Diversity and Evolution of Viral Pathogen Community in Cave Nectar Bats (Eonycteris spelaea)

 , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Collection for NGS Library Preparation
2.2. NGS Data Analysis
2.3. Sample Collection and PCR Assays for Detection of Specific Viruses
2.4. Phylogenetic Analysis
3. Results
3.1. Next Generation Sequencing Analysis
3.2. Adenoviridae
3.3. Flaviviridae
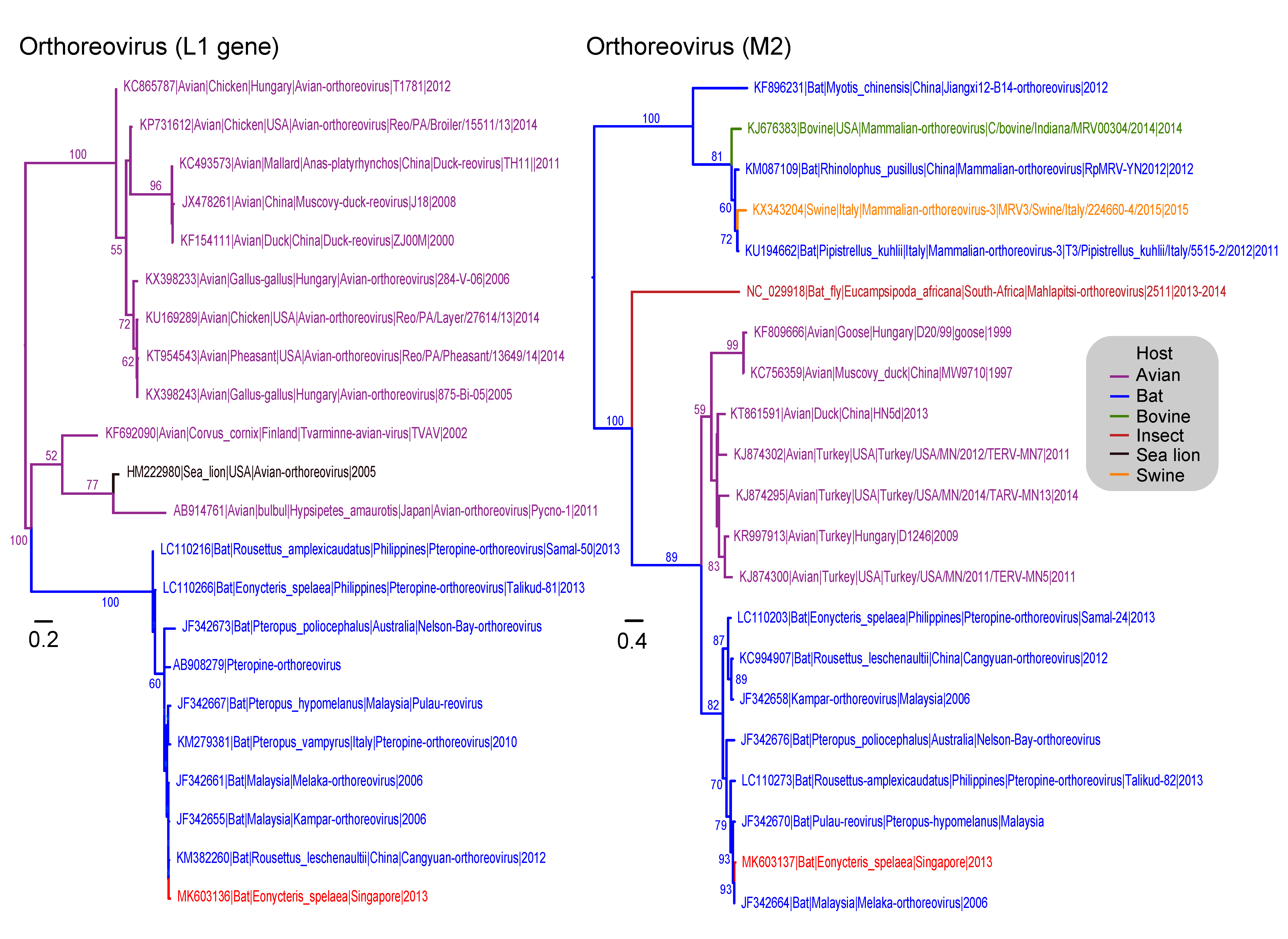
3.4. Reoviridae
3.5. Papillomaviridae
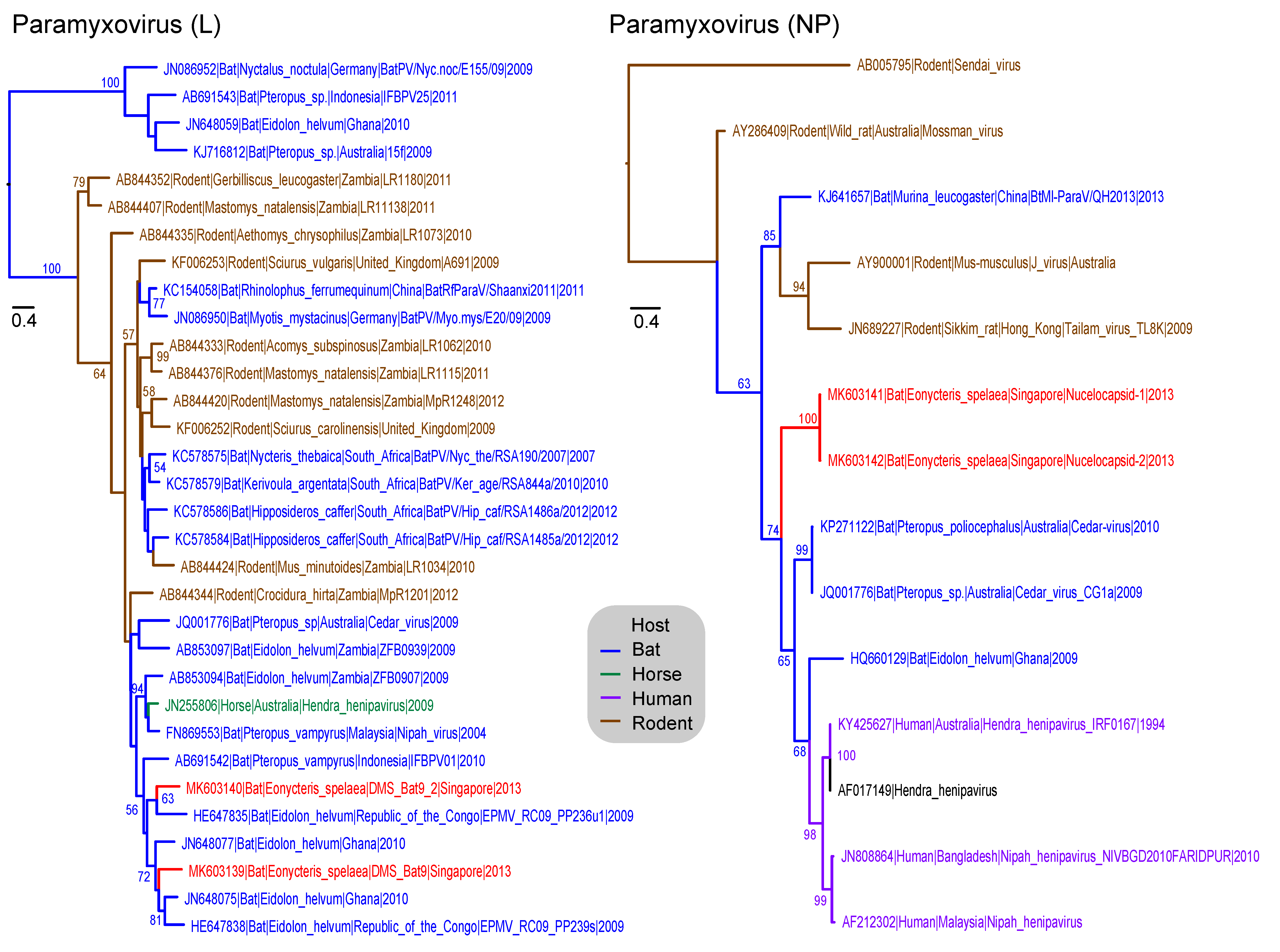
3.6. Paramyxoviridae
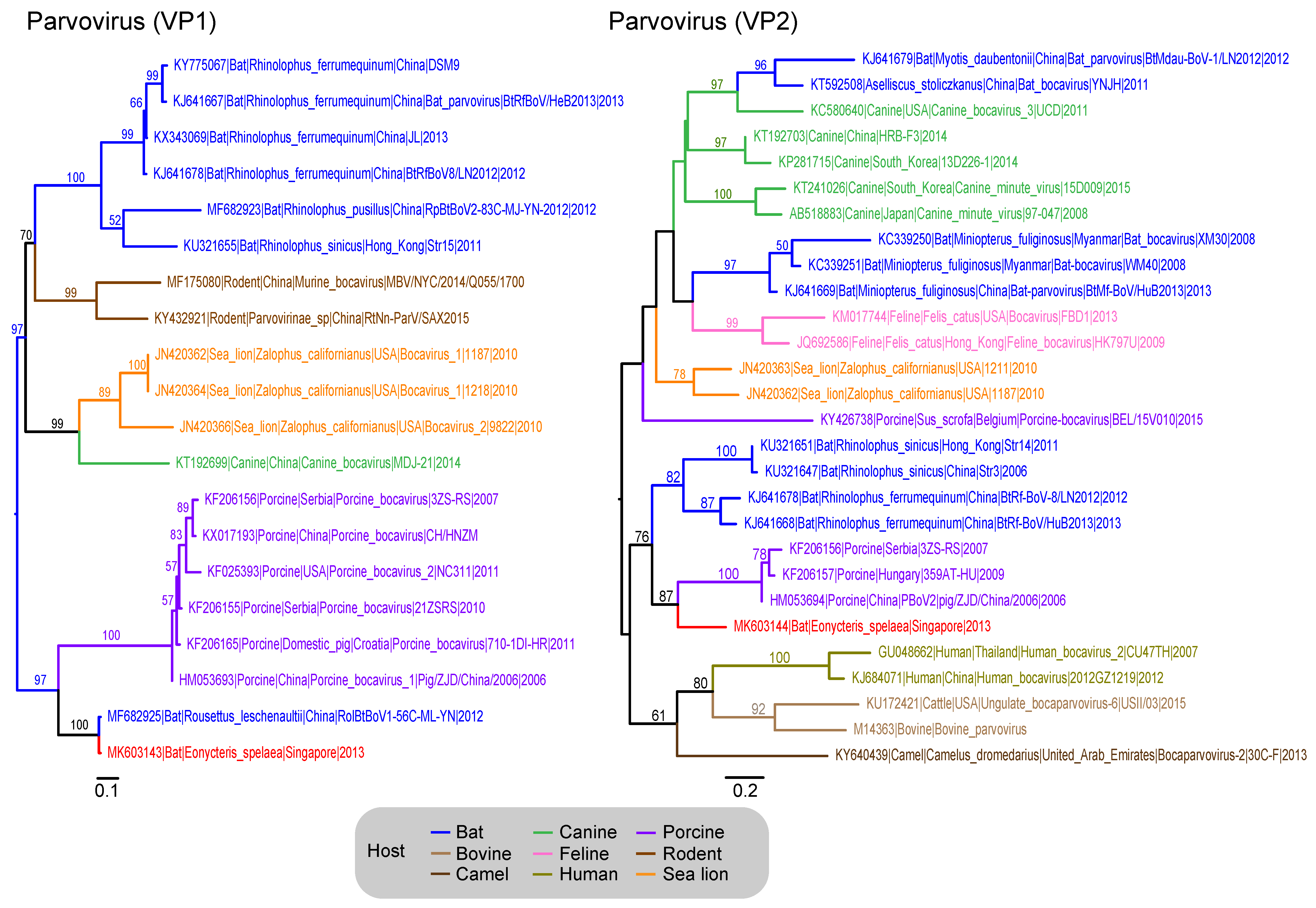
3.7. Parvoviridae
3.8. Picornaviridae
3.9. Polyomaviridae
3.10. Additional Viruses Detected
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerenwinkel, N.; Gunthard, H.F.; Roth, V.; Metzner, K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol. 2012, 3, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanDevanter, D.R.; Warrener, P.; Bennett, L.; Schultz, E.R.; Coulter, S.; Garber, R.L.; Rose, T.M. Detection and analysis of diverse herpesviral species by consensus primer PCR. J. Clin. Microbiol. 1996, 34, 1666–1671. [Google Scholar] [PubMed]
- Vijgen, L.; Moes, E.; Keyaerts, E.; Li, S.; Van Ranst, M. A pancoronavirus RT-PCR assay for detection of all known coronaviruses. Methods Mol. Biol. 2008, 454, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Prachayangprecha, S.; Schapendonk, C.M.; Koopmans, M.P.; Osterhaus, A.D.; Schurch, A.C.; Pas, S.D.; Van der Eijk, A.A.; Poovorawan, Y.; Haagmans, B.L.; Smits, S.L. Exploring the potential of next-generation sequencing in detection of respiratory viruses. J. Clin. Microbiol. 