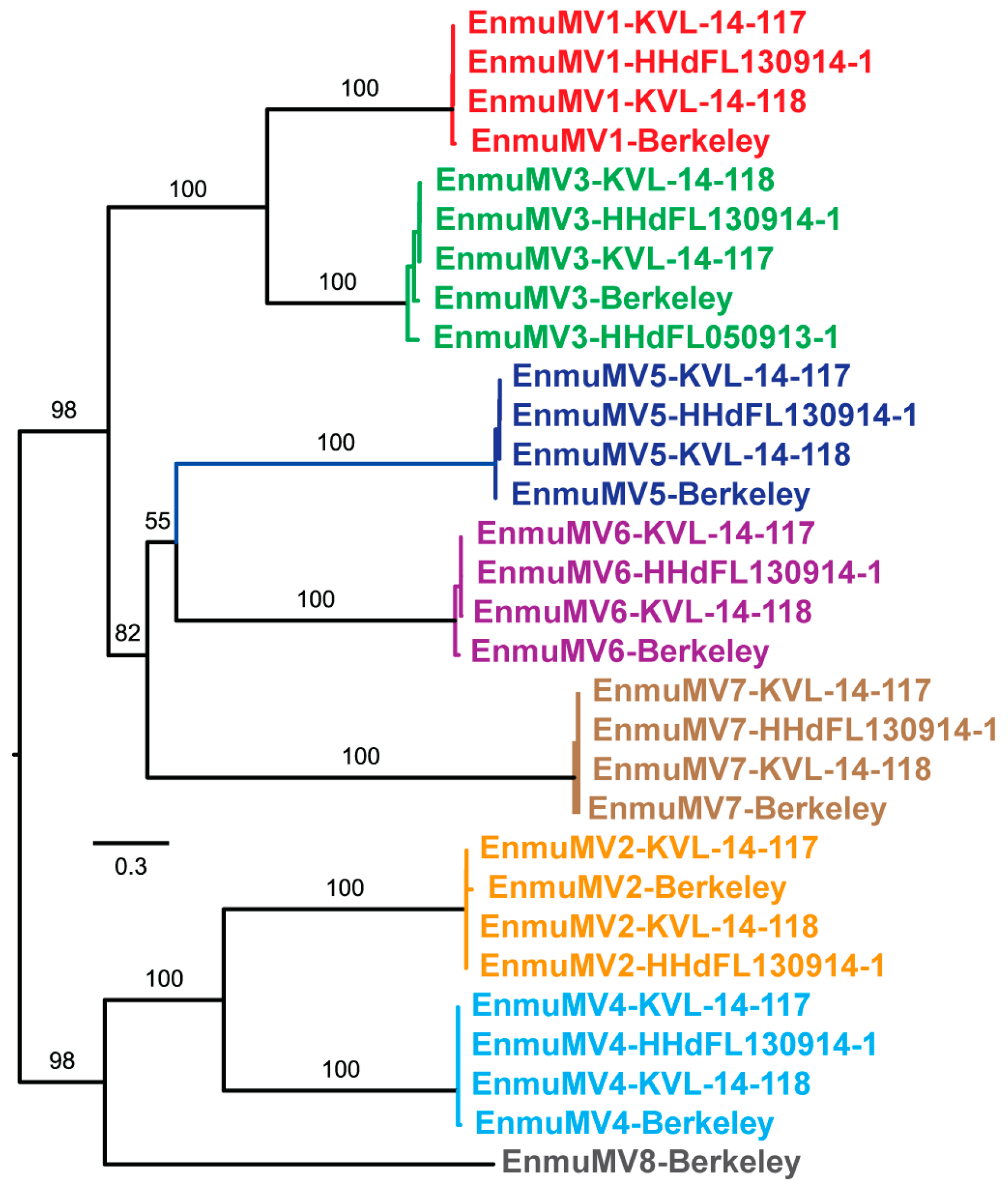
Features of the mitovirus-like contigs described above led us to suspect that there are multiple new mitovirus species represented by these sequences. To address this possibility, we performed pairwise comparisons of the nt sequences using EMBOSS Needleall. These comparisons grouped the sequences into eight distinct clusters, with pairwise identities >86% within each cluster and <50% between any two clusters (Figure S2). The degree of divergence between these clusters, as well as the low identity scores with previously reported mitoviruses found in the BLASTP searches described above (≤42%), led us to conclude that these eight clusters represent eight new mitovirus species. Phylogenetic analyses involving the E. muscae mitoviruses alone corroborated this conclusion by showing the distribution of the 30 viruses into eight well-delimited clades (Figure 2). In addition, by examining which members of each clade derive from which E. muscae isolate, we found that one of the E. muscae isolates (Berkeley; from fruit fly) harbors strains of all eight of the new mitovirus species, three of the E. muscae isolates (KVL-14-117, KVL-14-118, and HHdFL130914-1; from house flies) harbor strains of even of the new mitovirus species, and the remaining one isolate (HHdFL050913-1; from cabbage fly) harbors a strain of only one of the new mitovirus species (Figure 1 and Figure 2). We suggest the names of these new species to be “Entomophthora muscae mitovirus 1” through “Entomophthora muscae mitovirus 8” and further suggest the abbreviations EnmuMV1 through EnmuMV8 for the common names of these viruses. Six of these eight viruses (EnmuMV1, EnmuMV2, EnmuMV4, EnmuMV5, EnmuMV6, and EnmuMV7) are represented by four strains each, one of these viruses (EnmuMV3) is represented by five strains, and the remaining one of these viruses (EnmuMV8) is represented by only one strain in the results presented here. In fact, the few short mitovirus-like contigs assembled from fly-grown E. muscae cabbage fly isolate HHdFL040913-2 are sufficient to identify it as a sixth strain of EnmuMV3, most closely related to EnmuMV3-HHdFL050913-1 (95% nt identity), i.e., from another cabbage fly isolate of E. muscae.
