
High Uric Acid Ameliorates Indoxyl Sulfate-Induced Endothelial Dysfunction and Is Associated with Lower Mortality among Hemodialysis Patients


Abstract
:1. Introduction
2. Results
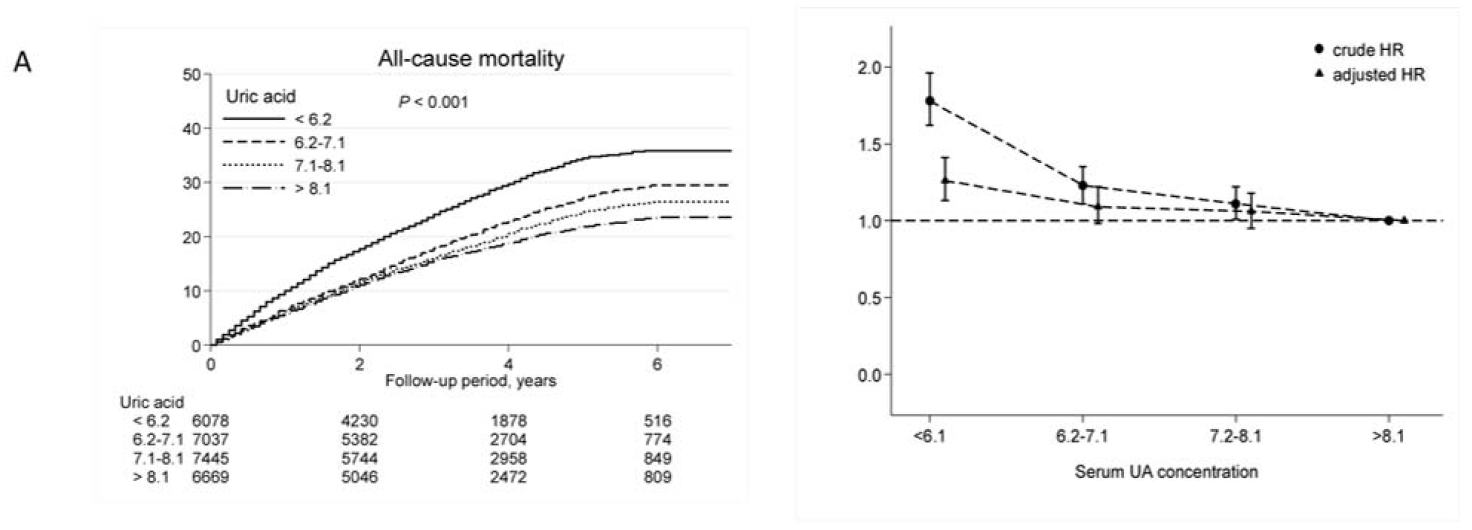
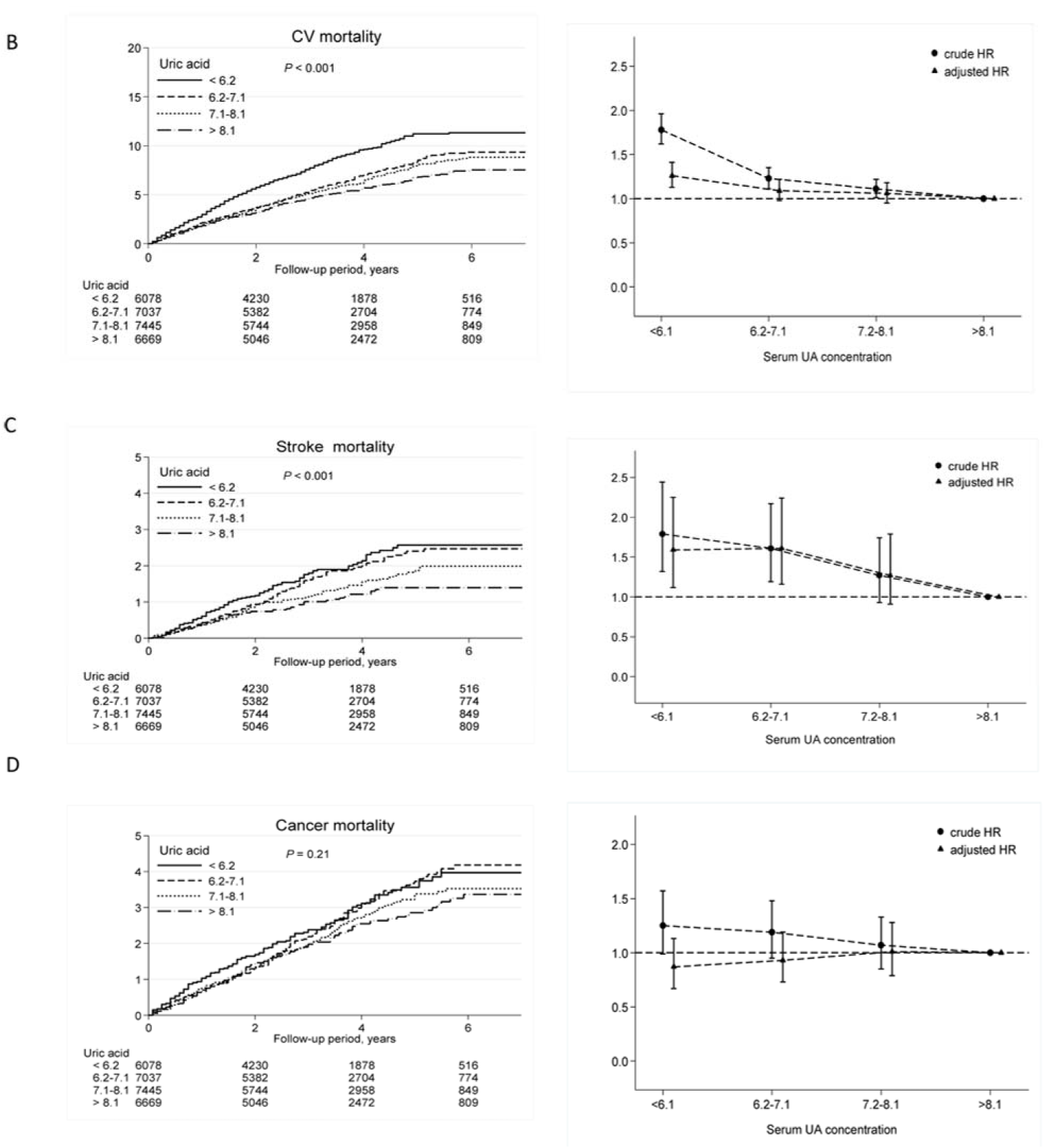
2.1. High UA Levels Are Associated with Lower CV and All-Cause Mortality
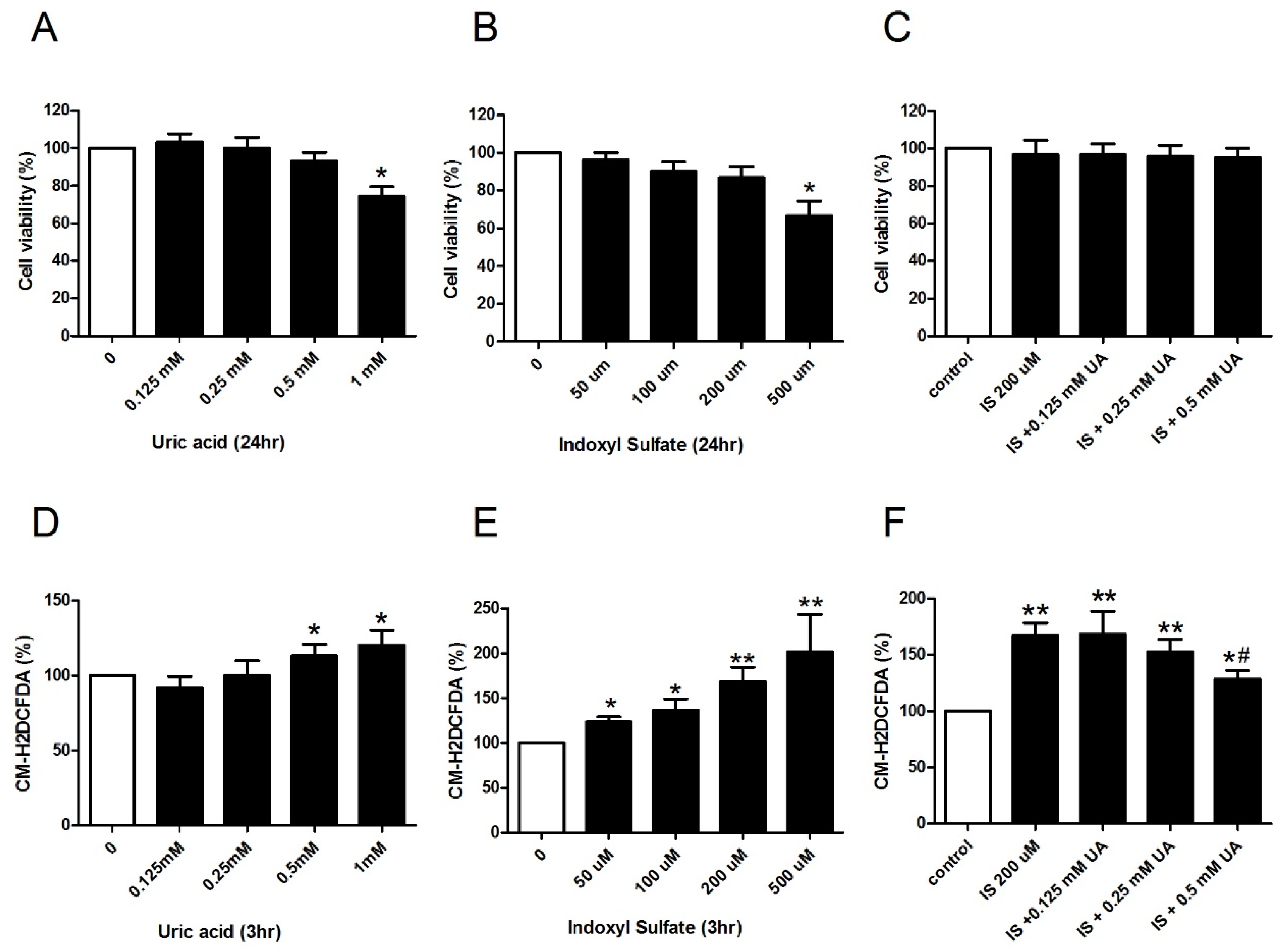
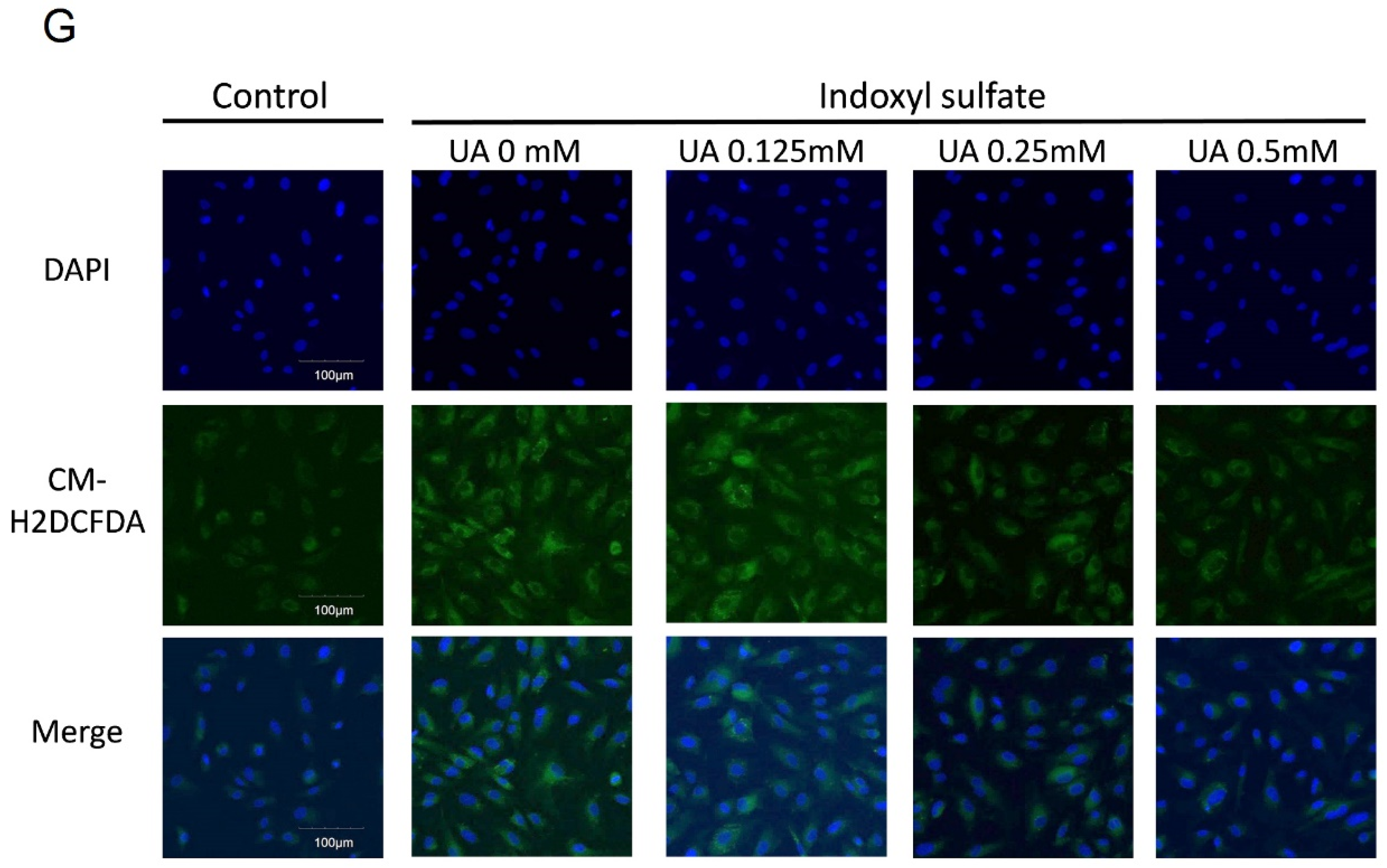
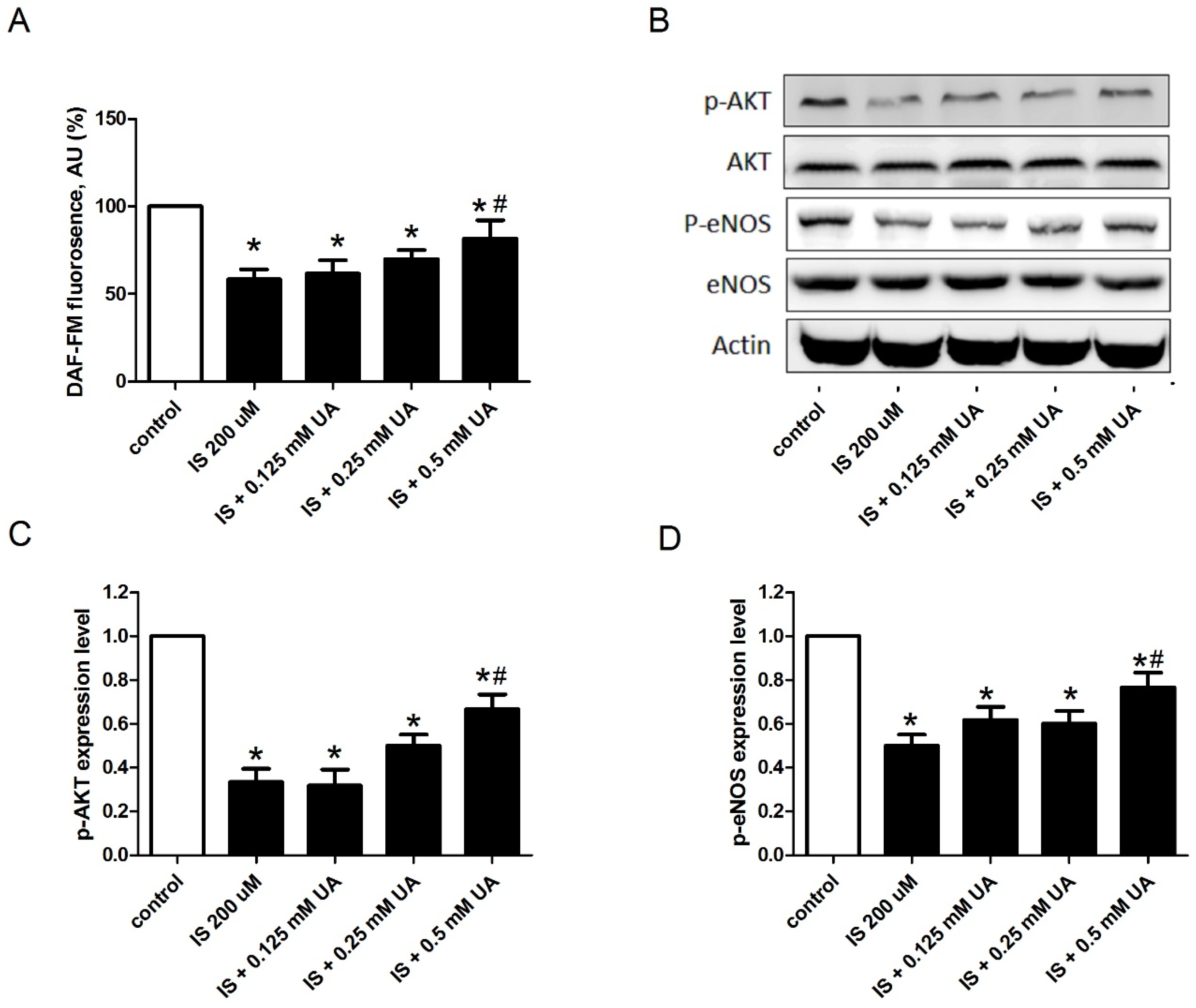
2.2. UA Ameliorates IS-Induced Endothelial Dysfunction In Vitro
2.3. UA Attenuates IS-Impaired Endothelial NO Production
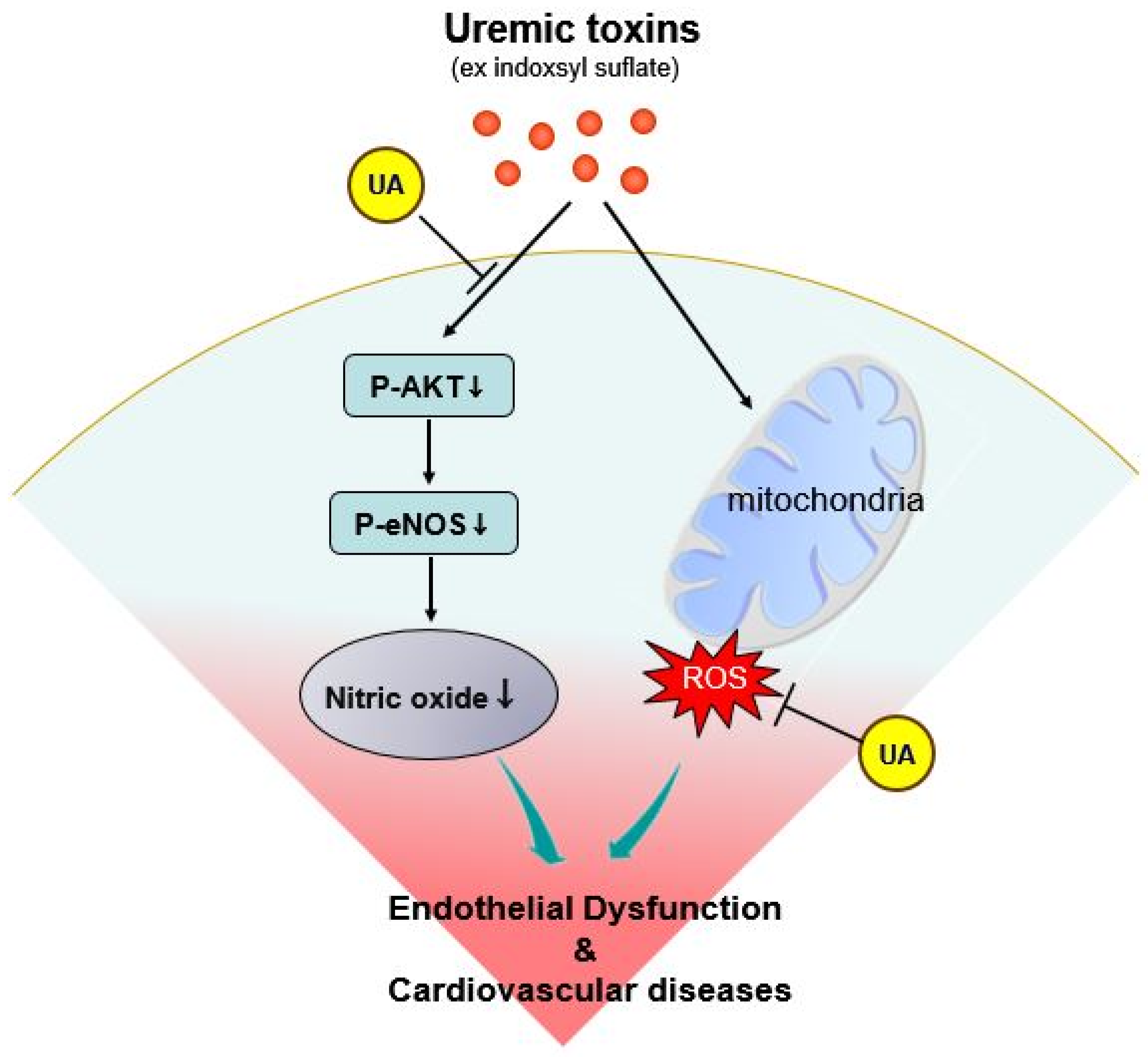
3. Discussion
3.1. Main Findings
3.2. Comparison with Previous Studies
3.3. Potential Mechanisms
3.4. Limits and Strengths
4. Conclusions
5. Materials and Methods
5.1. Data Sources
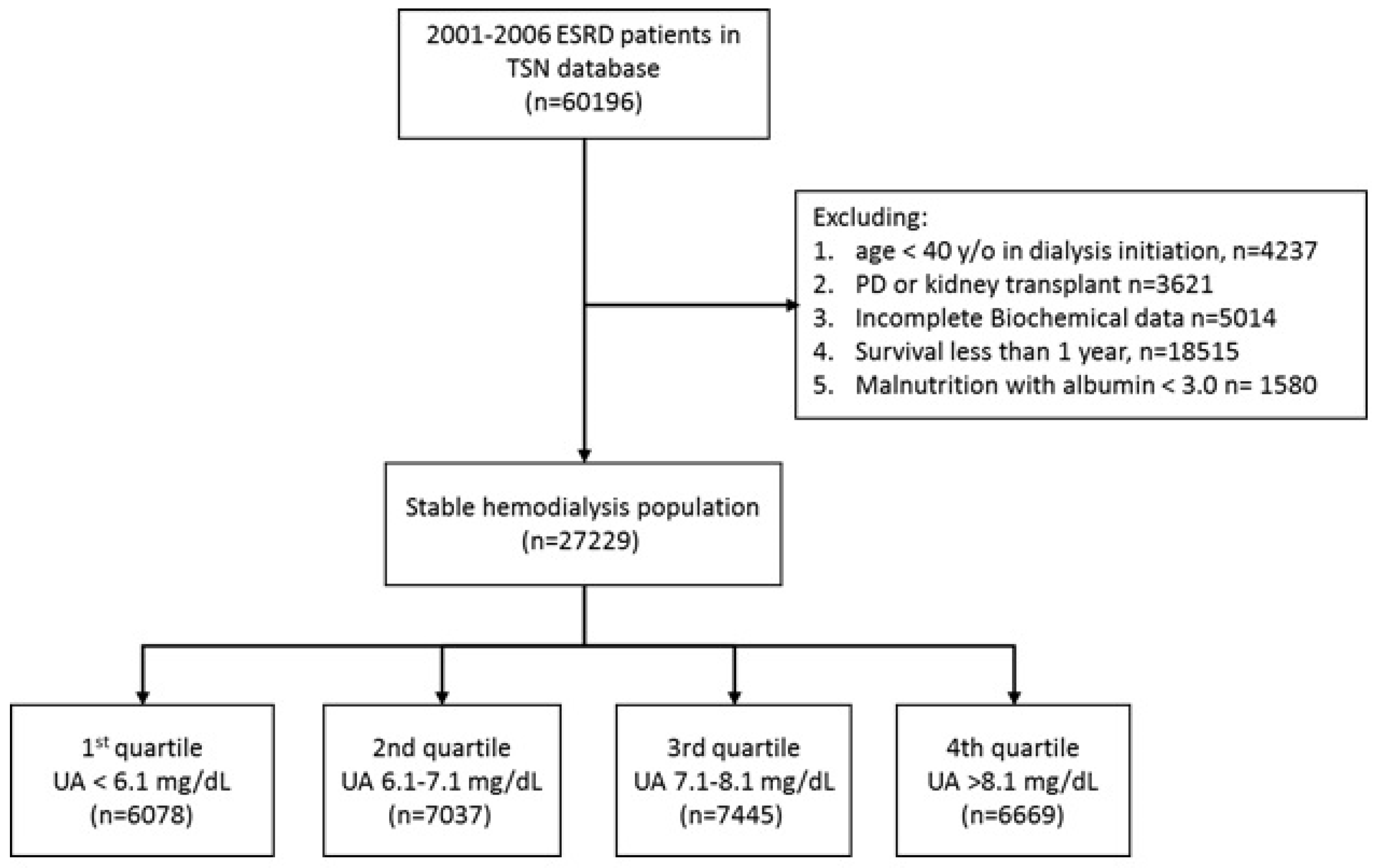
5.2. Study Participants and Follow Up
5.3. Cell Culture and Preparation of UA and Indoxyl Sulfate
5.4. Determining Oxidative Stress And Nitric Oxide In Vitro
5.5. Western Blotting
5.6. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lanaspa, M.A.; Tapia, E.; Soto, V.; Sautin, Y.; Sanchez-Lozada, L.G. Uric acid and fructose: Potential biological mechanisms. Semin. Nephrol. 2011, 31, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Bardin, T. Gout. Lancet 2010, 375, 318–328. [Google Scholar] [CrossRef]