2014, 52, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Volpicella, M.; Leoni, C.; Costanza, A.; Fanizza, I.; Placido, A.; Ceci, L.R. Genome walking by next generation sequencing approaches. Biology 2012, 1, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; McElhinney, L.M.; Ellis, R.J.; Horton, D.L.; Wise, E.L.; Leech, S.L.; David, D.; De Lamballerie, X.; Fooks, A.R. Next generation sequencing of viral RNA genomes. BMC Genomics 2013, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Bialasiewicz, S.; McVernon, J.; Nolan, T.; Lambert, S.B.; Zhao, G.; Wang, D.; Nissen, M.D.; Sloots, T.P. Detection of a divergent Parainfluenza 4 virus in an adult patient with influenza like illness using next-generation sequencing. BMC Infect. Dis. 2014, 14, 275. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012, 93, 1853–1868. [Google Scholar] [CrossRef] [Green Version]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Li, Z.; Yang, F.; Zheng, J.; Feng, Y.; Guo, H.; Li, Y.; Wang, Y.; Su, N.; Zhang, F.; et al. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS ONE 2013, 8, e61950. [Google Scholar] [CrossRef]
- Wang, J.; Moore, N.E.; Murray, Z.L.; McInnes, K.; White, D.J.; Tompkins, D.M.; Hall, R.J. Discovery of novel virus sequences in an isolated and threatened bat species, the New Zealand lesser short-tailed bat (Mystacina tuberculata). J. Gen. Virol. 2015, 96, 2442–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.E.; Crameri, S.; Wang, L.F.; et al. Metagenomic study of the viruses of African straw-coloured fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yinda, C.K.; Zeller, M.; Conceicao-Neto, N.; Maes, P.; Deboutte, W.; Beller, L.; Heylen, E.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent reassortant bat rotaviruses in Cameroon, without evidence of zoonosis. Sci. Rep. 2016, 6, 34209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Li, M.; Li, L.; Monagin, C.; Chmura, A.A.; Schneider, B.S.; Epstein, J.H.; Mei, X.; Shi, Z.; Daszak, P.; et al. Evidence for retrovirus and paramyxovirus infection of multiple bat species in china. Viruses 2014, 6, 2138–2154. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Feng, Y.; Zhang, H.; Xu, L.; Yang, W.; Zhang, Y.; Li, X.; Tu, C. Filovirus RNA in fruit bats, China. Emerg. Infect. Dis. 2015, 21, 1675–1677. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Hutson, A.M.; Mickleburgh, S.P. Microchiropteran Bats: Global Status Survey and Conservation Action Plan; IUCN: Gland, Switzerland, 2001; Volume 56. [Google Scholar]
- Kunz, T.H.; Fenton, M.B. Bat Ecology; University of Chicago Press: Chicago, IL, USA, 2005. [Google Scholar]
- Thomas, S.P.; Suthers, R.A. The physiology and energetics of bat flight. J. Exp. Biol. 1972, 57, 317–335. [Google Scholar]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Cui, J.; Irving, A.T.; Wang, L.F. Unique loss of the PYHIN Gene family in bats amongst mammals: Implications for Inflammasome sensing. Sci. Rep. 2016, 6, 21722. [Google Scholar] [CrossRef]
- Zhou, P.; Tachedjian, M.; Wynne, J.W.; Boyd, V.; Cui, J.; Smith, I.; Cowled, C.; Ng, J.H.; Mok, L.; Michalski, W.P.; et al. Contraction of the type I IFN locus and unusual constitutive expression of IFN-alpha in bats. Proc. Natl. Acad. Sci. USA 2016, 113, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, Y.; Shen, X.; Goh, G.; Zhu, Y.; Cui, J.; Wang, L.F.; Shi, Z.L.; Zhou, P. Dampened STING-dependent interferon activation in bats. Cell Host Microbe 2018, 23, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Francis, C.; Rosell-Ambal, G.; Tabaranza, B.; Carino, P.; Helgen, K.; Molur, S.; Srinivasulu, C. Eonycteris Spelaea. Available online: http://dx.doi.org/10.2305/IUCN.UK.2008.RLTS.T7787A12850087.en (accessed on 4 June 2018).