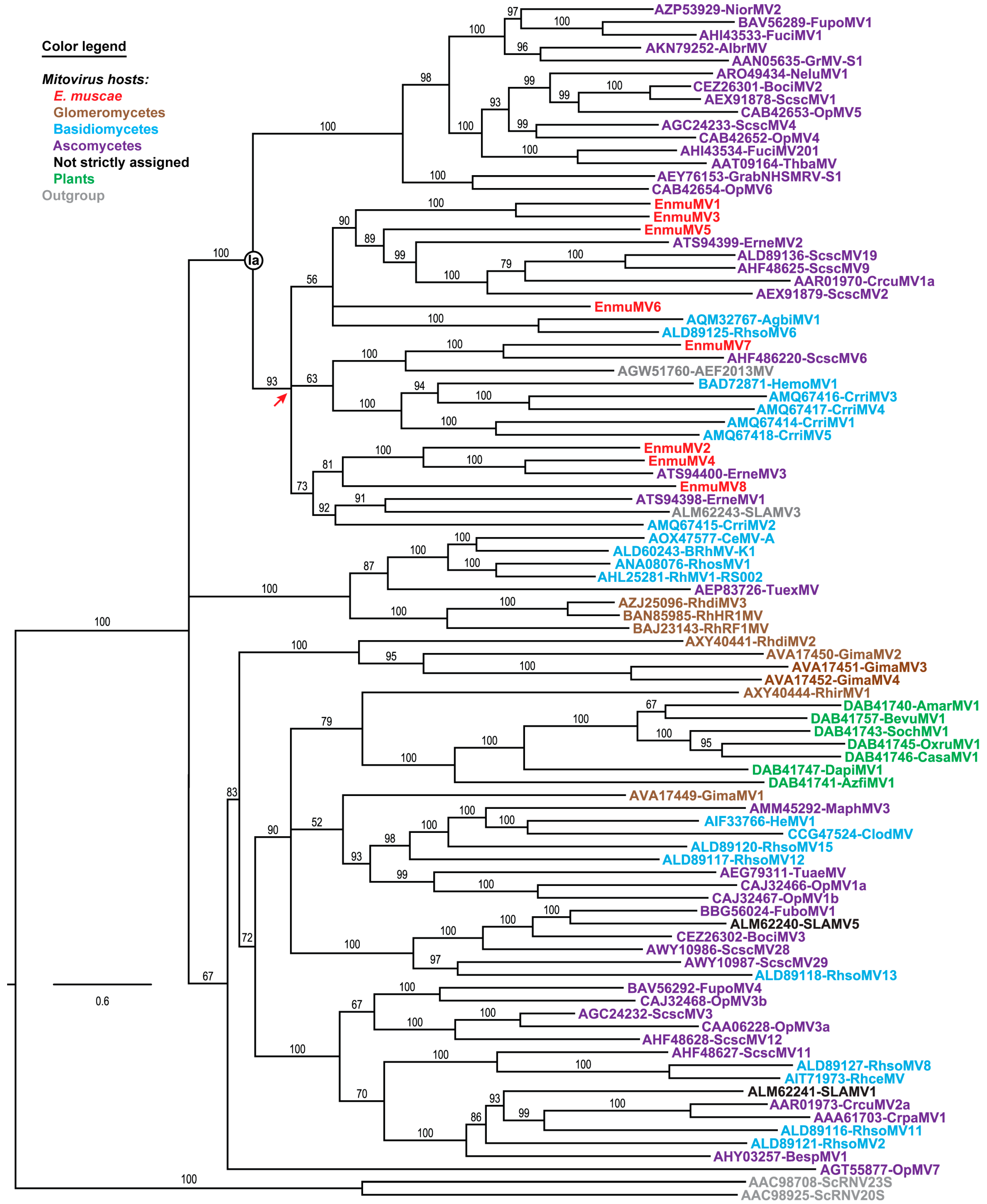
Phylogenetic analyses were next performed to ascertain the positions of the eight E. muscae mitoviruses relative to others in current genus Mitovirus. A representative set of 83 previously reported, full-length mitovirus RdRp sequences were obtained from the Protein database at NCBI and then used in multiple sequence alignments along with the eight mitovirus RdRp sequences from E. muscae isolate Berkeley, plus two RdRp sequences from members of current genus Narnavirus as an outgroup. Each multiple sequence alignment was analyzed using the ModelFinder module within IQ-TREE to identify the best-fit substitution model, which was then directly applied for maximum-likelihood phylogenetic analyses within IQ-TREE. As shown by the representative phylogram in Figure 3, the results reveal that EnmuMV1–EnmuMV8 (red) all cluster within the same discernible subclade of current genus Mitovirus, within a portion of the major clade previously designated Ia. This clustering suggests that all eight of these mitoviruses from E. muscae shared a most recent common ancestor, from which they all diverged, near the root of this subclade (red arrow in Figure 3), i.e., fairly early in mitovirus evolution but still well removed from the root.
By aligning the sequences of the different strains within each mitovirus species from E. muscae, we observed that the coding sequences for each species are marked by a total absence of indels among the strains, such that RdRp length is conserved within each species (Figure 1). One minor exception is that in EnmuMV2-Berkeley, there is a slightly earlier stop codon (UAA) resulting from a C-to-U substitution relative to the other EnmuMV2 strains, such that the RdRp length of EnmuMV2-Berkeley is reduced by 5 aa. In contrast, within the NTRs, several small indels are seen in the alignments among certain strains from the same species (EnmuMV1, EnmuMV2, EnmuMV3, and EnmuMV7), and especially in their 5´ NTRs (Figure S3). The putative functions of the NTRs in RNA replication, translation, etc., are thus presumably able to accommodate small indels in some positions better than the functions of the coding region and/or the encoded RdRp of each species.
3.4. UGA(Trp) Codons in Mitochondrial Core Genes of E. muscae
A previous report has revealed that UGA(Trp) is a rarely used codon in mitochondrial core genes of many basal fungi [4], including Zancudomyces (Smittium) culisetae from subphylum Kickxellomycotina, phylum Zoopagomycota, the only member of this basal phylum for which a complete mitochondrial genome sequence had been annotated as such at NCBI at that time (accession NC_006837.1) [41]. Simplistically, one might therefore expect UGA(Trp) to be a rarely used codon also in mitochondrial genes of subphylum Entomophthoromycotina, phylum Zoopagomycota. The evidence presented above for numerous UGA(Trp) codons in the mitovirus sequences from E. muscae, however, run counter to this expectation. To address this possible discrepancy, we sought to determine whether UGA(Trp) codons are found, too, in mitochondrial core genes of E. muscae. We first performed TBLASTN searches for transcript contigs representing mitochondrial core genes in TSA database entries for subphylum Entomophthoromycotina, using the deduced sequences of 14 mitochondrial core proteins from Z. culisetae as queries. Strong hits, all from the E. muscae transcriptome study BioProject PRJEB10825 [25], were obtained for 12 of the mitochondrial core genes: atp6, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad5, and nad6.
