RNA-Seq-Based Transcriptome Analysis of Aflatoxigenic Aspergillus flavus in Response to Water Activity

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Growth Conditions
2.2. Growth Assessment and Aflatoxin Analysis
2.3. cDNA Preparation and Illumina Sequencing
2.4. Clean Reads and Sequence Assembly
2.5. Annotation and Analysis of Unigenes
2.6. Identification and Analysis of Differentially Expressed Genes
2.7. Availability of Supporting Data
3. Results and Discussion
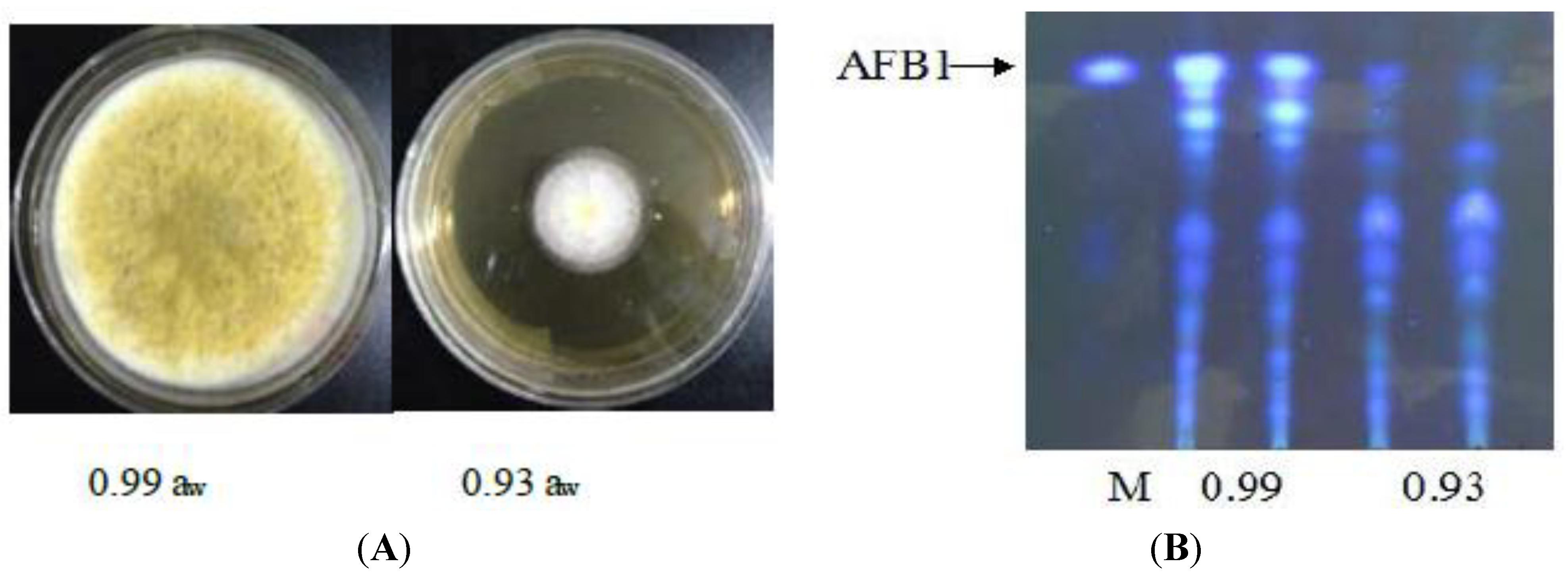
3.1. Effect of Water Activity on Growth and Aflatoxin Production of A. flavus

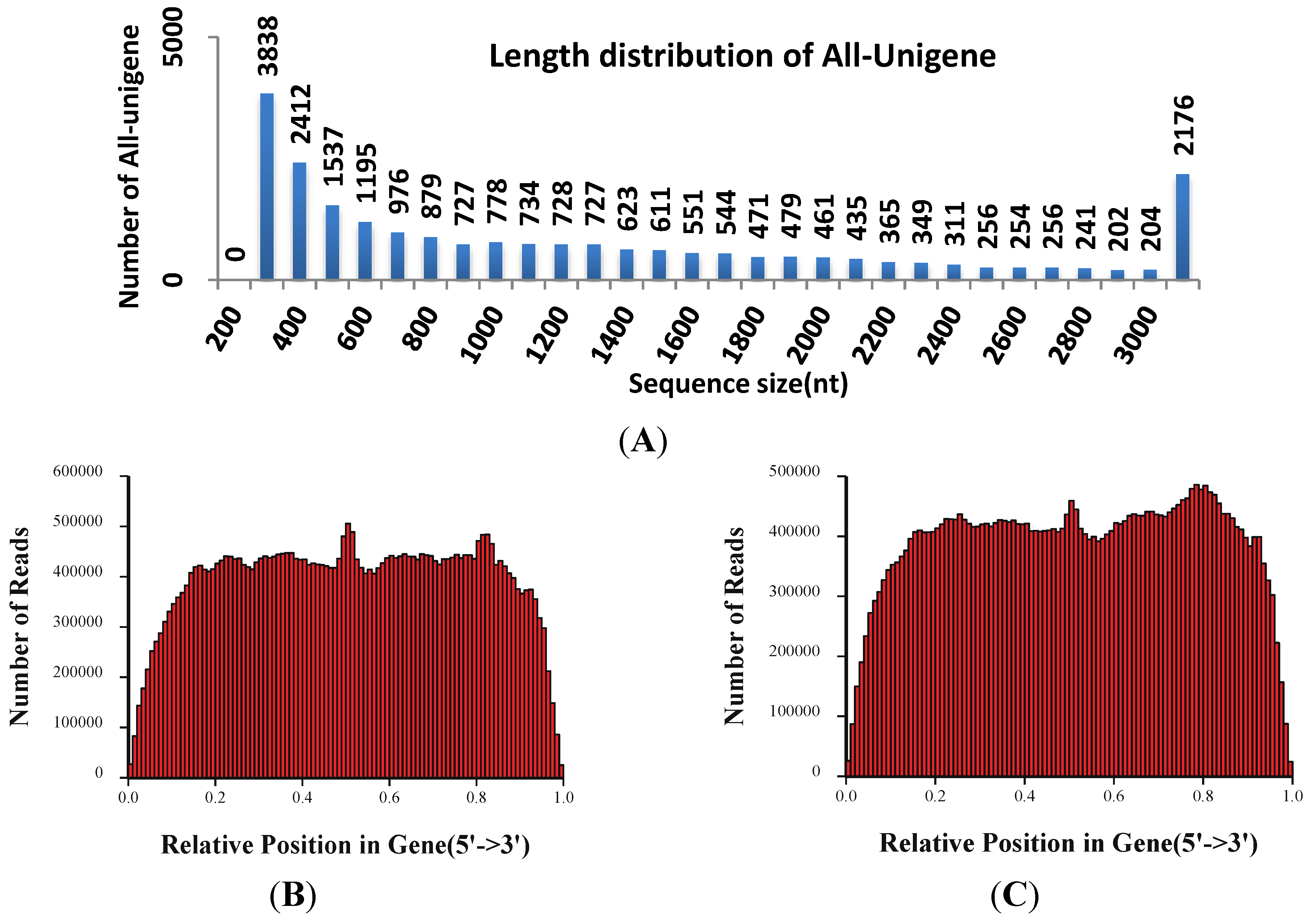
3.2. Illumina Sequencing and Reads Assembling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Treatments | Total number | Mean length (Nt) | N50 |
|---|---|---|---|---|
| Contigs | 0.93 aw | 29,420 | 663 | 1705 |
| 0.99 aw | 30,619 | 653 | 1623 | |
| Unigenes | 0.93 aw | 25,190 | 1004 | 1740 |
| 0.99 aw | 24,991 | 1073 | 1829 |
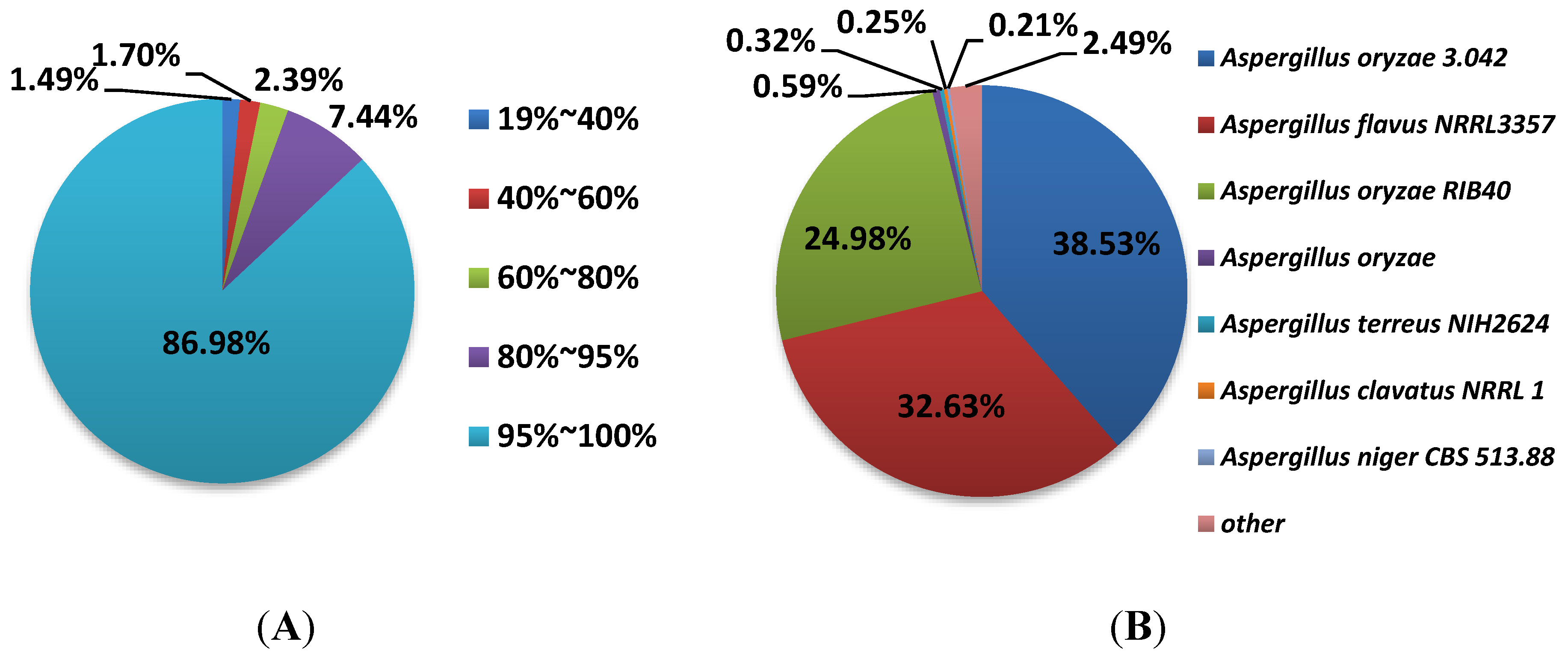
3.3. Annotation and Analysis of All-Unigenes

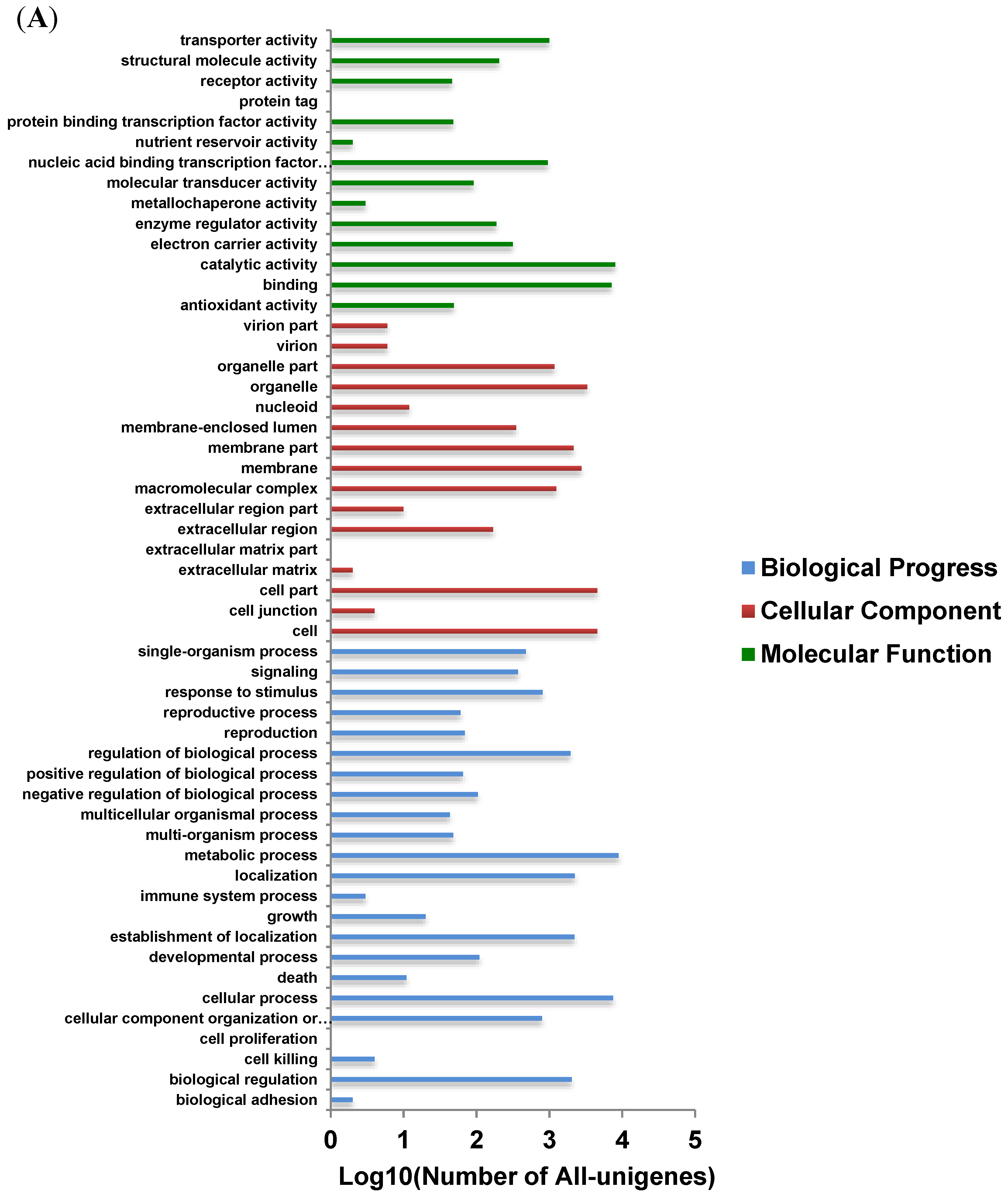
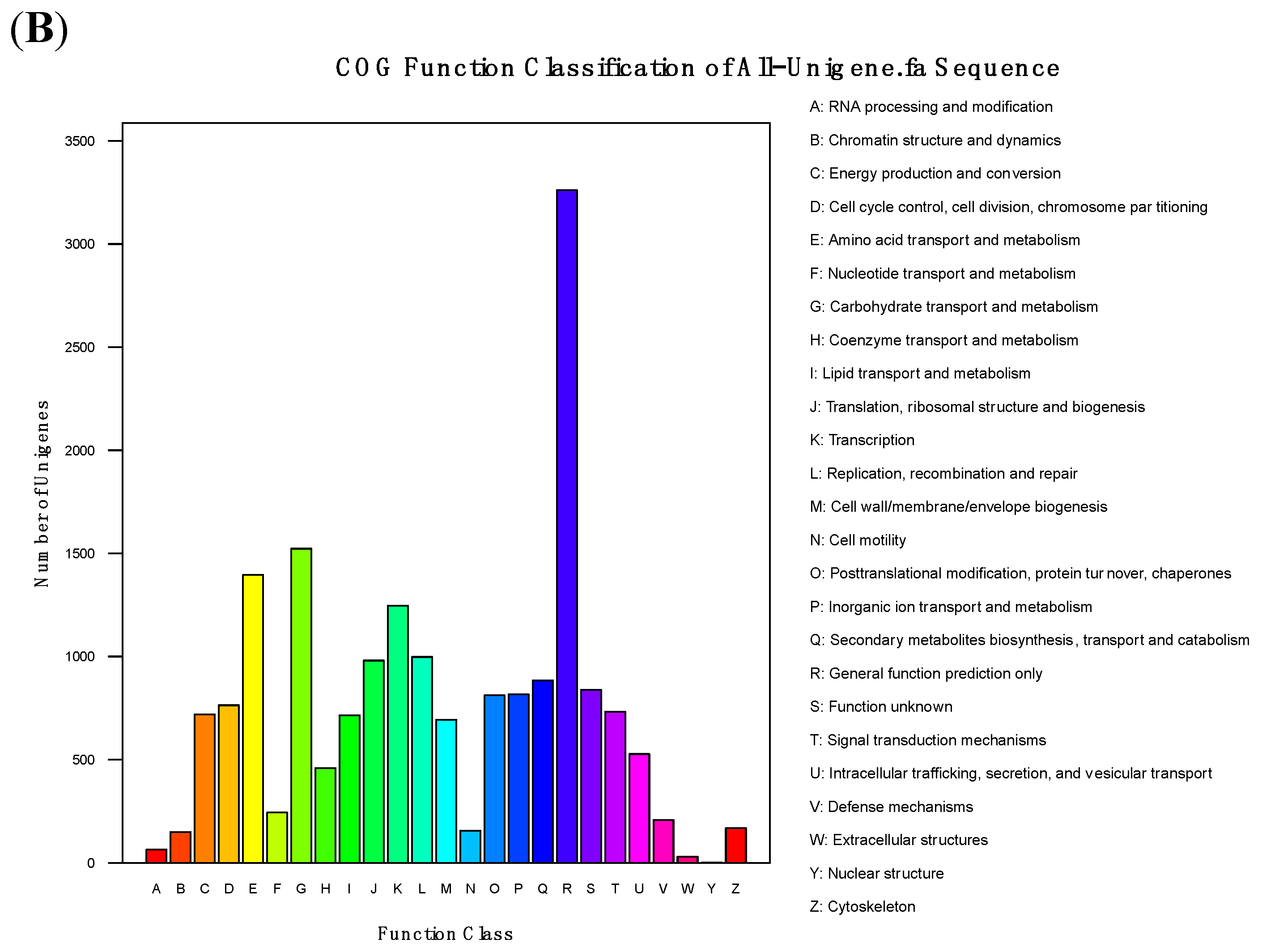
3.4. Functional Analysis and Classification of All-Unigenes

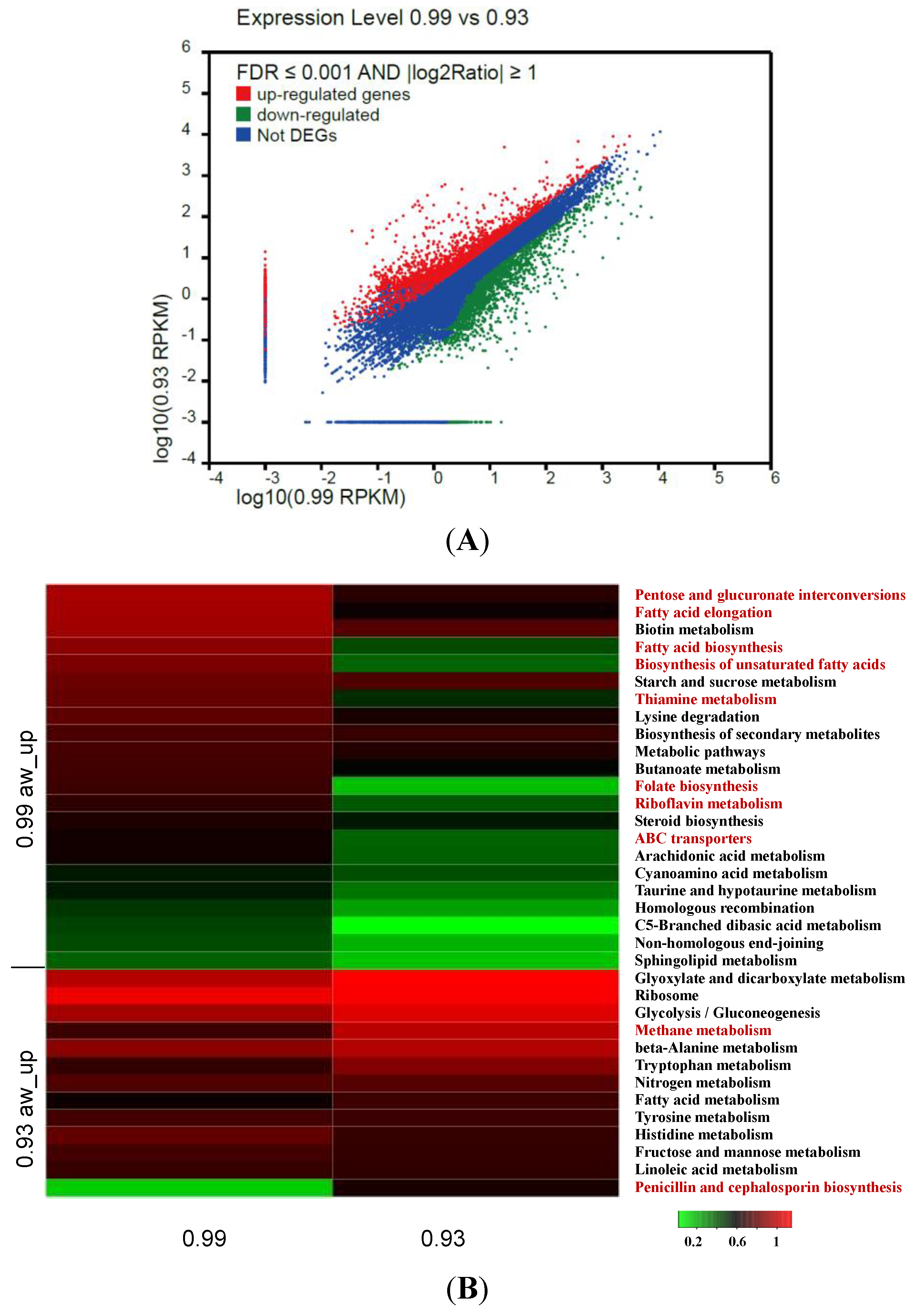
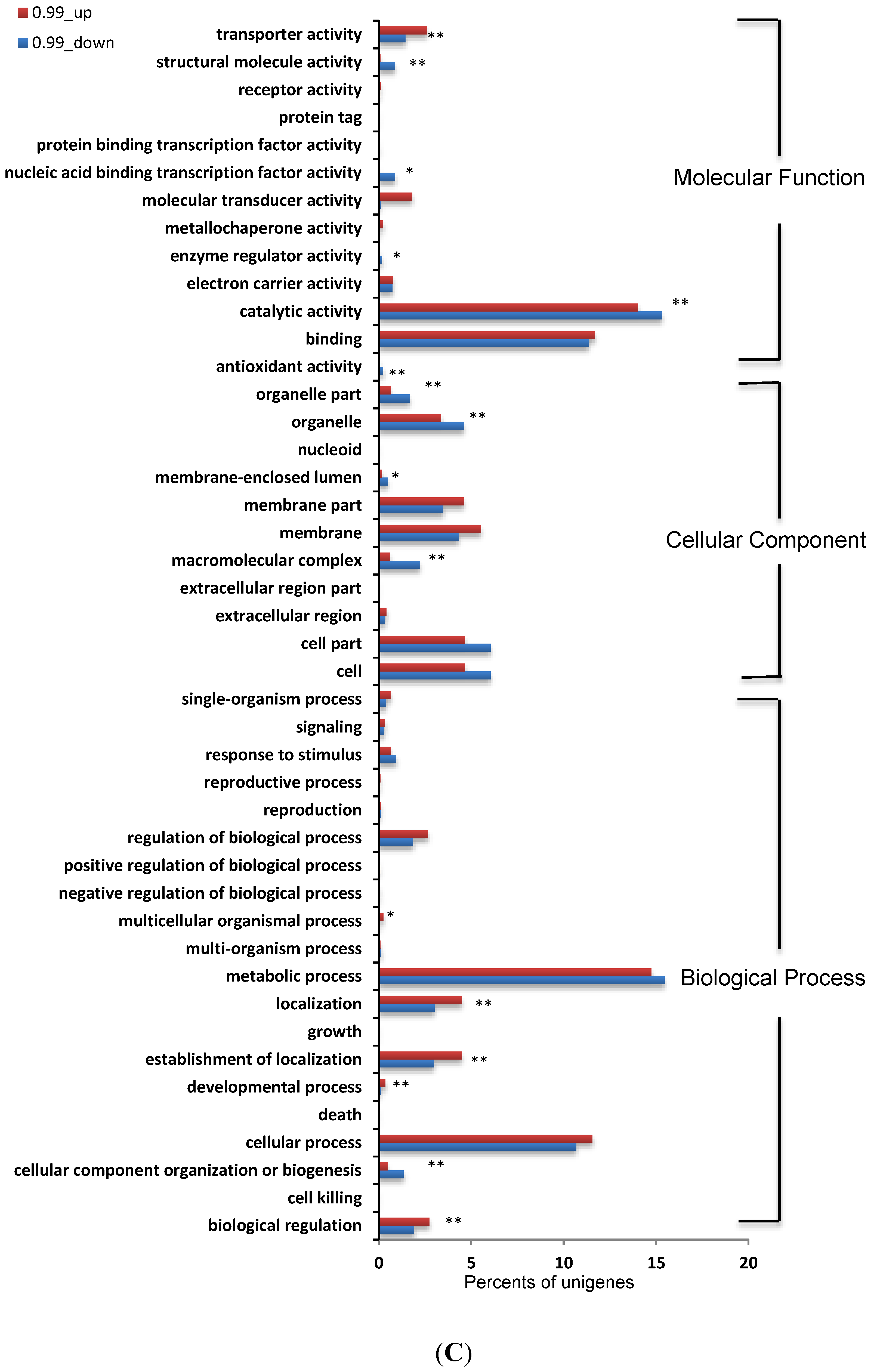
3.5. Identification and Analysis of DEGs


3.6. Analysis of DEGs Involved in Aflatoxin Biosynthesis
3.7. Analysis of DEGs Involved in Development


| Gene | ref_ID | Function | 0.99_RPKM | 0.93_RPKM | Log2 (0.99_RPKM/0.93_RPKM) | Changes (*) |
|---|---|---|---|---|---|---|
| aflF | XP_002379954 | Dehydrogenase | 5.2 | 0.1 | 6.3 | Up |
| aflU | XP_002379953 | P450 monooxygenase | 16.3 | 1.1 | 3.9 | Up |
| aflG | XP_002379937 | Cytochrome P450 monooxygenase | 50.6 | 4.2 | 3.6 | Up |
| aflNa | XP_002379938 | Hypothetical protein | 121.4 | 17.5 | 2.8 | Up |
| aflT | XP_002379952 | Transmembrane protein | 10.4 | 1.7 | 2.6 | Up |
| aflQ | XP_002379931 | Cytochrome P450 monooxigenase | 163.3 | 28.3 | 2.5 | Up |
| aflJ | XM_002379902 | Esterase | 6.1 | 1.3 | 2.2 | Up |
| aflI | XP_002379934 | Cytochrome P450 monooxygenase | 1.1 | 0.3 | 1.7 | Up |
| aflMa | XP_002379940 | Hypothetical protein | 11.9 | 4.1 | 1.5 | Up |
| aflYb | XP_002379924 | Putative hexose transporter | 5.9 | 2.2 | 1.4 | Up |
| aflYd | XP_002379922 | Sugar regulator | 2.8 | 1.1 | 1.4 | Up |
| aflX | XP_002379927 | Monooxygenase oxidase | 6.4 | 2.4 | 1.4 | Up |
| aflB | XP_002379947 | Fatty acid synthase beta subunit | 149.9 | 72.2 | 1.1 | Up |
| aflW | XP_002379928 | Monooxygenase | 3.2 | 1.6 | 1.0 | Up |
| aflY | XP_002379926 | Hypothetical protein | 4.4 | 2.2 | 1.0 | Up |
| aflL | XP_002379936 | P450 monooxygenase | 4.6 | 2.4 | 0.9 | |
| aflN | XP_002379939 | Monooxygenase | 3.1 | 1.7 | 0.9 | |