- Bumrungsri, S.; Sripaoraya, E.; Chongsiri, T.; Sridith, K.; Racey, P.A. The pollination ecology of durian (Durio zibethinus, Bombacaceae) in southern Thailand. J. Trop. Ecol. 2009, 25, 85–92. [Google Scholar] [CrossRef]
- Bumrungsri, S.; Lang, D.; Harrower, C.; Sripaoraya, E.; Kitpipit, K.; Racey, P.A. The dawn bat, Eonycteris spelaea Dobson (Chiroptera: Pteropodidae) feeds mainly on pollen of economically important food plants in Thailand. Acta Chiropterol. 2013, 15, 95–104. [Google Scholar] [CrossRef]
- Mendenhall, I.H.; Borthwick, S.; Neves, E.S.; Low, D.; Linster, M.; Liang, B.; Skiles, M.; Jayakumar, J.; Han, H.; Gunalan, V.; et al. Identification of a lineage D Betacoronavirus in Cave Nectar Bats (Eonycteris spelaea) in Singapore and an overview of lineage D reservoir ecology in SE Asian Bats. Transbound Emerg. Dis. 2017, 64, 1790–1800. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. Available online: http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/ (accessed on 17 September 2015).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellehan, J.F., Jr.; Childress, A.L.; Marschang, R.E.; Johnson, A.J.; Lamirande, E.W.; Roberts, J.F.; Vickers, M.L.; Gaskin, J.M.; Jacobson, E.R. Consensus nested PCR amplification and sequencing of diverse reptilian, avian, and mammalian orthoreoviruses. Vet. Microbiol. 2009, 133, 34–42. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bininda-Emonds, O.R. transAlign: Using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinformat. 2005, 6, 156. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molec. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Molec. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Bonning, B.C.; Miller, W.A. Dicistroviruses. Ann. Rev. Entomol. 2010, 55, 129–150. [Google Scholar] [CrossRef]
- Laing, E.D.; Mendenhall, I.H.; Linster, M.; Low, D.H.W.; Chen, Y.; Yan, L.; Sterling, S.L.; Borthwick, S.; Neves, E.S.; Lim, J.S.L.; et al. Serologic evidence of fruit bat exposure to filoviruses, Singapore, 2011–2016. Emerg. Infect. Dis. 2018, 24, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, I.H.; Skiles, M.M.; Neves, E.S.; Borthwick, S.A.; Low, D.H.W.; Liang, B.; Lee, B.P.Y.; Su, Y.C.F.; Smith, G.J.D. Influence of age and body condition on astrovirus infection of bats in Singapore: An evolutionary and epidemiological analysis. One Health 2017, 4, 27–33. [Google Scholar] [CrossRef]
- Queen, K.; Shi, M.; Anderson, L.J.; Tong, S. Other bt-borne viruses. In Bats and Viruses; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2015; Chapter 9; pp. 217–247. [Google Scholar]
- Yang, X.-L.; Zhang, Y.-Z.; Jiang, R.-D.; Guo, H.; Zhang, W.; Li, B.; Wang, N.; Wang, L.; Waruhiu, C.; Zhou, J.-H. Genetically diverse filoviruses in Rousettus and Eonycteris spp. bats, China, 2009 and 2015. Emerg. Infect. Dis. 2017, 23, 482. [Google Scholar] [CrossRef]
- Yob, J.M.; Field, H.; Rashdi, A.M.; Morrissy, C.; Van der Heide, B.; Rota, P.; Bin Adzhar, A.; White, J.; Daniels, P.; Jamaluddin, A.; et al. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg. Infect. Dis. 2001, 7, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Pankovics, P.; Gyongyi, Z.; Delwart, E.; Boros, A. Novel dicistrovirus from bat guano. Arch. Virol. 2014, 159, 3453–3456. [Google Scholar] [CrossRef] [PubMed]