We next performed BLASTP searches of the NR database to discern which of the TSA hits for the 12 mitochondrial core genes appear to have originated from a basal fungus. We then used those TSA accessions (see Materials and Methods for accession numbers) as queries to search the SRA libraries from which the mitovirus sequences from E. muscae were assembled as described above, beginning with the SRA transcriptome libraries from E. muscae isolate Berkeley (BioProject PRJNA435715) [24]. Using the matching sequence reads that we identified, we were then ultimately able to assemble complete coding sequences for all 12 of these mitochondrial core genes from E. muscae isolate Berkeley (Figure 4). Additional BLASTP searches of the NR database strongly supported the conclusion that the mitochondrial protein sequences deduced from these contigs derived from a member of subphylum Entomophthoromycotina, phylum Zoopagomycota, given that the top-scoring one or two hit(s) for each protein consistently derived from Conidiobolus species C. coronatus or C. heterosporus, the latter for which a complete mitochondrial genome sequence has only recently been deposited and annotated at NCBI (accession MK049352.1) [43] (Table 1). Moreover, phylogenetic analyses using the concatenated sequences of these 12 mitochondrial proteins from E. muscae, along with those from other representative fungi, placed the E. muscae proteins adjacent to those of C. heterosporus within the Zoopagomycota clade (Figure S4).
To provide additional evidence that these transcript contigs from E. muscae isolate Berkeley represent the respective mitochondrial genes of E. muscae, we used the contigs to search the SRA transcriptome libraries from the four other E. muscae isolates KVL-14-117, KVL-14-118, HHdFL130914-1, and HHdFL050913-1. The results summarized in Figure 4 reflect that nearly identical coding sequences for all 12 mitochondrial genes were assembled from sequence reads from all four other E. muscae isolates. Complete coding sequences were assembled for eight to ten of these genes from each isolate and partial coding sequences for the other genes (Figure 4), with single-nt mismatches at only a few nt positions between any two isolates. These findings provide further strong support for the conclusion that these 12 mitochondrial coding sequences indeed derive from E. muscae.
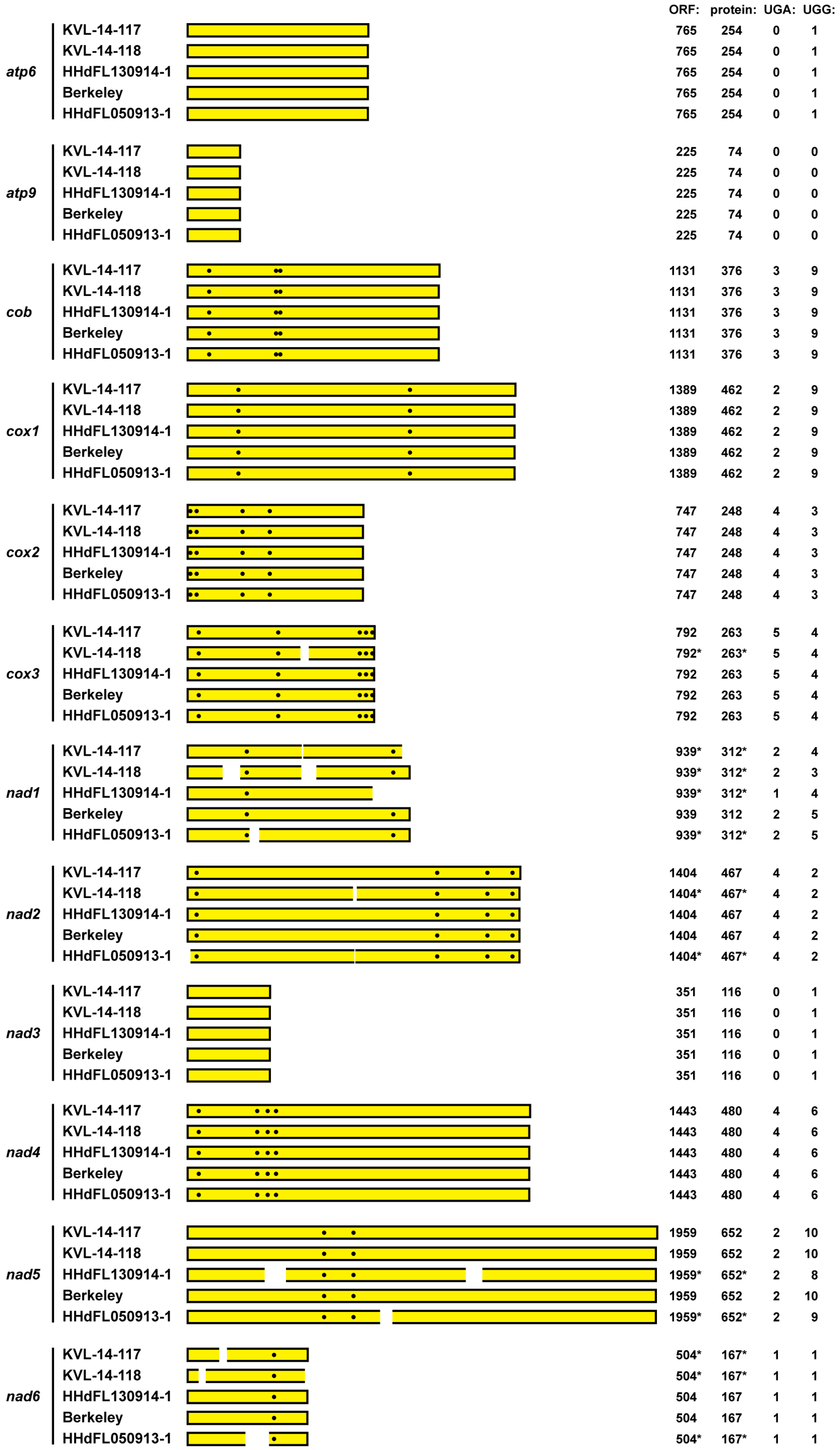
We lastly examined the 12 mitochondrial coding sequences from E. muscae for UGA (Trp) codons. At least one and as many as five UGA (Trp) codons each are found in nine of these sequences, namely, in all but those for atp6, atp9, and nad3, which also contain no or only one UGG (Trp) codon each (Figure 4). Notably as well, these UGA (Trp) codons are conserved among the sequences assembled from the five different E. muscae isolates (Figure 4). Overall in these coding sequences, 35% of the Trp codons are UGA (vs. 65% UGG). We therefore conclude that UGA (Trp) is not a rarely used codon in the mitochondrial core genes of E. muscae, consistent with our findings for the mitovirus sequences from this fungus. Our initial generalization that UGA (Trp) might be expected to be a rarely used codon in the mitochondria of all basal fungi thus appears to have been incorrect.