- Feig, D.I.; Kang, D.H.; Johnson, R.J. Uric acid and cardiovascular risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, A.C.; Miname, M.H.; Santos, R.D. Uric acid: A marker of increased cardiovascular risk. Atherosclerosis 2009, 202, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Papezikova, I.; Pekarova, M.; Kolarova, H.; Klinke, A.; Lau, D.; Baldus, S.; Lojek, A.; Kubala, L. Uric acid modulates vascular endothelial function through the down regulation of nitric oxide production. Free Radic. Res. 2013, 47, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, S.; Krotova, K.; Hu, H.; Baylis, C.; Johnson, R.J.; Block, E.R.; Patel, J. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1183–C1190. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.P.; Pai, M.F.; Peng, Y.S.; Chiang, C.K.; Ho, T.I.; Hung, K.Y. Serum uric acid levels show a ‘j-shaped’ association with all-cause mortality in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Lee, A.L.; Winters, T.J.; Tam, E.; Jaleel, M.; Stenvinkel, P.; Johnson, R.J. Low serum uric acid level is a risk factor for death in incident hemodialysis patients. Am. J. Nephrol. 2009, 29, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.E.; Johnson, R.J.; Garcia-Lopez, E.; Qureshi, A.R.; Molinaei, H.; Carrero, J.J.; Heimburger, O.; Barany, P.; Axelsson, J.; Lindholm, B.; et al. J-shaped mortality relationship for uric acid in CKD. Am. J. Kidney Dis. 2006, 48, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Latif, W.; Karaboyas, A.; Tong, L.; Winchester, J.F.; Arrington, C.J.; Pisoni, R.L.; Marshall, M.R.; Kleophas, W.; Levin, N.W.; Sen, A.; et al. Uric acid levels and all-cause and cardiovascular mortality in the hemodialysis population. Clin. J. Am. Soc. Nephrol. 