| aflV | XP_002379929 | Cytochrome P450 monooxygenase | 2.1 | 1.2 | 0.8 | |
| aflE | XP_002379942 | NOR reductase dehydrogenase | 6.5 | 4.1 | 0.7 | |
| aflM | XP_002379941 | Ketoreductase | 14.0 | 10.8 | 0.4 | |
| aflYa | XP_002379925 | NADH oxidase | 2.1 | 1.6 | 0.4 | |
| aflS | XP_002379945 | Pathway regulator | 0.6 | 0.5 | 0.3 | |
| aflP | XP_002379932 | O-methyltransferase A | 4.7 | 4.2 | 0.2 | |
| aflK | XP_002379930 | VERB synthase | 11.5 | 11.2 | 0.0 | |
| aflR | XM_002379905 | Transcription activator | 0.1 | 0.1 | 0.0 | |
| aflLa | XP_002379935 | Hypothetical protein | 1.4 | 1.6 | −0.1 | |
| aflO | XP_002379933 | O-methyltransferase B | 6.7 | 7.7 | −0.2 | |
| aflH | XP_002379944 | Short chain alcohol dehydrogenase | 4.4 | 5.5 | −0.3 | |
| aflA | XP_002379948 | Fatty acid synthase alpha subunit | 4.7 | 6.0 | −0.3 | |
| aflCa | XP_002379950 | Hypothetical protein | 0.4 | 0.6 | −0.4 | |
| aflD | XP_002379949 | Reductase | 0.4 | 0.6 | −0.4 | |
| aflYc | XP_002379923 | Glucosidase | 19.9 | 26.2 | −0.4 | |
| aflC | XP_002379951 | Polyketide synthase | 4.8 | 7.7 | −0.7 |
| Gene | ref_ID | Function | 0.99_RPKM | 0.93_RPKM | Log2 (0.99_RPKM/0.93_RPKM) | Changes (*) |
|---|---|---|---|---|---|---|
| rgsA | gi|259484767|tpe|CBF81270.1| | G protein regulator | ND | ND | ||
| nsdD | gi|259485893|tpe|CBF83303.1| | DNA binding protein | ND | ND | ||
| flbC | gi|259487830|tpe|CBF86815.1| | Putative zinc finger protein | 135.1 | 5.7 | 4.6 | Up |
| cryA | gi|40747330|gb|EAA66486.1| | Hypothetical protein | 0.2 | 0 | 3.2 | Up |
| MAT1-1 | gi|259486330|tpe|CBF84081.1| | Mating type alpha box protein | 1.1 | 0.1 | 3.2 | Up |
| gprB | gi|34482020|tpg|DAA01795.1| | Pheromone receptor | 9.9 | 1.7 | 2.5 | Up |
| abr1 | gi|6090821|gb|AAF03353.1| | Brown 1 | 2.8 | 0.7 | 1.9 | Up |
| brnA | gi|134081843|emb|CAK42098.1| | Unnamed protein product | 2.8 | 0.7 | 1.9 | Up |
| MAT1-2 | gi|259482427|tpe|CBF76901.1| | Mating type HMG-box protein | 8.6 | 2.5 | 1.8 | Up |
| brlA | gi|259488735|tpe|CBF88417.1| | Regulatory protein | 7.7 | 2.3 | 1.8 | Up |
| lreA | gi|259485576|tpe|CBF82714.1| | GATA-factor | 6.6 | 2.9 | 1.2 | Up |
| stuA | gi|259480005|tpe|CBF70741.1| | Cell pattern formation-associated protein | 3.8 | 1.8 | 1.1 | Up |
| medA | gi|259479562|tpe|CBF69898.1| | Medusa | 6.1 | 3 | 1.1 | Up |
| tpsB | gi|1488038|gb|AAB05869.1| | Trehalose-6-phosphate synthase | 91.5 | 47.4 | 0.9 | |
| tpsA | gi|3170246|gb|AAC18060.1| | Trehalose-6-phosphate synthase subunit 1 | 91.5 | 47.4 | 0.9 | |
| steA | gi|259487683|tpe|CBF86542.1| | Transcription factor | 25 | 15 | 0.8 | |
| gprA | gi|34482022|tpg|DAA01796.1| | Pheromone receptor | 4.4 | 2.8 | 0.7 | |
| gprD | gi|259485627|tpe|CBF82810.1| | Integral membrane protein | 11.3 | 7 | 0.7 | |