4. Discussion
The eight mitovirus species newly identified in this report are the first to be discovered in a fungal host from phylum Zoopagomycota (subphylum Entomophthoromycotina). Before this, the most phylogenetically basal fungi [1,2] that had been found to harbor mitoviruses were ones from phylum Mucoromycota (subphylum Glomeromycotina) [5,26,27,28]. The mitoviruses in Entomophthora muscae might have been acquired by horizontal transmission, perhaps from some more apically diverging fungi given their juxtaposition with ascomycete and basidiomycete mitoviruses in the Figure 3 tree. The explanation that we favor, however, is that an early ancestor of the E. muscae mitoviruses first entered the fungal lineage in an ancestral species that predated the divergence of phylum Zoopagomycota from the three more apically diverging phyla Mucoromycota, Basidiomycota, and Ascomycota, and was then transmitted largely by vertical means over the subsequent course of fungal evolution, including to E. muscae. See Figure S4 for a fungal tree of life based on mitochondrial proteins, which shows a similar pattern of progressive divergence of the fungal phyla to that previously shown for nuclear products [1,2], and which was similarly shown for fungal mitochondrial proteins by Nie et al. [43] in their recent report on the complete mitochondrial genome sequence of entomophthoroid fungus Conidiobolus heterosporus. The reciprocal possibility, i.e., that E. muscae mitoviruses have been horizontally transmitted to some more apically diverging fungi, must also be considered given their juxtapositions in the Figure 3 tree. For example, note the apparent proximities of Erysiphe necator (ascomycete) mitovirus 1 (ATS94400) to EnmuMV4 and Sclerotinia sclerotiorum (ascomycete) mitovirus 6 (AHF48622) to EnmuMV7.
Four of the E. muscae isolates whose mitoviruses were characterized in this study were each found to contain strains of either seven or eight different mitovirus species. Although this large number might seem odd, similar findings have been made in other fungi. For example, strains of at least seven different mitovirus species have been found in the same isolate, Ld, of the Dutch elm disease fungus Ophiostoma novo-ulmi (phylum Ascomycota) [39,44,45]; strains of six different mitovirus species have been found in the same isolate, AG2-2-IV, of Rhizoctonia solani (phylum Basidiomycota) [46]; and strains of five different mitovirus species have been found in the same isolate, BC-u3, of the white pine blister rust fungus Cronartium ribicola (phylum Basidiomycota) [47]. Such multiple co-infections raise interesting questions about possible interactions among the different viruses in each isolate and whether there are selective forces or circumstances that favor multiple co-infections. The fungus E. muscae is multinucleate throughout all stages of its life cycle, and both protoplast cells and asexual spores may contain up to 20 nuclei each [25]. Although mitochondrial morphology and membrane potential dynamics are considered independent of nuclear cycle state [48], the consistent presence of multiple nuclei might facilitate a heterogeneous intracellular environment conducive to the presence of multiple mitoviruses. Another, more trivial possibility relates to the fact that the E. muscae isolates whose sequence reads were analyzed for this report originated not from individual spores, but from conidial showers from infected flies, meaning that there remains a possibility that these isolates are mixtures of E. muscae strains, with differences in mitovirus content between strains.
The findings in this report, including both mitovirus and mitochondrial coding sequences, suggest that the mitochondrial protein synthesis machinery in E. muscae can translate codon UGA (Trp) more efficiently than can that in many other basal fungi [4]. In this regard, E. muscae mitochondria are more like those of most members of the more apically diverging phyla Ascomycota and Basidiomycota. As noted in Results, UGA (Trp) has been shown to be a rarely used mitochondrial codon in Zancudomyces culistae (subphylum Kickxellomycotina, phylum Zoopagomycota; no UGA(Trp) codons in the 12 mitochondrial core-gene coding sequences examined here), such that there appears to be some variation in mitochondrial UGA (Trp) codon usage across members of phylum Zoopagomycota. In the recently reported mitochondrial genome sequence of Conidiobolus heterosporus (subphylum Entomophthoromycotina, phylum Zoopagomycota) [43], we find that UGA (Trp) is also somewhat uncommonly used (6 UGA (Trp) vs. 62 UGG (Trp) codons in the same 12 mitochondrial coding sequences, 8.8%), even though C. heterosporus encodes a mitochondrial tRNA sequence with anticodon UCA, which should allow for more efficient translation of UGA (Trp). A complete mitochondrial genome sequence for E. muscae, and for other members of subphylum Entomophthoromycotina, may be helpful for understanding why UGA (Trp) codons are more commonly used in the mitochondria of E. muscae than in those of related species C. heterosporus (same order, Entomophorales; different families, Entomophthoraceae and Ancylistaceae, respectively).
The RNA samples that were analyzed for E. muscae isolates KVL-14-118, Berkeley, and HHdFL050913-1 in the original transcriptome studies (BioProjects PRJEB10825 and PRJNA435715) [24,25] were obtained from infected host flies. One might therefore have some concern that the mitovirus sequences identified from those samples could have originated from the flies, or from a different fly symbiont, rather than from E. muscae itself. Importantly, however, the samples from E. muscae isolate KVL-14-117 analyzed in the original transcriptome studies (BioProject PRJNA372837) [25] and then further employed here were obtained from media-grown cultures of the fungus, serving to allay such possible concern. Indeed, the general absence of fly-derived sequence reads in the SRA libraries from media-grown E. muscae isolate KVL-14-117 [25] was confirmed in this study by use of several fly-derived mitochondrial gene queries. In addition, for E. muscae isolate Berkeley, the original transcriptome study (BioProject PRJNA435715) [24] included a number of samples from flies not inoculated with E. muscae, and the SRA libraries from those control samples contain no or very few sequence reads matching the E. muscae mitoviruses (Table S4). Moreover, the SRA libraries from the samples from inoculated flies in that study contain increasing numbers of sequence reads matching the E. muscae mitoviruses at increasing times after inoculation with E. muscae isolate Berkeley, consistent with the growth of E. muscae in those flies following inoculation (Table S4). Both of these observations provide strong evidence that the mitoviruses are associated with E. muscae isolate Berkeley itself, not the fly hosts. Lastly, we have been recently able to confirm the presence of all seven mitovirus sequences in new transcriptome data from conidial samples of E. muscae isolate HHdFL130914-1, which will be described in detail in a future article [49] and also provides important evidence for vertical transmission of the E. muscae mitoviruses via conidiospores. The consistency of sequence findings that we obtained from the different E. muscae isolates, for both mitovirus and mitochondrial coding sequences and despite these isolates having been originally obtained from three different species of host flies, also strongly supports the conclusion that these sequences originated from E. muscae.
