Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum

Abstract
:1. Introduction

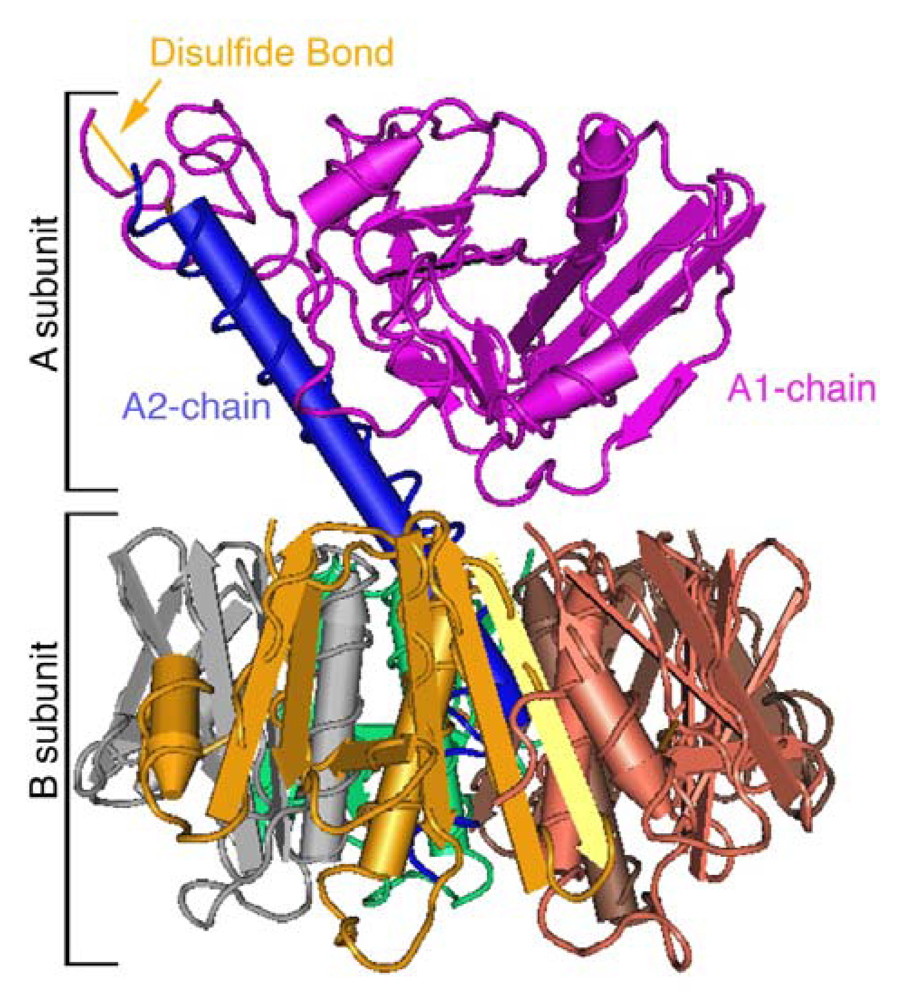
2. Structure and Function

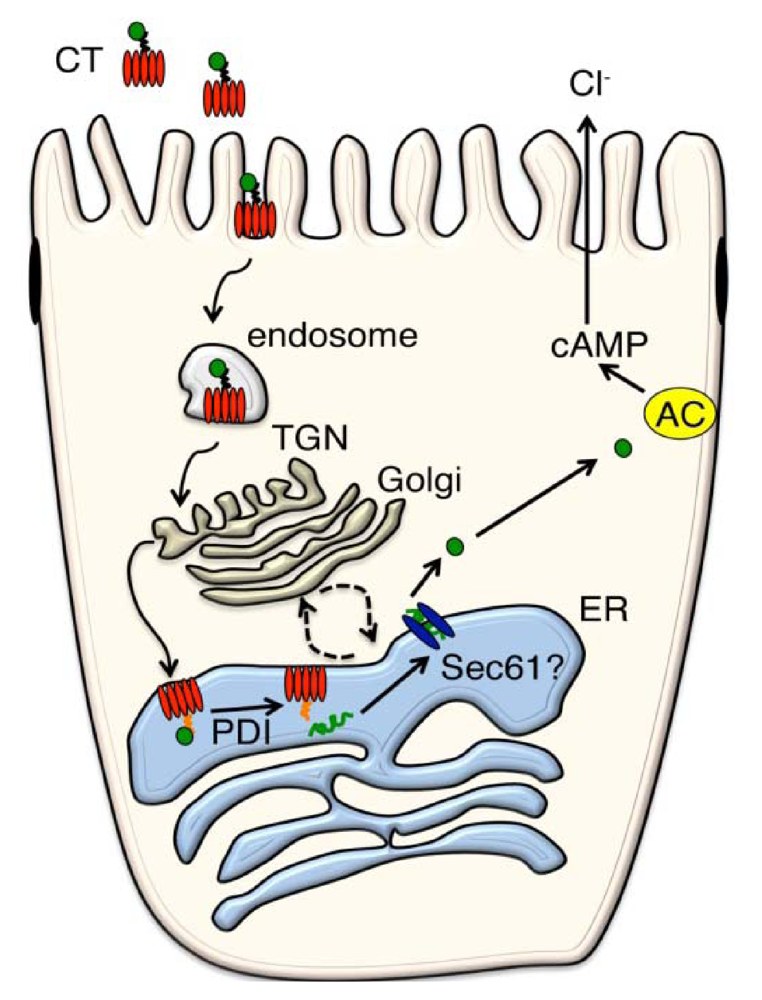
3. Retrograde from the PM to the ER
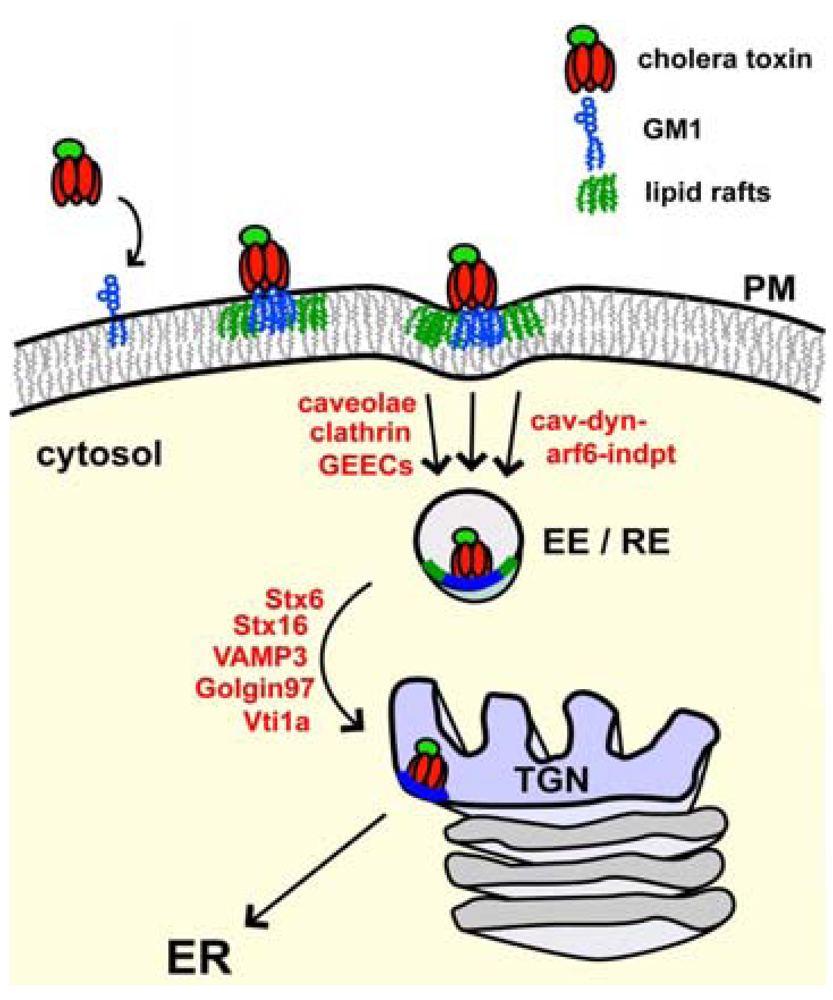
3.1. Binding and Entry via the PM

3.2. Endocytosis and Trafficking Back to the ER
4. From ER to cytosol
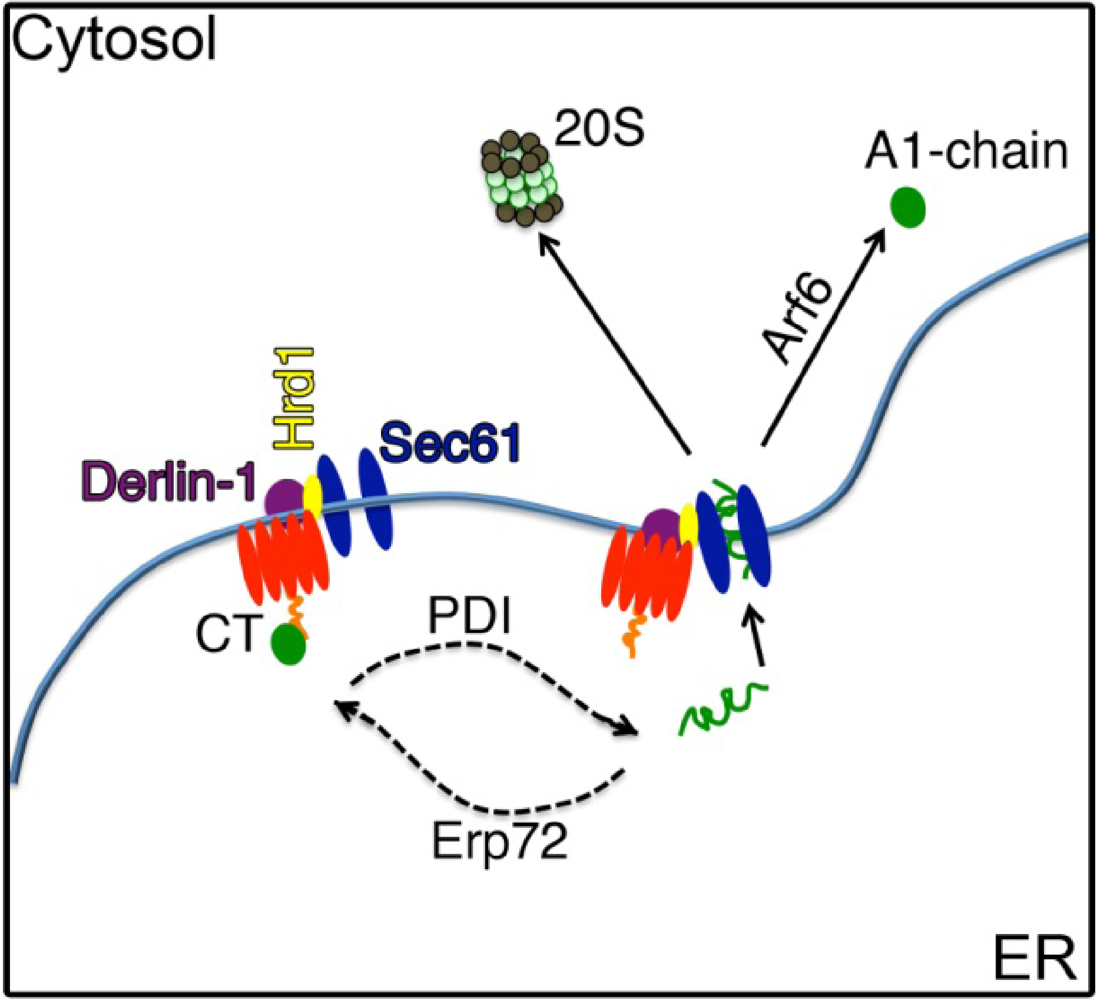
4.1. The A1-chain Hijacks the ERAD Pathway
4.2. Escape from the ER into the Cytosol
4.3. Cytosolic Factors

5. Summary

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host factor | Site of Action | Cellular Function |
|---|---|---|
| Trafficking factors | ||
| Ganglioside GM1 | Host cell membranes: PM, endosomes, Golgi, ER | Lipid receptor for toxin binding and trafficking |
| Lipid rafts | Host cell membranes | Putative small dynamic membrane microdomains that self assemble by phase separation of membrane lipids to form structures with functions in trafficking and signal transduction |
| Clathrin | PM, endosome, Golgi | Protein coat for some forms of endocytosis and vesicle budding |
| ARF 1- 6 | PM, endosome, Golgi | Small GTPases involved in coat formation and membrane traffic. The ARF family was discovered by their ability to act as co-factors for the ADP-ribosylation activity of the CT A1-chain |
| Syntaxin 6 and 16 | early endosome | Component of protein complex involved in fusion of vesicles moving from early endosome to TGN |
| Golgin97 | TGN | Tethering factor for vesicles moving from early endosome to TGN |
| Retromer | endosome | Complex of proteins involved in transport of vesicles form early endosome to TGN |
| Rab 6A' | endosome, TGN | Small GTPases involved in sorting vesicles retrograde from early endosome and TGN to ER |
| VAMP3, Vti1a | TGN | Components of protein complex involved in fusion of vesicles moving from early endosome to TGN |
| SNX1 and 2 | early/recycling endosome | Retromer components that contain phosphatidyl inositol binding and membrane curvature sensing BAR domains. |