- Dacheux, L.; Cervantes-Gonzalez, M.; Guigon, G.; Thiberge, J.M.; Vandenbogaert, M.; Maufrais, C.; Caro, V.; Bourhy, H. A preliminary study of viral metagenomics of French bat species in contact with humans: Identification of new mammalian viruses. PloS ONE 2014, 9, e87194. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Hondo, E.; Terakawa, J.; Kiso, Y.; Nakaichi, N.; Endoh, D.; Sakai, K.; Morikawa, S.; Mizutani, T. Isolation of novel adenovirus from fruit bat (Pteropus dasymallus yayeyamae). Emerg. Infect. Dis. 2008, 14, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, X.; Zhang, H.; Zhou, P.; Zhu, Y.; Zhang, Y.; Yuan, J.; Wang, L.F.; Shi, Z. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 2010, 84, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.; Rupprecht, C.; Olson, J.; Ksiazek, T.; Rollin, P.; Niezgoda, M.; Goldsmith, C.; An, U.; Nichol, S. Isolation of Kaeng Khoi virus from dead Chaerephon plicata bats in Cambodia. J. Gen. Virol. 2003, 84, 2685–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.; Witkowski, P.T.; Auste, B.; Nowak, K.; Weber, N.; Fahr, J.; Mombouli, J.-V.; Wolfe, N.D.; Drexler, J.F.; Drosten, C. Hantavirus in bat, sierra leone. Emerg. Infect. Dis. 2012, 18, 159. [Google Scholar] [CrossRef]
- Mourya, D.; Yadav, P.; Basu, A.; Shete, A.; Patil, D.; Zawar, D.; Majumdar, T.; Kokate, P.; Sarkale, P.; Raut, C. Malsoor virus, a novel bat Phlebovirus is closely related to STFS and Heartland viruses. J. Virol. 2014, 88, 3605–3609. [Google Scholar] [CrossRef] [PubMed]
- De Lamballerie, X.; Crochu, S.; Billoir, F.; Neyts, J.; De Micco, P.; Holmes, E.C.; Gould, E.A. Genome sequence analysis of Tamana bat virus and its relationship with the genus Flavivirus. J. Gen. Virol. 2002, 83, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Tajima, S.; Takasaki, T.; Matsuno, S.; Nakayama, M.; Kurane, I. Genetic characterization of Yokose virus, a flavivirus isolated from the bat in Japan. Virology 2005, 332, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaun, J.; Klein, J.; Hebrard, G. A new virus, Phnom-Penh bat virus, isolated in Cambodia from a short-nosed fruit bat, Cynopterus brachyotis angulatus. Ann. Microbiol. (Inst. Pasteur) 1974, 125, 485–495. [Google Scholar]
- Janoska, M.; Vidovszky, M.; Molnar, V.; Liptovszky, M.; Harrach, B.; Benko, M. Novel adenoviruses and herpesviruses detected in bats. Vet. J. 2011, 189, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurth, A.; Kohl, C.; Brinkmann, A.; Ebinger, A.; Harper, J.A.; Wang, L.F.; Muhldorfer, K.; Wibbelt, G. Novel paramyxoviruses in free-ranging European bats. PLoS ONE 2012, 7, e38688. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.E.; Marsh, G.A. Bat Paramyxoviruses. In Bats and Viruses; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2015; Chapter 4; pp. 99–126. [Google Scholar]
- Tse, H.; Tsang, A.K.L.; Tsoi, H.W.; Leung, A.S.P.; Ho, C.C.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.Y. Identification of a Novel Bat Papillomavirus by Metagenomics. PLoS ONE 2012, 7, e43986. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, R.; Gottschling, M.; Wibbelt, G.; Bravo, I.G. Multiple evolutionary origins of bat papillomaviruses. Vet. Microbiol. 2013, 165, 51–60. [Google Scholar] [CrossRef]
- Li, Y.; Ge, X.; Hon, C.C.; Zhang, H.; Zhou, P.; Zhang, Y.; Wu, Y.; Wang, L.F.; Shi, Z. Prevalence and genetic diversity of adeno-associated viruses in bats from China. J. Gen. Virol. 2010, 91, 2601–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canuti, M.; Eis-Huebinger, A.M.; Deijs, M.; De Vries, M.; Drexler, J.F.; Oppong, S.K.; Muller, M.A.; Klose, S.M.; Wellinghausen, N.; Cottontail, V.M.; et al. Two novel parvoviruses in frugivorous New and Old World bats. PLoS ONE 2011, 6, e29140. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Lai, K.K.; Huang, Y.; Yip, C.C.; Shek, C.T.; Lee, P.; Lam, C.S.; Chan, K.H.; Yuen, K.Y. Complete genome analysis of three novel picornaviruses from diverse bat species. J. Virol. 2011, 85, 8819–8828. [Google Scholar] [CrossRef] [PubMed]