By convention, each ORF diagrammed in Figure 1 and Figure 4 is considered to start with the first in-frame AUG codon. Because an alternative start codon might be used in some cases, however, it remains possible that some of these ORFs may be longer or shorter at their 5´ ends than suggested here. In this regard, we observed in particular that the cox1 ORF of E. muscae contains a conserved in-frame GUG codon well-upstream of the first in-frame AUG (see Data S2), which would extend that ORF and its encoded protein considerably, to 1587 nt and 528 aa, respectively, bringing those lengths more in line with those of C. heterosporus [43] and most other organisms. For the mitoviruses, considering the strains with a longer region of sequence between the first in-frame AUG and the first upstream in-frame stop codon (see Figure 1), use of an alternative start codon upstream of the first in-frame AUG seems most likely for EnmuMV4, EnmuMV5, and EnmuMV8. Reciprocally, considering those viruses with shorter 5´ NTRs preceding the first in-frame AUG (see Figure 1), use of a start codon downstream of the first in-frame AUG seems most probable for EnmuMV6. Apparently notable in these regards, use of an alternative start codon upstream of the first in-frame AUG in EnmuMV4, EnmuMV5, and EnmuMV8 could provide for several additional residues near the RdRp N-terminus that would be conserved among all of the mitovirus strains reported here, and use of the second in-frame AUG codon to initiate translation in EnmuMV6 would retain these conserved residues (Figure S5). We therefore consider it likely that the first in-frame AUG is not the true start codon for RdRp translation in several of these viruses. In particular, for EnmuMV4, EnmuMV5, and EnmuMV8, we propose that an upstream AUU start codon is used instead, and for EnmuMV6, we propose that the second in-frame AUG codon is used instead (see Figure S3, Figure S5, and Data S2).
Coyle et al. [50] recently identified three SRA transcriptome libraries derived from mixtures of wild-caught flies, at least some of which were apparently harboring E. muscae when their RNAs were sampled (SRX955881, SRX955902, and SRX1711976; from BioProjects PRJNA277921 and PRJNA318834 [12]). Upon performing MegaBLAST searches of those libraries using the sequences of EnmuMV1–8 as queries, we found that two of the libraries, SRX955902 and SRX1711976, include sequence reads matching the E. muscae mitoviruses. Specifically, SRX955902 includes reads matching all eight of these viruses, and SRX1711976 includes reads matching four of these viruses (EnmuMV1, EnmuMV3, EnmuMV6, and EnmuMV7) (Table S5). Only the reads matching EnmuMV7 from SRX1711976 were sufficient in coverage to allow assembly of a complete coding sequence for this virus, which turns out to correspond (99.9% nt-sequence identity) to Hubei narna-like virus 25 (GenBank KX883546; 88.7–91.0% pairwise nt-sequence identity with the four EnmuMV7 strains from E. muscae detailed above). Thus, although Hubei narna-like virus 25 has been previously reported as an insect mitovirus [12], it is now shown to derive more likely from the fly-associated entomophthoroid fungus E. muscae or perhaps a related fungus. These findings provide further support for the conclusion that these and perhaps other yet-to-be-discovered mitoviruses are widespread in naturally occurring populations of E. muscae.
The behavioral effects of E. muscae on host flies are fascinating and are indeed the subjects of ongoing genetic and neuroscientific investigations in Drosophila [24]. Interestingly, a distinct plus-strand RNA virus from E. muscae, an iflavirus (order Picornavirales), has been recently reported by Coyle et al. [50] and speculated to contribute to the manipulation of fly-host behaviors by E. muscae. The E. muscae mitoviruses that we describe here might also be considered as possible contributors in that regard. On a related note, two novel mitoviruses were recently identified in the entomopathogenic fungus Ophiocordyceps sinensis (phylum Ascomycota) [51], which causes behavioral perturbations and death of its caterpillar hosts. In this case, the dying caterpillar, which had been feeding as normal on plant roots underground, appears to be induced by the infecting fungus to crawl into a position and orientation relative to the soil surface that is ideal for growth of the fungal fruiting body aboveground, promoting efficient dispersal of the fungal spores [52]. These effects are clearly analogous to those of E. muscae in flies, and it is fascinating to speculate that again in this case the fungal mitoviruses might in some way contribute to the manipulation of insect host behaviors by the entomopathogenic fungus.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4915/11/4/351/s1, Figure S1: Multiple sequence alignment of deduced RdRp sequences of mitoviruses from E. muscae isolate Berkeley, Figure S2: Sequence identity scores from pairwise alignments of E. muscae mitovirus sequences, Figure S3: Multiple sequence alignments of 5´ and 3´ NTR sequences of E. muscae mitoviruses, Figure S4: Fungal tree of life based on mitochondrial core-protein sequences from representative species, Figure S5: Multiple sequence alignment of N-terminal regions of deduced RdRp sequences of E. muscae mitoviruses, Table S1: Contig assembly values for coding-complete mitovirus sequences from E. muscae, Table S2: Contig assembly values for mitochondrial core-gene coding sequences from E. muscae, Table S3: SRA accessions used for contig assemblies in this study, Table S4: Mitovirus-matching reads in samples from fruit flies infected or not with E. muscae isolate Berkeley per Elya et al. [24], Table S5: Mitovirus-matching reads in samples from SRA transcriptome libraries derived from mixtures of wild-caught flies apparently infected with E. muscae per Coyle et al. [50], Table S6: Mitovirus RdRp sequences used for phylogenetic analyses, Data S1: E. muscae mitovirus sequences, Data S2: E. muscae mitochondrial coding sequences.
Author Contributions
Conceptualization, M.L.N. and H.J.D.; Investigation, M.L.N., H.J.D., A.R.M., I.V.G., and H.H.D.F.L.; Methodology, M.L.N., H.J.D., A.R.M., I.V.G., and H.H.D.F.L.; Validation, M.L.N., H.J.D., A.R.M., I.V.G., and H.H.D.F.L.; Writing—Original Draft Preparation, M.L.N.; Writing—Review & Editing, M.L.N., H.J.D., A.R.M., I.V.G., and H.H.D.F.L.
Funding
This research was funded in part by NIH Grant T32 AI007245 to the Ph.D. program in Virology at Harvard University (A.R.M.). The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 (I.V.G.). H.H.D.F.L. was supported by the Independent Research Fund Denmark and the Villum Foundation (Grant No. 10122).