2011, 6, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, J.C.; Coresh, J.; Powe, N.R.; Levey, A.S.; Fink, N.E.; Martin, A.; Klag, M.J. Traditional cardiovascular disease risk factors in dialysis patients compared with the general population: The choice study. J. Am. Soc. Nephrol. 2002, 13, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the american heart association councils on kidney in cardiovascular disease, high blood pressure research, clinical cardiology, and epidemiology and prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Yoshida, M. Protein-bound uremic toxins: New culprits of cardiovascular events in chronic kidney disease patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Jhawar, S.; Singh, P.; Torres, D.; Ramirez-Valle, F.; Kassem, H.; Banerjee, T.; Dolgalev, I.; Heguy, A.; Zavadil, J.; Lowenstein, J. Functional genomic analysis identifies indoxyl sulfate as a major, poorly dialyzable uremic toxin in end-stage renal disease. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Ha, S.K. Uric acid puzzle: Dual role as anti-oxidantand pro-oxidant. Electrolyte Blood Press. 2014, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. P-cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [PubMed]
- Smith, A.R.; Hagen, T.M. Vascular endothelial dysfunction in aging: Loss of Akt-dependent endothelial nitric oxide synthase phosphorylation and partial restoration by (R)-alpha-lipoic acid. Biochem. Soc. Trans. 2003, 31, 1447–1449. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Nakagawa, T.; Feng, L.; Watanabe, S.; Han, L.; Mazzali, M.; Truong, L.; Harris, R.; Johnson, R.J. A role for uric acid in the progression of renal disease. J. Am. Soc. Nephrol. 2002, 13, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Vinik, O.; Wechalekar, M.D.; Falzon, L.; Buchbinder, R.; van der Heijde, D.M.; Bombardier, C. Treatment of asymptomatic hyperuricemia for the prevention of gouty arthritis, renal disease, and cardiovascular events: A systematic literature review. J. Rheumatol. Suppl. 2014, 92, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Givertz, M.M.; Anstrom, K.J.; Redfield, M.M.; Deswal, A.; Haddad, H.; Butler, J.; Tang, W.H.; Dunlap, M.E.; LeWinter, M.M.; Mann, D.L.; et al. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: The xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation 2015, 131, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Gouri, A.; Dekaken, A.; Bentorki, A.A.; Touaref, A.; Yakhlef, A.; Kouicem, N. Serum uric acid level and cardiovascular risks in hemodialysis patients: An algerian cohort study. Clin. Lab. 2014, 60, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Serafini, M.; Colic Baric, I.; Hazen, S.L.; Klein, S. Effect of plasma uric acid on antioxidant