| lreB | gi|259481867|tpe|CBF75789.1| | GATA-factor | 76.3 | 51.6 | 0.6 | |
| rosA | gi|259484624|tpe|CBF81007.1| | Repressor of sexual development | 47.1 | 30.7 | 0.6 | |
| nosA | gi|259487198|tpe|CBF85681.1| | NosA protein | 47.1 | 30.7 | 0.6 | |
| wetA | gi|259487296|tpe|CBF85858.1| | Regulatory protein | 5.3 | 3.6 | 0.6 | |
| phnA | gi|259489726|tpe|CBF90234.1| | Phosducin-like protein | 33.1 | 21.7 | 0.6 | |
| arp2 | gi|6090729|gb|AAF03314.1| | Tetrahydroxynaphthalene reductase | 14 | 10.8 | 0.4 | |
| kapA | gi|259487521|tpe|CBF86262.1| | Karyopherin alpha | 281.4 | 221.4 | 0.4 | |
| arp1 | gi|2555060|gb|AAC49843.1| | Scytalone dehydratase | 63 | 52.6 | 0.3 | |
| nsdC | gi|259481122|tpe|CBF74364.1| | NSDC | 8 | 6.5 | 0.3 | |
| schA | gi|259481151|tpe|CBF74417.1| | CAMP-dependent protein kinase-like | 2.9 | 2.4 | 0.3 | |
| abaA | gi|167998|gb|AAA33286.1| | AbaA protein | 6 | 5.7 | 0.1 | |
| pkaA | gi|259479481|tpe|CBF69742.1| | CAMP-dependent protein kinase | 23.3 | 21.1 | 0.1 | |
| mpkB | gi|259481736|tpe|CBF75537.1| | Mitogen-activated protein kinase | 26.4 | 25.2 | 0.1 | |
| flbB | gi|259483861|tpe|CBF79600.1| | bZIP-type transcription factor | 48 | 43.4 | 0.1 | |
| fphA | gi|259486541|tpe|CBF84471.1| | Phytochrome | 10.1 | 9 | 0.1 | |
| steC | gi|259487662|tpe|CBF86503.1| | MAPKK kinase | 17.3 | 16.3 | 0.1 | |
| veA | gi|259488644|tpe|CBF88249.1| | Mutant VeA1 protein | 32.2 | 30.7 | 0.1 | |
| laeA | gi|259488911|tpe|CBF88745.1| | Methyltransferase | 19.3 | 17.5 | 0.1 | |
| flbE | gi|259489004|tpe|CBF88918.1| | Putative uncharacterized protein | 54.4 | 51.4 | 0.1 | |
| flbA | gi|259479939|tpe|CBF70620.1| | Developmental regulator | 18.4 | 18.4 | 0.0 | |
| gpgA | gi|259486344|tpe|CBF84107.1| | G protein gamma subunit | 248.6 | 247 | 0.0 | |
| sfaD | gi|259489728|tpe|CBF90238.1| | G-protein beta subunit | 78 | 77.2 | 0.0 | |
| abr2 | gi|6090815|gb|AAF03349.1| | Brown 2 | 6.8 | 7.5 | −0.2 | |
| gpaA | gi|27524346|emb|CAC81704.1| | GMP binding protein alpha subunit | 43.8 | 47.3 | −0.2 | |
| gpaB | gi|27524350|emb|CAC81805.1| | GMP binding protein alpha subunit | 1.6 | 1.9 | −0.2 | |
| pkaB | gi|67537094|ref|XP_662321.1| | Hypothetical protein | 0.6 | 0.7 | −0.2 | |
| gaoC | gi|83773752|dbj|BAE63877.1| | Unnamed protein product | 43.8 | 47.3 | −0.2 | |
| yA | gi|259480215|tpe|CBF71142.1| | Laccase-1 Precursor | 6.8 | 7.5 | −0.2 | |
| ganB | gi|259488687|tpe|CBF88328.1| | G protein alpha subunit | 1.6 | 1.9 | −0.2 | |
| fadA | gi|259489081|tpe|CBF89057.1| | GMP binding protein subunit alpha | 43.8 | 47.3 | −0.2 | |
| velB | gi|259489398|tpe|CBF89638.1| | VelB | 15.1 | 16.6 | −0.2 | |
| flbD | gi|259489501|tpe|CBF89824.1| | Putative uncharacterized protein | 9.9 | 11.4 | −0.2 | |