| Vps26, 29 and 35 | early/recycling endosome | Retromer components important for cargo selection, such as Shiga toxin and the mannose-6-phosphate receptor |
| ERAD factors | ||
| Protein Disulfide Isomerase (PDI) | ER lumen | Disulfide bond isomerase and protein chaperone, unfolds and dissociates the A1 chain for the B-subunit |
| ER Protein 72 (Erp72) | ER lumen | PDI-like molecule with counteracting function to refold the A1-chain |
| ER oxidase 1 (Ero1) | ER lumen | ER oxidase that oxidizes PDI to release the A1-chain |
| Heavy chain binding protein (BiP) | ER lumen | HSP70 chaperone with major functions in protein folding and ERAD |
| Sec 61 translocon | ER membrane | Core component of the translocon that ribosomes dock with to allow for translocation of membrane and secreted proteins into the ER during biosynthesis. It is also a candidate for the protein conducting channel in ERAD. |
| Derlin-1 | ER membrane | Component of the core Hrd1 complex required for retro-translocation of lumenal ERAD substrates. It is also a candidate for the protein conducting channel in ERAD. |
| Hrd1 | ER membrane | ER membrane ubiquitin E3 ligase forming central component of a protein complex involved in retro-translocation of ER lumenal and membrane ERAD substrates. It is also a candidate for the protein conducting channel in ERAD. |
| gp78 | ER membrane | ER membrane ubiquitin E3 ligase forming central component of a protein complex involved in retro-translocation of ER lumenal and membrane ERAD substrates. |
| Ubiquitin-conjugating enzyme (Ube) | Cytosol | Ubiquitin E2 ligase: Enzyme required before the E3 ligases in the pathway of conjugating ubiquitin to primarily lysine residues on proteins. |
| Ube2g2 | Cytosol, ER associated | Ubiquitin E2 ligase: Enzyme required before the E3 ligases in the pathway of conjugating ubiquitin to primarily lysine residues on proteins. |