- Fagrouch, Z.; Sarwari, R.; Lavergne, A.; Delaval, M.; De Thoisy, B.; Lacoste, V.; Verschoor, E.J. Novel polyomaviruses in South American bats and their relationship to other members of the family Polyomaviridae. J. Gen. Virol. 2012, 93, 2652–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, V.; Dumonceaux, T.; Dubois, J.; Willis, C.; Nadin-Davis, S.; Severini, A.; Wandeler, A.; Lindsay, R.; Artsob, H. Detection of polyoma and corona viruses in bats of Canada. J. Gen. Virol. 2009, 90, 2015–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Shi, M.; Conrardy, C.; Kuzmin, I.V.; Recuenco, S.; Agwanda, B.; Alvarez, D.A.; Ellison, J.A.; Gilbert, A.T.; Moran, D. Discovery of diverse polyomaviruses in bats and the evolutionary history of the Polyomaviridae. J. Gen. Virol. 2013, 94, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.L.; Nordhausen, R.; Garner, M.M.; Huckabee, J.R.; Johnson, S.; Wohrle, R.D.; Davidson, W.B.; Wilkins, K.; Li, Y.; Doty, J.B. Novel poxvirus in big brown bats, northwestern United States. Emerg. Infect. Dis. 2013, 19, 1002. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, M.A.; Tu, S.-L.; Pang, S.; De Ridder, T.; Jackson, B.; Upton, C. Genomic characterization of a novel poxvirus from a flying fox: Evidence for a new genus? J. Gen. Virol. 2016, 97, 2363–2375. [Google Scholar] [CrossRef]
- Pritchard, L.I.; Chua, K.B.; Cummins, D.; Hyatt, A.; Crameri, G.; Eaton, B.T.; Wang, L.F. Pulau virus; A new member of the Nelson Bay orthoreovirus species isolated from fruit bats in Malaysia. Arch. Virol. 2006, 151, 229–239. [Google Scholar] [CrossRef]
- Thalmann, C.M.; Cummins, D.M.; Yu, M.; Lunt, R.; Pritchard, L.I.; Hansson, E.; Crameri, S.; Hyatt, A.; Wang, L.F. Broome virus, a new fusogenic Orthoreovirus species isolated from an Australian fruit bat. Virology 2010, 402, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Kurth, A. Bat Reoviruses. In Bats and Viruses; Wang, L.-F., Cowled, C., Eds.; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2015; Chapter 8; pp. 203–215. [Google Scholar]
- Yang, X.L.; Tan, B.; Wang, B.; Li, W.; Wang, N.; Luo, C.M.; Wang, M.N.; Zhang, W.; Li, B.; Peng, C.; et al. Isolation and identification of bat viruses closely related to human, porcine, and mink orthoreoviruses. J. Gen. Virol. 2015, 96, 3525–3531. [Google Scholar] [CrossRef]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T.; Bowen, R.A.; Cryan, P.M.; McCracken, G.F.; O’Shea, T.J.; Peel, A.J.; Gilbert, A.; Webb, C.T.; Wood, J.L. Ecology of zoonotic infectious diseases in bats: Current knowledge and future directions. Zoonoses Public Health 2013, 60, 2–21. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Muller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef]
- Drexler, J.F.; Geipel, A.; Konig, A.; Corman, V.M.; Van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D.; et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, J.F.; Seelen, A.; Corman, V.M.; Fumie Tateno, A.; Cottontail, V.; Melim Zerbinati, R.; Gloza-Rausch, F.; Klose, S.M.; Adu-Sarkodie, Y.; Oppong, S.K.; et al. Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae. J. Virol. 2012, 86, 9134–9147. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Schaulies, J. Cellular receptors for viruses: Links to tropism and pathogenesis. J. Gen. Virol. 2000, 81, 1413–1429. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Chen, Y.; Wong, E.Y.; Chan, K.-H.; Chen, H.; Zhang, L.; Xia, N.; Yuen, K.-Y. Rapid detection of MERS coronavirus-like viruses in bats: Pote1ntial for tracking MERS coronavirus transmission and animal origin. Emerg. Microb. Infect. 2018, 7, 18. [Google Scholar] [CrossRef]
- Widagdo, W.; Begeman, L.; Schipper, D.; Van Run, P.R.; Cunningham, A.A.; Kley, N.; Reusken, C.B.; Haagmans, B.L.; Van den Brand, J.M. Tissue distribution of the MERS-coronavirus receptor in bats. Sci. Rep. 2017, 7, 1193. [Google Scholar] [CrossRef]
- Letko, M.; Miazgowicz, K.; McMinn, R.; Seifert, S.N.; Sola, I.; Enjuanes, L.; Carmody, A.; Van Doremalen, N.; Munster, V. Adaptive evolution of MERS-CoV to Species variation in DPP4. Cell Rep. 2018, 24, 1730–1737. [Google Scholar] [CrossRef]