| alb1 | gi|3136092|gb|AAC39471.1| | Polyketide synthase | 13.9 | 17.4 | −0.3 | |
| chsC | gi|4519181|dbj|BAA75501.1| | Chitin synthase | 8.8 | 11 | −0.3 | |
| fwnA | gi|134078436|emb|CAL00851.1| | Unnamed protein product | 13.9 | 17.4 | −0.3 | |
| sfgA | gi|259480894|tpe|CBF73944.1| | SfgA | 0.8 | 0.9 | −0.3 | |
| treB | gi|2827392|gb|AAB99831.1| | Neutral trehalase | 43.2 | 62.1 | −0.5 | |
| pptA | gi|134080185|emb|CAK46165.1| | Unnamed protein product | 38.3 | 54.6 | −0.5 | |
| cyaA | gi|259481514|tpe|CBF75105.1| | Adenylate cyclase | 1.6 | 2.4 | −0.5 | |
| vosA | gi|259487318|tpe|CBF85898.1| | VosA | 4.3 | 6.2 | −0.5 | |
| ppoB | gi|259479464|tpe|CBF69709.1| | Fatty acid oxygenase | 15.4 | 27.7 | −0.7 | |
| ppoC | gi|259482096|tpe|CBF76249.1| | Fatty acid oxygenase | 15.4 | 27.7 | −0.7 | |
| ppoA | gi|259487326|tpe|CBF85912.1| | Fatty acid oxygenase | 15.4 | 27.7 | −0.7 | |
| rasA | gi|259489610|tpe|CBF90024.1| | Ras-like protein | 1.1 | 1.8 | −0.7 | |
| chsA | gi|465390|dbj|BAA04806.1| | Chitin synthase | 8.7 | 19.6 | −1.3 | Down |
| fluG | gi|259482332|tpe|CBF76713.1| | FluG | 11.8 | 34.5 | −1.7 | Down |
| chiB | gi|259485098|tpe|CBF81882.1| | Class V chitinase | 0.1 | 0.1 | −1.7 | Down |
| rolA | gi|28875529|dbj|BAC65230.1| | Hydrophobin putative | 1.5 | 6.9 | −2.3 | Down |
| rodB | gi|70996676|ref|XP_753093.1| | Conidial hydrophobin | 1.5 | 6.9 | −2.3 | Down |
| rodA | gi|259482991|tpe|CBF77990.1| | Rodlet protein | 1.5 | 6.9 | −2.3 | Down |
| ganA | gi|259485962|tpe|CBF83426.1| | G protein alpha subunit homolog | ND | ND |
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gonçalves, S.S.; Cano, J.F.; Stchigel, A.M.; Melo, A.S.; Godoy-Martinez, P.C.; Correa, B.; Guarro, J. Molecular phylogeny and phenotypic variability of clinical and environmental strains of Aspergillus flavus. Fungal Biol. 2012, 116, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Amaike, S.; Keller, N.P. Aspergillus flavus . Annu. Rev. Phytopathol. 2011, 49, 107–133. [Google Scholar] [CrossRef]
- Hua, S.S.T.; McAlpin, C.E.; Chang, P.-K.; Sarreal, S.B.L. Characterization of aflatoxigenic and non-aflatoxigenic Aspergillus flavus isolates from Pistachio. Mycotoxin Res. 2012, 28, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Roebuck, B.D.; Wogan, G.N.; Groopman, J.D. Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci. 2011, 120, S28–S48. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, A.; Atoui, A.; Rizk, T.; Lteif, R.; Kallassy, M.; Lebrihi, A. Differentiation between Aspergillus flavus and Aspergillus parasiticus from pure culture and aflatoxin-contaminated grapes using PCR-RFLP analysis of aflR-aflJ intergenic spacer. J. Food Sci. 2011, 76, M247–M253. [Google Scholar]