| AAA-ATPase p97 | Cytosol | AAA-ATPase involved in chaperone function, proteasomal degradation. It is key for retro-translocation of most ERAD substrates, perhaps providing the driving force for the retro-translocation reaction itself. However, it is not required for retro-translocation of the CT A1-chain. |
References
- Tsai, B.; Rodighiero, C.; Lencer, W.I.; Rapoport, T. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 2001, 104, 937–948. [Google Scholar] [CrossRef]
- Nichols, B.J.; Kenworthy, A.K.; Polishchuk, R.S.; Lodge, R.; Roberts, T.H.; Hirschberg, K.; Phair, R.D.; Lippincott-Schwartz, J. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 2001, 153, 529–541. [Google Scholar]
- Richards, A.A.; Stang, E.; Pepperkok, R.; Parton, R.G. Inhibitors of COP-mediated Transport and Cholera Toxin Action Inhibit Simian Virus 40 Infection. Mol. Biol. Cell 2002, 13, 1750–1764. [Google Scholar] [CrossRef]
- Fujinaga, Y.; Wolf, A.A.; Rodigherio, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera toxin from the plasma membrane to the ER. Mol. Biol. Cell 2003, 14, 4783–4793. [Google Scholar]
- Feng, Y.; Jadhav, A.P.; Rodighiero, C.; Fujinaga, Y.; Kirchhausen, T.; Lencer, W.I. Retrograde transport of cholera toxin from the plasma membrane to the endoplasmic reticulum requires the trans-Golgi network but not the Golgi apparatus in Exo2-treated cells. EMBO Rep. 2004, 5, 596–601. [Google Scholar]
- Winkeler, A.; Godderz, D.; Herzog, V.; Schmitz, A. BiP-dependent export of cholera toxin from endoplasmic reticulum-derived microsomes. FEBS Lett. 2003, 554, 439–442. [Google Scholar] [CrossRef]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef]
- Dixit, G.; Mikoryak, C.; Hayslett, T.; Bhat, A.; Draper, R.K. Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp. Biol. Med. (Maywood) 2008, 233, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, K.M.; Forster, M.L.; Lencer, W.I.; Tsai, B. Derlin-1 facilitates the retro-translocation of cholera toxin. Mol. Biol. Cell 2008, 19, 877–884. [Google Scholar]
- Bernardi, K.M.; Williams, J.M.; Kikkert, M.; van Voorden, S.; Wiertz, E.J.; Ye, Y.; Tsai, B. The E3 ubiquitin ligases hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol. Biol. Cell 2010, 21, 140–151. [Google Scholar]