- Rose, T.M.; Schultz, E.R.; Henikoff, J.G.; Pietrokovski, S.; McCallum, C.M.; Henikoff, S. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 1998, 26, 1628–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, C.; Braxton, C.; Charlebois, R.; Deyati, A.; Duncan, P.; La Neve, F.; Malicki, H.; Ribrioux, S.; Rozelle, D.; Michaels, B. Considerations for Optimization of High-throughput sequencing bioinformatics pipelines for virus detection. Viruses 2018, 10, 528. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.-P.; Yang, X.-L.; Ge, X.-Y.; Zhang, W.; Li, B.; Xie, J.-Z.; Shen, X.-R.; Zhang, Y.-Z.; Wang, N. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T. Biannual birth pulses allow filoviruses to persist in bat populations. Proc. Biol. Sci. Royal Soc. 2015, 282, 20142591. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Ng, J.H.; Zhu, F.; Chionh, Y.T.; Chia, W.N.; Mendenhall, I.H.; Lee, B.P.-H.; Irving, A.T.; Wang, L.-F. Exploring the genome and transcriptome of the cave nectar bat Eonycteris spelaea with PacBio long-read sequencing. GigaScience 2018, 7, giy116. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Family (Genus) | Gene | Initial Sequence Alignment | Down Sampled Sequence Alignment | Down Sampled Alignment Length (bp) |
|---|---|---|---|---|
| Adenoviridae | DNA Polymerase | 227 | 37 | 885 |
| Flaviviridae | E–Envelope | 398 | 45 | 333 |
| NS5–Non-structural protein 5 | 600 | 47 | 231 | |
| Papillomaviridae | V3–minor capsid protein | 368 | 36 | 969 |
| Paramyxoviridae | N–Nucleoprotein | 15 | 14 | 720 |
| L–Polymerase | 743 | 32 | 471 | |
| Parvoviridae | VP1–Capsid | 27 | 20 | 528 |
| VP2–Capsid | 501 | 28 | 323 | |
| Picornaviridae | 3D–RNA polymerase | 1753 | 20 | 246 |
| Polyprotein | 40 | 23 | 813 | |
| Polyomaviridae | E1–major capsid protein | 289 | 26 | 441 |
| Reoviridae (Orthoreovirus) | M2–viral outer capsid proteins (ς1 and μ1c) | 149 | 21 | 624 |
| L2–core spike protein λ2 | 30 | 22 | 318 | |
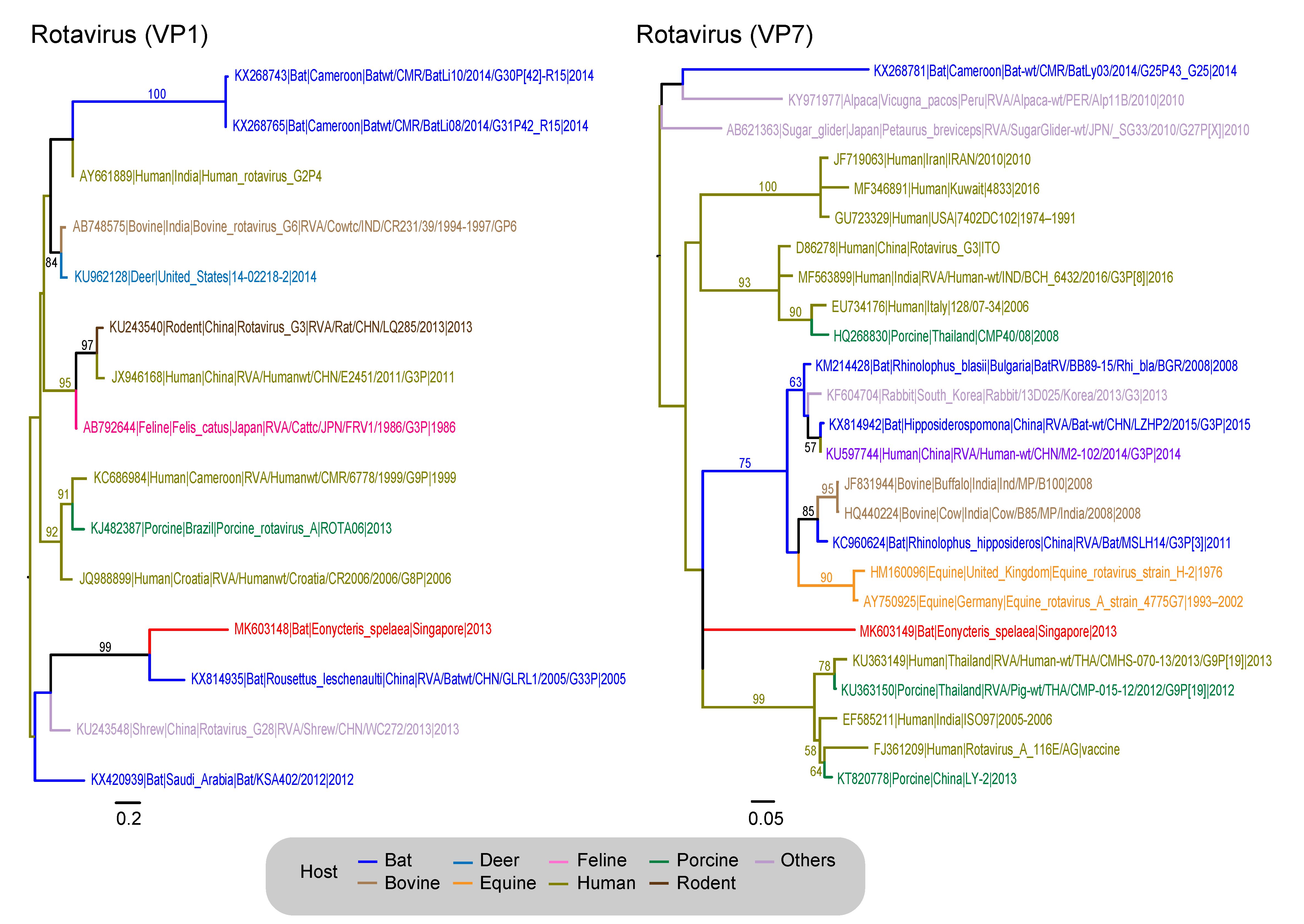
| Reoviridae (Rotavirus) | VP1–RNA-dependent RNA polymerase | 501 | 15 | 231 |
| VP7–outer capsid protein | 536 | 25 | 291 |
| Data set-Name | Total Reads in Data set | Total Reads Assigned by MEGAN | Eukaryotes | Mammalia | Arthropoda | Bacteria | Archaea | Fungi | Virus |
|---|---|---|---|---|---|---|---|---|---|
| Urine-MiSeq-25 | 5,126,632 | 1,527,375 | 1,154,068 | 1,010,668 | 583 | 222,953 | 204 | 8217 | 1606 |
| Urine-MiSeq-27 | 13,421,263 | 4,137,771 | 3,230,684 | 2,994,683 | 1201 | 548,911 | 497 | 20,496 | 3691 |
| Feces HiSeq | 68,584,413 | 3,993,465 | 2,178,408 | 69,136 | 53,546 | 546,757 | 68 | 675,967 | 21,856 |
| Feces MiSeq | 4,952,973 | 3,750,870 | 2,836,374 | 54,457 | 60,533 | 420,446 | 97 | 1,326,425 | 106,823 |
| Virus Family | Urine-MiSeq-25 Reads | Urine-MiSeq-27 Reads | Urine reads by Family (% of Total) | Feces-Hiseq Reads | Feces-MiSeq Reads | Fecal Reads by Family (% of Total) | Total Reads | Consensus Reads/Unassembled Reads |
|---|---|---|---|---|---|---|---|---|
| AdenoviridaeV | 171 | 115 | 286 (7.14%) | 31 | 43 | 74 (0.07%) | 360 (0.31%) | 28/17 |
| AlphaflexiviridaeP/F | - | - | - | - | 143 | 143 (0.13%) | 143 (0.12%) | 28 / 13 |
| AlphatetraviridaeA | - | - | - | - | 1 | 1 | 1 | - |
| AstroviridaeV | 50 | - | 50 (1.25%) | 6 | 8 | 14 (0.01%) | 64 (0.05%) | 6/3 |
| BaculoviridaeA | - | - | - | 1 | 2 | 3 | 3 | 0/3 |
| BetaflexiviridaeP/F | 4 | - | 4 (0.10%) | 9 | 14 | 23 (0.02%) | 27 (0.02%) | 7/9 |
| BunyaviridaeV | - | - | - | - | 1 | 1 | 1 | - |
| CaliciviridaeV | - | - | - | - | 9 | 9 (0.01%) | 9 (0.01%) | 4/1 |
| CarmotetraviridaeA | - | - | - | - | 34 | 34 (0.03%) | 34 (0.03%) | 5/0 |
| CaulimoviridaeA/P | - | - | - | 3 | 5 | 8 (0.01%) | 8 (0.01%) | 2/4 |
| ChrysoviridaeF | 15 | - | 15 (0.37%) | 3 | 3 | 6 (0.01%) | 21 (0.02%) | 5/2 |
| CoronaviridaeV | 170 | 440 | 610 (15.22%) | 46 | 87 | 133 (0.12%) | 743 (0.64%) | 52/21 |
| DicistroviridaeA | 120 | 705 | 825 (20.59%) | 292 | 65,061 | 65,353 (58.09%) | 66,178 (56.80%) | 2326/1546 |