- Leema, G.; Chou, D.-S.; Jesudasan, C.A.N.; Geraldine, P.; Thomas, P.A. Expression of genes of the aflatoxin biosynthetic pathway in Aspergillus flavus isolates from Keratitis. Mol. Vis. 2011, 17, 2889–2897. [Google Scholar] [PubMed]
- Adjovi, Y.C.; Bailly, S.; Gnonlonfin, B.J.; Tadrist, S.; Querin, A.; Sanni, A.; Oswald, I.P.; Puel, O.; Bailly, J.D. Analysis of the contrast between natural occurrence of toxigenic Aspergilli of the Flavi section and aflatoxin B1 in Cassava. Food Microbiol. 2014, 38, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Jamali, M.; Karimipour, M.; Shams-Ghahfarokhi, M.; Amani, A.; Razzaghi-Abyaneh, M. Expression of aflatoxin genes aflO (omtB) and aflQ (ordA) differentiates levels of aflatoxin production by Aspergillus flavus strains from soils of Pistachio orchards. Res. Microbiol. 2013, 164, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, D.; Cary, J.W.; Ehrlich, K.; Yu, J.; Cleveland, T.E. Understanding the genetics of regulation of aflatoxin production and Aspergillus flavus development. Mycopathologia 2006, 162, 155–166. [Google Scholar] [PubMed]
- Schmidt-Heydt, M.; Rüfer, C.E.; Abdel-Hadi, A.; Magan, N.; Geisen, R. The production of aflatoxin B1 or G1 by Aspergillus parasiticus at various combinations of temperature and water activity is related to the ratio of aflS to aflR expression. Mycotoxin Res. 2010, 26, 241–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgianna, D.R.; Payne, G.A. Genetic regulation of aflatoxin biosynthesis: From gene to genome. Fungal Genet. Biol. 2009, 46, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Heydt, M.; Magan, N.; Geisen, R. Stress induction of mycotoxin biosynthesis genes by abiotic factors. FEMS Microbiol. Lett. 2008, 284, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Alborch, L.; Bragulat, M.; Abarca, M.; Cabañes, F. Effect of water activity, temperature and incubation time on growth and ochratoxin A production by Aspergillus niger and Aspergillus carbonarius on maize kernels. Int. J. Food Microbiol. 2011, 147, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Mousa, W.; Ghazali, F.; Jinap, S.; Ghazali, H.; Radu, S. Modelling the effect of water activity and temperature on growth rate and aflatoxin production by two isolates of Aspergillus flavus on paddy. J. Appl. Microbiol. 2011, 111, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fedorova, N.D.; Montalbano, B.G.; Bhatnagar, D.; Cleveland, T.E.; Bennett, J.W.; Nierman, W.C. Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-Seq. FEMS Microbiol. Lett. 2011, 322, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hadi, A.; Schmidt-Heydt, M.; Parra, R.; Geisen, R.; Magan, N. A systems approach to model the relationship between aflatoxin gene cluster expression, environmental factors, growth and toxin production by Aspergillus flavus. J. R. Soc. Interface. 