Acknowledgments
We wish to acknowledge and thank those scientists who made this study possible by depositing their sequence reads and shotgun assemblies in the public databases. We also thank Carolyn Elya (Harvard University) for helpful discussions and permission to use her SRA data from NCBI.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Spatafora, J.W.; Chang, Y.; Benny, G.L.; Lazarus, K.; Smith, M.E.; Berbee, M.L.; Bonito, G.; Corradi, N.; Grigoriev, I.; Gryganskyi, A.; et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 2016, 108, 1028–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The fungal tree of life: From molecular systematics to genome-scale phylogenies. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Hillman, B.I.; Cai, G. The family Narnaviridae: Simplest of RNA viruses. Adv. Virus Res. 2013, 86, 149–176. [Google Scholar]
- Nibert, M.L. Mitovirus UGA(Trp) codon usage parallels that of host mitochondria. Virology 2017, 507, 96–100. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shimura, H.; Kitahara, R.; Masuta, C.; Ezawa, T. A novel virus-like double-stranded RNA in an obligate biotroph arbuscular mycorrhizal fungus: A hidden player in mycorrhizal symbiosis. Mol. Plant Microbe Interact. 2012, 25, 1005–1012. [Google Scholar] [CrossRef]
- Rogers, H.J.; Buck, K.W.; Brasier, C.M. A mitochondrial target for the double-stranded RNAs in diseased isolates of the fungus that causes dutch elm disease. Nature 1987, 329, 558–560. [Google Scholar] [CrossRef]
- Polashock, J.J.; Hillman, B.I. A small mitochondrial double-stranded (ds) RNA element associated with a hypovirulent strain of the chestnut blight fungus and ancestrally related to yeast cytoplasmic T and W dsRNAs. Proc. Natl. Acad. Sci. USA 1994, 91, 8680–8684. [Google Scholar] [CrossRef] [PubMed]
- Polashock, J.J.; Bedker, P.J.; Hillman, B.I. Movement of a small mitochondrial double-stranded RNA element of Cryphonectria parasitica: Ascospore inheritance and implications for mitochondrial recombination. Mol. Gen. Genet. 1997, 256, 566–571. [Google Scholar] [CrossRef]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef]
- Nerva, L.; Vigani, G.; Di Silvestre, D.; Ciuffo, M.; Forgia, M.; Chitarra, W.; Turina, M. Biological and molecular characterization of Chenopodium quinoa mitovirus 1 reveals a distinct sRNA response compared to cytoplasmic RNA viruses. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Chung, B.Y.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glücksman, E.; Wang, H.; Brown, T.D.; et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS ONE 2013, 8, e80720. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Bruenn, J.A. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003, 31, 1821–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vong, M.; Manny, A.R.; Smith, K.L.; Gao, W.; Nibert, M.L. Beta vulgaris mitovirus 1 in diverse cultivars of beet and chard. Virus Res. 2019, 265, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, L.; Li, G.; Jiang, D.; Ghabrial, S.A. Genome characterization of a debilitation-associated mitovirus infecting the phytopathogenic fungus Botrytis cinerea. Virology 2010, 406, 117–126. [Google Scholar] [CrossRef]
- Wu, M.D.; Zhang, L.; Li, G.Q.; Jiang, D.H.; Hou, M.S.; Huang, H.C. Hypovirulence and double-stranded RNA in Botrytis cinerea. Phytopathology 2007, 97, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ghabrial, S.A. Molecular characterization of two mitoviruses co-infecting a hypovirulent isolate of the plant pathogenic fungus Sclerotinia sclerotiorum. Virology 2012, 428, 77–85. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, S.; Liu, L.; Cheng, J.; Fu, Y.; Jiang, D.; Xie, J. A mitovirus related to plant mitochondrial gene confers hypovirulence on the phytopathogenic fungus Sclerotinia sclerotiorum. Virus Res. 2015, 197, 127–136. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. MBio 2018, 9. [Google Scholar] [CrossRef]
- Shaikh, N.; Hussain, K.A.; Petraitiene, R.; Schuetz, A.N.; Walsh, T.J. Entomophthoramycosis: A neglected tropical mycosis. Clin. Microbiol. Infect. 2016, 22, 688–694. [Google Scholar] [CrossRef]
- Vilela, R.; Mendoza, L. Human pathogenic Entomophthorales. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Araújo, J.P.; Hughes, D.P. Diversity of entomopathogenic fungi: Which groups conquered the insect body? Adv. Genet. 2016, 94, 1–39. [Google Scholar]
- Jensen, A.B.; Thomsen, L.; Eilenberg, J. Value of host range, morphological, and genetic characteristics within the Entomophthora muscae species complex. Mycol. Res. 2006, 110, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Elya, C.; Lok, T.C.; Spencer, Q.E.; McCausland, H.; Martinez, C.C.; Eisen, M. Robust manipulation of the behavior of Drosophila melanogaster by a fungal pathogen in the laboratory. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- De Fine Licht, H.H.; Jensen, A.B.; Eilenberg, J. Comparative transcriptomics reveal host-specific nucleotide variation in entomophthoralean fungi. Mol. Ecol. 2017, 26, 2092–2110. [Google Scholar] [CrossRef]
- Kitahara, R.; Ikeda, Y.; Shimura, H.; Masuta, C.; Ezawa, T. A unique mitovirus from Glomeromycota, the phylum of arbuscular mycorrhizal fungi. Arch. Virol. 2014, 159, 2157–2160. [Google Scholar] [CrossRef]
- Neupane, A.; Feng, C.; Feng, J.; Kafle, A.; Bücking, H.; Lee Marzano, S.Y. Metatranscriptomic analysis and in silico approach identified mycoviruses in the arbuscular mycorrhizal fungus Rhizophagus spp. Viruses 2018, 10, 707. [Google Scholar] [CrossRef] [PubMed]
- Turina, M.; Ghignone, S.; Astolfi, N.; Silvestri, A.; Bonfante, P.; Lanfranco, L. The virome of the arbuscular mycorrhizal fungus Gigaspora margarita reveals the first report of DNA fragments corresponding to replicating non-retroviral RNA viruses in fungi. Environ. Microbiol. 2018, 20, 2012–2025. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Przhibelskiy, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. bioRxiv 2018. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Becher, P.G.; Jensen, R.E.; Natsopoulou, M.E.; Verschut, V.; De Fine Licht, H.H. Infection of Drosophila suzukii with the obligate insect-pathogenic fungus Entomophthora muscae. J. Pest Sci. 2018, 91, 781–787. [Google Scholar] [CrossRef]
- Marienfeld, J.R.; Unseld, M.; Brandt, P.; Brennicke, A. Viral nucleic acid sequence transfer between fungi and plants. Trends Genet. 1997, 13, 260–261. [Google Scholar] [CrossRef]
- Hong, Y.; Cole, T.E.; Brasier, C.M.; Buck, K.W. Evolutionary relationships among putative RNA-dependent RNA polymerases encoded by a mitochondrial virus-like RNA in the Dutch elm disease fungus, Ophiostoma novo-ulmi, by other viruses and virus-like RNAs and by the Arabidopsis mitochondrial genome. Virology 1998, 246, 158–169. [Google Scholar] [CrossRef]