| EndornaviridaeP/F | - | - | - | - | 4 | 4 | 4 | 2/0 |
| FlaviviridaeV | 10 | 52 | 62 (1.55%) | - | 1 | 1 | 63 (0.05%) | 2/1 |
| HerpesviridaeV | 25 | - | 25 (0.62%) | 3 | 12 | 15 (0.01%) | 40 (0.03%) | 7/5 |
| IflaviridaeA | 8 | - | 8 (0.20%) | 46 | 578 | 624 (0.55%) | 632 (0.54%) | 79/21 |
| InoviridaeB | - | - | - | 1 | - | 1 | 1 | - |
| LeviviridaeB | - | - | - | - | 4 | 4 | 4 | 2/0 |
| LuteoviridaeP | - | - | - | - | 1 | 1 | 1 | - |
| MicroviridaeB | 17 | 117 | 134 (3.34%) | 3016 | 438 | 3454 (3.07%) | 3588 (3.08%) | 39/8 |
| MimiviridaePR | - | - | - | - | 1 | 1 | 1 | - |
| MyoviridaeB | 1 | - | 1 (0.02%) | 4029 | 1688 | 5717 (5.08%) | 5718 (4.91%) | 525/346 |
| NarnaviridaeF | - | - | - | 16 | 345 | 361 (0.32%) | 361 (0.31%) | 42/6 |
| NodaviridaeA | - | - | - | - | 117 | 117 (0.10%) | 117 (0.10%) | 13/10 |
| NudiviridaeA | - | - | - | 28 | 63 | 91 (0.08%) | 91 (0.08%) | 32/16 |
| PapillomaviridaeV | - | - | - | 3 | 9 | 12 (0.01%) | 12 (0.01%) | 3/3 |
| ParamyxoviridaeV | 14 | 34 | 48 (1.20%) | 3 | 6 | 9 (0.01%) | 57 (0.05%) | 6/1 |
| PartitiviridaeP/F | 12 | 129 | 141 (3.52%) | 12 | 22 | 34 (0.03%) | 175 (0.15%) | 12/11 |
| ParvoviridaeV | - | - | - | 203 | 421 | 624 (0.55%) | 624 (0.54%) | 55/21 |
| Genus: Parvovirinae | - | - | - | 187 | 370 | 557 (0.50%) | 557 (0.48%) | - |
| Genus: Denovirinae | - | - | - | 8 | 51 | 59 (0.05%) | 59 (0.05%) | - |
| PermutotetraviridaeA | - | - | - | 55 | 933 | 988 (0.88%) | 988 (0.85%) | 92/7 |
| PhycodnaviridaeP | 3 | 1 | 4 (0.10%) | - | 2 | 2 | 6 (0.01%) | 1/3 |
| PicobirnaviridaeV | - | - | - | - | 3 | 3 | 3 | 0/3 |
| PicornaviridaeV | - | - | - | 45 | 114 | 159 (0.14%) | 159 (0.14%) | 33/17 |
| PodoviridaeB | 26 | - | 26 (0.65%) | 4002 | 13,349 | 17,351 (15.42%) | 17,377 (14.91%) | 787/225 |
| PolydnaviridaeA | - | - | - | 4 | 6 | 10 (0.01%) | 10 (0.01%) | 3/4 |
| PolyomaviridaeV | 8 | - | 8 (0.20%) | 63 | 93 | 156 (0.14%) | 164 (0.14%) | 33/13 |
| PotyviridaeP | - | - | - | 44 | 15 | 59 (0.05%) | 59 (0.05%) | 13/12 |
| PoxviridaeV | 16 | - | 16 (0.40%) | 2 | 1 | 3 | 19 (0.02%) | 3/3 |
| ReoviridaeV | 1 | - | 1 (0.02%) | 95 | 76 | 171 (0.15%) | 172 (0.15%) | 42/22 |
| Genus: Orthoreovirus | 1 | - | 1 | 53 | 25 | 78 (0.07%) | 103 (0.09%) | - |
| Genus: Rotavirus | - | - | - | 24 | 39 | 63 (0.06%) | 102 (0.09%) | - |
| RetroviridaeV | 552 | 465 | 1017 (25.38%) | 95 | 39 | 134 (0.12%) | 1151 (0.99%) | 170/61 |
| RhabdoviridaeA | - | - | - | - | 11 | 11 (0.01%) | 11 (0.01%) | 4/2 |
| SecoviridaeP | - | 87 | 87 (2.17%) | - | 33 | 33 (0.03%) | 120 (0.10%) | 6/2 |
| SiphoviridaeB | 17 | 325 | 342 (8.54%) | 8839 | 7662 | 16,501 (14.67%) | 16,843 (14.46%) | 518/144 |
| TotiviridaeV/A/P | 16 | 281 | 297 (7.41%) | - | 47 | 47 (0.04%) | 344 (0.30%) | 23/12 |
| TymoviridaeP | - | - | - | 1 | 8 | 9 (0.01%) | 9 (0.01%) | 3/1 |
| VirgaviridaeP | - | - | - | - | 2 | 2 | 2 | 1/0 |
| Total Reads | 1256 | 2751 | 4007 | 20,996 | 116,518 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendenhall, I.H.; Wen, D.L.H.; Jayakumar, J.; Gunalan, V.; Wang, L.; Mauer-Stroh, S.; Su, Y.C.F.; Smith, G.J.D. Diversity and Evolution of Viral Pathogen Community in Cave Nectar Bats (Eonycteris spelaea). Viruses 2019, 11, 250. https://doi.org/10.3390/v11030250
Mendenhall IH, Wen DLH, Jayakumar J, Gunalan V, Wang L, Mauer-Stroh S, Su YCF, Smith GJD. Diversity and Evolution of Viral Pathogen Community in Cave Nectar Bats (Eonycteris spelaea). Viruses. 2019; 11(3):250. https://doi.org/10.3390/v11030250
Chicago/Turabian StyleMendenhall, Ian H, Dolyce Low Hong Wen, Jayanthi Jayakumar, Vithiagaran Gunalan, Linfa Wang, Sebastian Mauer-Stroh, Yvonne C.F. Su, and Gavin J.D. Smith. 2019. "Diversity and Evolution of Viral Pathogen Community in Cave Nectar Bats (Eonycteris spelaea)" Viruses 11, no. 3: 250. https://doi.org/10.3390/v11030250