2012, 9, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Heydt, M.; Abdel-Hadi, A.; Magan, N.; Geisen, R. Complex regulation of the aflatoxin biosynthesis gene cluster of Aspergillus flavus in relation to various combinations of water activity and temperature. Int. J. Food Microbiol. 2009, 135, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorni, P.; Camardo Leggieri, M.; Magan, N.; Battilani, P. Comparison of temperature and moisture requirements for sporulation of Aspergillus flavus sclerotia on natural and artificial substrates. Fungal Biol. 2012, 116, 637–642. [Google Scholar] [CrossRef]
- Lin, J.Q.; Zhao, X.X.; Zhi, Q.Q.; Zhao, M.; He, Z.M. Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genet. Biol. 2013, 56, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.K.; Scharfenstein, L.L.; Mack, B.; Ehrlich, K.C. Deletion of the Aspergillus flavus orthologue of A. nidulans fluG reduces conidiation and promotes production of sclerotia but does not abolish aflatoxin biosynthesis. Appl. Environ. Microbiol. 2012, 78, 7557–7563. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Waalwijk, C.; de Wit, P.J.; Tang, D.; van der Lee, T. RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yao, J.; Wang, X.; Guo, H.; Duan, D. Transcriptome sequencing and comparative analysis of Saccharina japonica (Laminariales, Phaeophyceae) under blue light induction. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 138–148. [Google Scholar]
- Samapundo, S.; Devlieghere, F.; Geeraerd, A.; De Meulenaer, B.; Van Impe, J.; Debevere, J. Modelling of the individual and combined effects of water activity and temperature on the radial growth of Aspergillus flavus and A. parasiticus on corn. Food Microbiol. 2007, 24, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Manavathu, E.K.; Chandrasekar, P.H. Aspergillus flavus: An emerging non-fumigatus Aspergillus species of significance. Mycoses 2009, 52, 206–222. [Google Scholar] [PubMed]
- Frealle, E.; Aliouat-Denis, C.-M.; Delhaes, L.; Hot, D.; Dei-Cas, E. Transcriptomic insights into the oxidative response of stress-exposed Aspergillus fumigatus. Curr. Pharm. Des. 2013, 19, 3713–3737. [Google Scholar] [PubMed]
- Novodvorska, M.; Hayer, K.; Pullan, S.T.; Wilson, R.; Blythe, M.J.; Stam, H.; Stratford, M.; Archer, D.B. Transcriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-sequencing. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, M.; Miskei, M.; Karányi, Z.; Lenkey, B.; Pócsi, I.; Emri, T. Transcriptome changes initiated by carbon starvation in Aspergillus nidulans. Microbiology 2013, 159, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L.; Mack, B.; Yu, J.; Ehrlich, K.C. Transcriptomic profiles of Aspergillus flavus CA42, a strain that produces small sclerotia, by decanal treatment and after recovery. Fungal Genet. Biol. 2014, 68, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Payne, G.A.; Nierman, W.C.; Machida, M.; Bennett, J.W.; Campbell, B.C.; Robens, J.F.; Bhatnagar, D.; Dean, R.A.; Cleveland, T.E. Aspergillus flavus genomics as a tool for studying the mechanism of aflatoxin formation. Food Addit. Contam. 2008, 25, 1152–1157. [Google Scholar] [CrossRef]
- Medina, M.L.; Kiernan, U.A.; Francisco, W.A. Proteomic analysis of rutin-induced secreted proteins from Aspergillus flavus. Fungal Genet. Biol. 2004, 41, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, C.M.; Wilson, D.; Linz, J.E.; Townsend, C.A. Demonstration of the catalytic roles and evidence for the physical association of type I fatty acid synthases and a polyketide synthase in the biosynthesis of aflatoxin B1. Chem. Biol. 1996, 3, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Bhatnagar, D.; Cleveland, T.E. Completed sequence of aflatoxin pathway gene cluster in Aspergillus parasiticus. FEBS Lett. 2004, 564, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Skory, C.D.; Linz, J.E. Cloning of a gene associated with aflatoxin B1 biosynthesis in Aspergillus parasiticus. Curr. Genet. 1992, 21, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chang, P.-K.; Ehrlich, K.C.; Cary, J.W.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Linz, J.E.; Woloshuk, C.P.; Bennett, J.W. Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 2004, 70, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Yabe, K.; Nakajima, H. Enzyme reactions and genes in aflatoxin biosynthesis. Appl. Microbiol. Biotechnol. 2004, 64, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Carbone, I.; Ramirez-Prado, J.H.; Jakobek, J.L.; Horn, B.W. Gene duplication, modularity and adaptation in the evolution of the aflatoxin gene cluster. BMC Evol. Biol. 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, K.C. Predicted roles of the uncharacterized clustered genes in aflatoxin biosynthesis. Toxins 2009, 1, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hadi, A.; Carter, D.; Magan, N. Temporal monitoring of the nor-1 (aflD) gene of Aspergillus flavus in relation to aflatoxin B1 production during storage of peanuts under different water activity levels. J. Appl. Microbiol. 2010, 109, 1914–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayram, Ö.; Braus, G.H. Coordination of secondary metabolism and development in fungi: The velvet family of regulatory proteins. FEMS Microbiol. Rev. 2012, 36, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Krijgsheld, P.; Bleichrodt, R.; van Veluw, G.; Wang, F.; Müller, W.; Dijksterhuis, J.; Wösten, H. Development in Aspergillus. Stud. Mycol. 