capacity, oxidative stress, and insulin sensitivity in obese subjects. Diabetes 2014, 63, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.J.; Iribarren, C.; Gross, M.D.; Comstock, G.W.; Cutler, R.G. Uric acid and serum antioxidant capacity: A reaction to atherosclerosis? Atherosclerosis 2000, 148, 131–139. [Google Scholar] [CrossRef]
- Hayashi, S.; Fujiwara, S.; Noguchi, T. Evolution of urate-degrading enzymes in animal peroxisomes. Cell Biochem. Biophys. 2000, 32, 123–129. [Google Scholar] [CrossRef]
- Sohal, R.S. Role of oxidative stress and protein oxidation in the aging process. Free Radic. Biol. Med. 2002, 33, 37–44. [Google Scholar] [CrossRef]
- Glantzounis, G.K.; Tsimoyiannis, E.C.; Kappas, A.M.; Galaris, D.A. Uric acid and oxidative stress. Curr. Pharm. Des. 2005, 11, 4145–4151. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Spitsin, S.V.; Scott, G.S.; Mikheeva, T.; Zborek, A.; Kean, R.B.; Brimer, C.M.; Koprowski, H.; Hooper, D.C. Comparison of uric acid and ascorbic acid in protection against EAE. Free Radic. Biol. Med. 2002, 33, 1363–1371. [Google Scholar] [CrossRef]
- Johnson, R.J.; Titte, S.; Cade, J.R.; Rideout, B.A.; Oliver, W.J. Uric acid, evolution and primitive cultures. Semin. Nephrol. 2005, 25, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Obesity, oxidative stress, and fibrosis in chronic kidney disease. Kidney Int. Suppl. 