- Schmitz, A.; Herrgen, H.; Winkeler, A.; Herzog, V. Cholera toxin is exported from microsomes by the sec61p complex. J. Cell Biol. 2000, 148, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Spangler, B.D. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microb Rev. 1992, 56, 622–647. [Google Scholar]
- Falnes, P.O.; Sandvig, K. Penetration of protein toxins into cells. Curr. Opin. Cell Biol. 2000, 12, 407–413. [Google Scholar] [CrossRef]
- Sixma, T.K.; Pronk, S.E.; Kalk, K.H.; Wartna, E.S.; van Zanten, B.A.; Witholt, B.; Hol, W.G. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E.coli. Nature 1991, 351, 371–377. [Google Scholar] [PubMed]
- Zhang, R.G.; Scott, D.L.; Westbrook, M.L.; Nance, S.; Spangler, B.D.; Shipley, G.G.; Westbrook, E.M. The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 1995, 251, 563–573. [Google Scholar] [CrossRef]
- Panasiewicz, M.; Domek, H.; Hoser, G.; Kawalec, M.; Pacuszka, T. Structure of the ceramide moiety of GM1 ganglioside determines its occurrence in different detergent-resistant membrane domains in HL-60 cells. Biochemistry 2003, 42, 6608–6619. [Google Scholar]
- Kaiser, H.J.; Lingwood, D.; Levental, I.; Sampaio, J.L.; Kalvodova, L.; Rajendran, L.; Simons, K. Order of lipid phases in model and plasma membranes. Proc. Natl. Acad. Sci. USA 2009, 106, 16645–16650. [Google Scholar]
- Cicuta, P.; Keller, S.L.; Veatch, S.L. Diffusion of liquid domains in lipid bilayer membranes. J. Phys. Chem. B 2007, 111, 3328–3331. [Google Scholar]
- Sharma, P.; Varma, R.; Sarasij, R.C.; Ira; Gousset, K.; Krishnamoorthy, G.; Rao, M.; Mayor, S. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell 2004, 116, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, C.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.N.; Hein, B.; von Middendorff, C.; Schonle, A.; Hell, S.W. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 2009, 457, 1159–1162. [Google Scholar]