2013, 74, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Kwon, N.J.; Garzia, A.; Espeso, E.A.; Ugalde, U.; Yu, J.H. FlbC is a putative nuclear C2H2 transcription factor regulating development in Aspergillus nidulans. Mol. Microbiol. 2010, 77, 1203–1219. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-H.; Rosén, S.; Adams, T.H. Extragenic suppressors of loss-of-function mutations in the Aspergillus FlbA regulator of G-protein signaling domain protein. Genetics 1999, 151, 97–105. [Google Scholar] [PubMed]
- Etxebeste, O.; Herrero-García, E.; Araújo-Bazán, L.; Rodríguez-Urra, A.B.; Garzia, A.; Ugalde, U.; Espeso, E.A. The bZIP-type transcription factor FlbB regulates distinct morphogenetic stages of colony formation in Aspergillus nidulans. Mol. Microbiol. 2009, 73, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Arratia-Quijada, J.; Sánchez, O.; Scazzocchio, C.; Aguirre, J. FlbD, a Myb transcription factor of Aspergillus nidulans, is uniquely involved in both asexual and sexual differentiation. Eukaryot. Cell 2012, 11, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Garzia, A.; Etxebeste, O.; Herrero-Garcia, E.; Fischer, R.; Espeso, E.A.; Ugalde, U. Aspergillus nidulans FlbE is an upstream developmental activator of conidiation functionally associated with the putative transcription factor FlbB. Mol. Microbiol. 2009, 71, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Garzia, A.; Etxebeste, O.; Herrero-García, E.; Ugalde, U.; Espeso, E.A. The concerted action of bZip and cMyb transcription factors FlbB and FlbD induces brlA expression and asexual development in Aspergillus nidulans. Mol. Microbiol. 2010, 75, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Yu, J. Aspergillus flavus genomics: Gateway to human and animal health, food safety, and crop resistance to diseases. Rev. Iberoam Micol. 2005, 22, 194–202. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Guo, Z.; Zhong, H.; Wang, S.; Yang, W.; Liu, Y.; Wang, S. RNA-Seq-Based Transcriptome Analysis of Aflatoxigenic Aspergillus flavus in Response to Water Activity. Toxins 2014, 6, 3187-3207. https://doi.org/10.3390/toxins6113187
Zhang F, Guo Z, Zhong H, Wang S, Yang W, Liu Y, Wang S. RNA-Seq-Based Transcriptome Analysis of Aflatoxigenic Aspergillus flavus in Response to Water Activity. Toxins. 2014; 6(11):3187-3207. https://doi.org/10.3390/toxins6113187
Chicago/Turabian StyleZhang, Feng, Zhenni Guo, Hong Zhong, Sen Wang, Weiqiang Yang, Yongfeng Liu, and Shihua Wang. 2014. "RNA-Seq-Based Transcriptome Analysis of Aflatoxigenic Aspergillus flavus in Response to Water Activity" Toxins 6, no. 11: 3187-3207. https://doi.org/10.3390/toxins6113187