2014, 4, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Chang, C.H.; Sun, M.F.; Hsu, S.F.; Weng, C.S. DPP-4 inhibitor attenuates toxic effects of indoxyl sulfate on kidney tubular cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Adelibieke, Y.; Shimizu, H.; Saito, S.; Mironova, R.; Niwa, T. Indoxyl sulfate counteracts endothelial effects of erythropoietin through suppression of akt phosphorylation. Circ. J. 2013, 77, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Hwang, S.J. Incidence, prevalence and mortality trends of dialysis end-stage renal disease in Taiwan from 1990 to 2001: The impact of national health insurance. Nephrol. Dial. Transplant. 2008, 23, 3977–3982. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Hsieh, F.Y.; Chen, Y.H.; Lin, C.C.; Kuan, I.I.; Wang, S.H.; Wu, C.C.; Chien, H.F.; Lin, F.Y.; Chen, Y.L. Atorvastatin induces thrombomodulin expression in the aorta of cholesterol-fed rabbits and in tnfalpha-treated human aortic endothelial cells. Histol. Histopathol. 2009, 24, 1147–1159. [Google Scholar] [PubMed]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | UA Level (mg/dL) | p Value | |||
|---|---|---|---|---|---|
| <6.2 | 6.2–7.1 | 7.1–8.1 | >8.1 | ||
| n | 6078 | 7037 | 7445 | 6669 | |
| Age, years | 66.3 (11.7) | 63.5 (11.2) | 62.3 (11.2) | 61.0 (10.8) | <0.001 |
| Age group, n (%) | <0.001 | ||||
| 40–64 years | 2490 (41) | 3596 (51.1) | 4164 (55.9) | 4049 (60.7) | |
| 65–74 years | 1895 (31.2) | 2151 (30.6) | 2106 (28.3) | 1782 (26.7) | |
| 75+ years | 1693 (27.9) | 1290 (18.3) | 1175 (15.8) | 838 (12.6) | |