- Bruenn, J.A.; Warner, B.E.; Yerramsetty, P. Widespread mitovirus sequences in plant genomes. PeerJ 2015, 3, e876. [Google Scholar] [CrossRef]
- Seif, E.; Leigh, J.; Liu, Y.; Roewer, I.; Forget, L.; Lang, B.F. Comparative mitochondrial genomics in zygomycetes: Bacteria-like RNase P RNAs, mobile elements and a close source of the group I intron invasion in angiosperms. Nucleic Acids Res. 2005, 33, 734–744. [Google Scholar] [CrossRef]
- Schon, E.A. Appendix 4. Mitochondrial genetic codes in various organisms. Methods Cell Biol. 2007, 80, 831–833. [Google Scholar]
- Nie, Y.; Wang, L.; Cai, Y.; Tao, W.; Zhang, Y.J.; Huang, B. Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Appl. Microbiol. Biotechnol. 2018, 103, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Dover, S.L.; Cole, T.E.; Brasier, C.M.; Buck, K.W. Multiple mitochondrial viruses in an isolate of the Dutch Elm disease fungus Ophiostoma novo-ulmi. Virology 1999, 258, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.; Coutts, R.H.A.; Brasier, C.M.; Buck, K.W. Sequence of RNA-dependent RNA polymerase genes provides evidence for three more distinct mitoviruses in Ophiostoma novo-ulmi isolate Ld. Virus Genes 2006, 33, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Bartholomäus, A.; Wibberg, D.; Winkler, A.; Pühler, A.; Schlüter, A.; Varrelmann, M. Deep sequencing analysis reveals the mycoviral diversity of the virome of an avirulent isolate of Rhizoctonia solani AG-2-2 IV. PLoS ONE 2016, 11, e0165965. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Chan, D.; Xiang, Y.; Williams, H.; Li, X.R.; Sniezko, R.A.; Sturrock, R.N. Characterization of five novel mitoviruses in the white pine blister rust fungus Cronartium ribicola. PLoS ONE 2016, 11, e0154267. [Google Scholar] [CrossRef] [PubMed]
- Gerstenberger, J.P.; Occhipinti, P.; Gladfelter, A.S. Heterogeneity in mitochondrial morphology and membrane potential is independent of the nuclear division cycle in multinucleate fungal cells. Eukaryot. Cell 2012, 11, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Natsopoulou, M.E.; Nunn, A.; Hansen, A.N.; Veselská, T.; Gryganskyi, A.; Groth, M.; Felder, M.; Kolarík, M.; Grigoriev, I.; Voigt, K.; et al. Dual RNA-Seq analysis during infection of house flies with the insect-pathogenic fungus Entomophthora muscae reveals unique molecular interactions between host and pathogen. Manuscript in preparation.
- Coyle, M.C.; Elya, C.N.; Bronski, M.J.; Eisen, M.B. Entomophthovirus: An insect-derived iflavirus that infects a behavior manipulating fungal pathogen of dipterans. bioRxiv 2018. [Google Scholar] [CrossRef]
- Gilbert, K.; Holcomb, E.E.; Allscheid, R.L.; Carrington, J. Discovery of new mycoviral genomes within publicly available fungal transcriptomic datasets. bioRxiv 2019. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, E.W.; Wang, C.S.; Li, Y.L.; Liu, X.Z. Ophiocordyceps sinensis, the flagship fungus of China: Terminology, life strategy and ecology. Mycology 2012, 3, 2–10. [Google Scholar]
Figure 1.
Summary diagram of mitovirus sequences from E. muscae isolates. Mitovirus strains from five different E. muscae isolates, representing eight apparent new mitovirus species, are labeled at left. The RdRp-encoding open reading frame ORF is shown as a colored box within each assembled RNA sequence (see matching colors in Figure 2), starting with the first in-frame AUG codon by convention. The position of the first in-frame stop codon upstream of the proposed AUG start codon is shown as a colored line. Positions of UGA (Trp) codons in each RdRp ORF are shown as white dots. RNA sequences are aligned with respect to the catalytic motif GDD in each deduced RdRp sequence. Positions of two small gaps in sequencing coverage in the assembled EnmuMV2-KVL-14-118 sequence are shown as white breaks in the boxes; asterisks indicate inferred values consequent to these coverage gaps. For each sequence, lengths of the 5´ NTR, 3´ NTR, and overall RNA sequence are labeled in nt; length of the RdRp sequence in aa; and UGA and UGG codons in raw counts.
Figure 1.
Summary diagram of mitovirus sequences from E. muscae isolates. Mitovirus strains from five different E. muscae isolates, representing eight apparent new mitovirus species, are labeled at left. The RdRp-encoding open reading frame ORF is shown as a colored box within each assembled RNA sequence (see matching colors in Figure 2), starting with the first in-frame AUG codon by convention. The position of the first in-frame stop codon upstream of the proposed AUG start codon is shown as a colored line. Positions of UGA (Trp) codons in each RdRp ORF are shown as white dots. RNA sequences are aligned with respect to the catalytic motif GDD in each deduced RdRp sequence. Positions of two small gaps in sequencing coverage in the assembled EnmuMV2-KVL-14-118 sequence are shown as white breaks in the boxes; asterisks indicate inferred values consequent to these coverage gaps. For each sequence, lengths of the 5´ NTR, 3´ NTR, and overall RNA sequence are labeled in nt; length of the RdRp sequence in aa; and UGA and UGG codons in raw counts.

Figure 2.
Maximum-likelihood phylogenetic analysis of E. muscae mitoviruses. Deduced RdRp sequences were aligned using MAFFT-L-INS-i. The best-fit (per BIC score) aa-substitution model used for the phylogenetic analysis shown here was LG+I+G4. The tree is displayed as a midpoint-rooted rectangular phylogram with branch support values from UFboot (1000 replicates) shown in %; support values for distal branches have been deleted for visual clarity. Scale bar indicates average number of substitutions per alignment position. Labels of different colors highlight the clustering of strains of eight different mitovirus species suggested by these results, as well as by the results in Figure 1 and Figure S2.
Figure 2.
Maximum-likelihood phylogenetic analysis of E. muscae mitoviruses. Deduced RdRp sequences were aligned using MAFFT-L-INS-i. The best-fit (per BIC score) aa-substitution model used for the phylogenetic analysis shown here was LG+I+G4. The tree is displayed as a midpoint-rooted rectangular phylogram with branch support values from UFboot (1000 replicates) shown in %; support values for distal branches have been deleted for visual clarity. Scale bar indicates average number of substitutions per alignment position. Labels of different colors highlight the clustering of strains of eight different mitovirus species suggested by these results, as well as by the results in Figure 1 and Figure S2.

Figure 3.