| Male, n (%) | 2543 (41.8) | 3284 (46.7) | 3761 (50.5) | 3549 (53.2) | <0.001 |
| DM, n (%) | 2575 (42.4) | 2957 (42) | 3050 (41) | 2609 (39.1) | <0.001 |
| Kt/V | 1.7 (0.3) | 1.6 (0.3) | 1.6 (0.3) | 1.6 (0.3) | <0.001 |
| Hematocrit, % | 29.8 (3.3) | 30.1 (3.2) | 30.2 (3.3) | 30.3 (3.6) | <0.001 |
| Ferritin, ng/L | 574 (481) | 546 (470) | 537 (493) | 544 (514) | <0.001 |
| TSAT, % | 33.2 (14.6) | 33.2 (13.6) | 32.9 (13.9) | 32.3 (14.1) | <0.001 |
| Serum Ca, mg/dl | 9.3 (0.8) | 9.3 (0.7) | 9.3 (0.7) | 9.3 (0.8) | 0.20 |
| Serum P, mg/dl | 4.2 (1.2) | 4.7 (1.2) | 5.0 (1.2) | 5.5 (1.4) | <0.001 |
| Ca*P | 33.7 (13.3) | 34.3 (15.6) | 33.0 (17.9) | 29.4 (20.4) | <0.001 |
| iPTH, pg/mL | 142 (165) | 171 (181) | 192 (205) | 228 (236) | <0.001 |
| Uric Acid (mg/dL) | Events | IR | c. HR (95% CI) | a. HR (95% CI) |
|---|---|---|---|---|
| All-cause mortality | ||||
| <6.2 | 1627 | 66.3 | 1.68 (1.56–1.81) | 1.20 (1.10–1.31) |
| 6.2–7.1 | 1511 | 49.0 | 1.23 (1.14–1.33) | 1.09 (1.01–1.19) |
| 7.1–8.1 | 1444 | 43.9 | 1.10 (1.02–1.19) | 1.06 (0.97–1.15) |
| >8.1 | 1155 | 39.8 | 1.0 (reference) | 1.0 (reference) |
| CV related mortality | ||||
| <6.2 | 1073 | 43.7 | 1.78 (1.62–1.96) | 1.26 (1.13–1.41) |
| 6.2–7.1 | 935 | 30.3 | 1.23 (1.11–1.35) | 1.09 (0.98–1.22) |
| 7.1–8.1 | 902 | 27.4 | 1.11 (1.01–1.22) | 1.06 (0.95–1.18) |
| >8.1 | 716 | 24.7 | 1.0 (reference) | 1.0 (reference) |
| Stroke mortality | ||||
| <6.2 | 101 | 4.1 | 1.79 (1.32–2.44) | 1.59 (1.12–2.25) |
| 6.2–7.1 | 115 | 3.7 | 1.61 (1.19–2.17) | 1.61 (1.16–2.24) |
| 7.1–8.1 | 97 | 2.9 | 1.27 (0.93–1.74) | 1.27 (0.91–1.79) |
| >8.1 | 67 | 2.3 | 1.0 (reference) | 1.0 (reference) |
| Cancer mortality | ||||
| <6.2 | 146 | 5.9 | 1.25 (0.99–1.57) | 0.87 (0.67–1.13) |
| 6.2–7.1 | 177 | 5.7 | 1.19 (0.95–1.48) | 0.93 (0.73–1.19) |
| 7.1–8.1 | 170 | 5.2 | 1.07 (0.85–1.33) | 1.01 (0.79–1.28) |
| >8.1 | 140 | 4.8 | 1.0 (reference) | 1.0 (reference) |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, W.-L.; Li, S.-Y.; Liu, J.-S.; Huang, P.-H.; Lin, S.-J.; Hsu, C.-C.; Lin, Y.-P.; Tarng, D.-C. High Uric Acid Ameliorates Indoxyl Sulfate-Induced Endothelial Dysfunction and Is Associated with Lower Mortality among Hemodialysis Patients. Toxins 2017, 9, 20. https://doi.org/10.3390/toxins9010020
Hsu W-L, Li S-Y, Liu J-S, Huang P-H, Lin S-J, Hsu C-C, Lin Y-P, Tarng D-C. High Uric Acid Ameliorates Indoxyl Sulfate-Induced Endothelial Dysfunction and Is Associated with Lower Mortality among Hemodialysis Patients. Toxins. 2017; 9(1):20. https://doi.org/10.3390/toxins9010020
Chicago/Turabian StyleHsu, Wei-Liang, Szu-Yuan Li, Jia-Sin Liu, Po-Hsun Huang, Shing-Jong Lin, Chih-Cheng Hsu, Yao-Ping Lin, and Der-Cherng Tarng. 2017. "High Uric Acid Ameliorates Indoxyl Sulfate-Induced Endothelial Dysfunction and Is Associated with Lower Mortality among Hemodialysis Patients" Toxins 9, no. 1: 20. https://doi.org/10.3390/toxins9010020