Maximum-likelihood phylogenetic analysis of E. muscae mitoviruses and representative other mitoviruses. Deduced RdRp sequences were aligned using MAFFT-L-INS-i. The best-fit substitution model (per BIC score) used for the phylogenetic analysis shown here was LG + F + R8. The site proportions and rates for the FreeRate model were (0.0224, 0.0123) (0.0271, 0.0755), (0.0627, 0.2087), (0.1161, 0.4202), (0.1784, 0.6877), (0.2655, 1.0570), (0.2163, 1.4069), and (0.1115, 2.0462). The tree is displayed as a rectangular phylogram rooted on a set of two members of current genus Narnavirus included as outgroup. Branch support values from UFboot (1000 replicates) are shown in %; branches with <50% support have been collapsed to the preceding node. Scale bar indicates average number of substitutions per alignment position. Previously identified clade Ia of current genus Mitovirus is labeled. Red arrow, most recent common ancestor shared by all eight E. muscae mitoviruses. Virus names matching the sequence accession numbers and abbreviations are provided in Table S6. Color-coding: mitoviruses from E. muscae (subphylum Entomophthoromycotina, phylum Zoopagomycota), red; members of subphylum Glomeromycotina, phylum Mucoromycota, brown; members of phylum Basidiomycota, cyan; members of phylum Ascomycota, purple; not assigned to a specific fungal host, black; plants, green; and outgroup, gray.
Figure 3.
Maximum-likelihood phylogenetic analysis of E. muscae mitoviruses and representative other mitoviruses. Deduced RdRp sequences were aligned using MAFFT-L-INS-i. The best-fit substitution model (per BIC score) used for the phylogenetic analysis shown here was LG + F + R8. The site proportions and rates for the FreeRate model were (0.0224, 0.0123) (0.0271, 0.0755), (0.0627, 0.2087), (0.1161, 0.4202), (0.1784, 0.6877), (0.2655, 1.0570), (0.2163, 1.4069), and (0.1115, 2.0462). The tree is displayed as a rectangular phylogram rooted on a set of two members of current genus Narnavirus included as outgroup. Branch support values from UFboot (1000 replicates) are shown in %; branches with <50% support have been collapsed to the preceding node. Scale bar indicates average number of substitutions per alignment position. Previously identified clade Ia of current genus Mitovirus is labeled. Red arrow, most recent common ancestor shared by all eight E. muscae mitoviruses. Virus names matching the sequence accession numbers and abbreviations are provided in Table S6. Color-coding: mitoviruses from E. muscae (subphylum Entomophthoromycotina, phylum Zoopagomycota), red; members of subphylum Glomeromycotina, phylum Mucoromycota, brown; members of phylum Basidiomycota, cyan; members of phylum Ascomycota, purple; not assigned to a specific fungal host, black; plants, green; and outgroup, gray.

Figure 4.
Summary diagram of mitochondrial coding sequences from E. muscae isolates. Mitochondrial core genes from each E. muscae isolate are labeled at left. The protein-encoding ORF of the respective transcript is shown as a yellow box to represent each assembled RNA sequence, starting with the first in-frame AUG codon by convention. Positions of UGA(Trp) codons in each ORF are shown as white dots. Positions of gaps or truncations in the assembled sequences are indicated by white breaks in the boxes; asterisks indicate inferred values consequent to these breaks. For each sequence, length of the ORF is shown in nt, length of the encoded protein in aa, and UGA and UGG codons in raw counts.
Figure 4.
Summary diagram of mitochondrial coding sequences from E. muscae isolates. Mitochondrial core genes from each E. muscae isolate are labeled at left. The protein-encoding ORF of the respective transcript is shown as a yellow box to represent each assembled RNA sequence, starting with the first in-frame AUG codon by convention. Positions of UGA(Trp) codons in each ORF are shown as white dots. Positions of gaps or truncations in the assembled sequences are indicated by white breaks in the boxes; asterisks indicate inferred values consequent to these breaks. For each sequence, length of the ORF is shown in nt, length of the encoded protein in aa, and UGA and UGG codons in raw counts.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Top NR hits for deduced mitochondrial core-protein sequences derived from E. muscae.
| Gene a | Accession no. | Fungal Species | Sub–Phy b | E-Value c |
|---|---|---|---|---|
| atp6 | AZZ06694.1 | Conidiobolus heterosporus | Ent–Zoo | 2e−115 |
| KXN65649.1 | Conidiobolus coronatus | Ent–Zoo | 1e−102 | |
| atp9 | KXN65653.1 | Conidiobolus coronatus | Ent–Zoo | 6e−38 |
| AZZ06722.1 | Conidiobolus heterosporus | Ent–Zoo | 1e−37 | |
| cob | AZZ06693.1 | Conidiobolus heterosporus | Ent–Zoo | 0 |
| cox1 | AZZ06707.1 | Conidiobolus heterosporus | Ent–Zoo | 0 |
| cox2 | AZZ06713.1 | Conidiobolus heterosporus | Ent–Zoo | 7e−142 |
| cox3 | AZZ06710.1 | Conidiobolus heterosporus | Ent–Zoo | 6e−169 |
| nad1 | AZZ06725.1 | Conidiobolus heterosporus | Ent–Zoo | 4e−169 |
| nad2 | AZZ06721.1 | Conidiobolus heterosporus | Ent–Zoo | 6e−135 |
| nad3 | AZZ06726.1 | Conidiobolus heterosporus | Ent–Zoo | 6e−47 |
| nad4 | AZZ06717.1 | Conidiobolus heterosporus | Ent–Zoo | 0 |
| nad5 | AZZ06724.1 | Conidiobolus heterosporus | Ent–Zoo | 0 |
| nad6 | AZZ06724.1 | Conidiobolus heterosporus | Ent–Zoo | 1e−34 |
| KXN65652.1 | Conidiobolus coronatus | Ent–Zoo | 3e−23 |
a Mitochondrial core genes; b Subphylum–Phylum: Ent–Zoo, Entomophthoromycotina–Zoopagomycota; c Top one or two hits in terms of E-value score are shown from searching the NR database with each mitochondrial protein sequence deduced from the apparent mitochondrial core-gene coding sequences from E. muscae isolate Berkeley.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nibert, M.L.; Debat, H.J.; Manny, A.R.; Grigoriev, I.V.; De Fine Licht, H.H. Mitovirus and Mitochondrial Coding Sequences from Basal Fungus Entomophthora muscae. Viruses 2019, 11, 351. https://doi.org/10.3390/v11040351
AMA Style
Nibert ML, Debat HJ, Manny AR, Grigoriev IV, De Fine Licht HH. Mitovirus and Mitochondrial Coding Sequences from Basal Fungus Entomophthora muscae. Viruses. 2019; 11(4):351. https://doi.org/10.3390/v11040351
Chicago/Turabian StyleNibert, Max L., Humberto J. Debat, Austin R. Manny, Igor V. Grigoriev, and Henrik H. De Fine Licht. 2019. "Mitovirus and Mitochondrial Coding Sequences from Basal Fungus Entomophthora muscae" Viruses 11, no. 4: 351. https://doi.org/10.3390/v11040351
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.