- Ewers, H.; Romer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; Oppenheim, A.; Schwarzmann, G.; Feizi, T.; Schwille, P.; Sens, P.; Helenius, A.; Johannes, L. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell Biol. 2010, 12, 11–18. [Google Scholar]
- Wolf, A.A.; Jobling, M.G.; Saslowsky, D.E.; Kern, E.; Drake, K.R.; Kenworthy, A.K.; Holmes, R.K.; Lencer, W.I. Attenuated endocytosis and toxicity of a mutant cholera toxin with decreased ability to cluster gm1. Infect Immun. 2008, 76, 1476–1484. [Google Scholar]
- Saslowsky, D.E.; Lencer, W.I. Conversion of apical plasma membrane sphingomyelin to ceramide attenuates the intoxication of host cells by cholera toxin. Cell Microbiol. 2008, 10, 67–80. [Google Scholar]
- Wolf, A.A.; Fujinaga, Y.; Lencer, W.I. Uncoupling of the cholera toxin-G(M1) ganglioside receptor complex from endocytosis, retrograde Golgi trafficking, and downstream signal transduction by depletion of membrane cholesterol. J. Biol. Chem. 2002, 277, 16249–16256. [Google Scholar] [PubMed]
- Smith, D.C.; Sillence, D.J.; Falguieres, T.; Jarvis, R.M.; Johannes, L.; Lord, J.M.; Platt, F.M.; Roberts, L.M. The association of shiga-like toxin with detergent-resistant membranes is modulated by glucosylceramide and is an essential requirement in the endoplasmic reticulum for a cytotoxic effect. Mol. Biol. Cell 2006, 17, 1375–1387. [Google Scholar]
- Orlandi, P.A.; Fishman, P.H. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 1998, 141, 905–915. [Google Scholar]
- Torgersen, M.L.; Skretting, G.; van Deurs, B.; Sandvig, K. Internalization of cholera toxin by different endocytic mechanisms. J. Cell Sci. 2001, 114, 3737–3747. [Google Scholar]
- Massol, R.H.; Larsen, J.E.; Fujinaga, Y.; Lencer, W.I.; Kirchhausen, T. Cholera toxin toxicity does not require functional Arf6- and dynamin-dependent endocytic pathways. Mol. Biol. Cell 2004, 15, 3631–3641. [Google Scholar]
- Montesano, R.; Roth, J.; Robert, A.; Orci, L. Non-coated membrane invaginations are involved in binding and internalization of cholera and tetanus toxins. Nature 1982, 296, 651–653. [Google Scholar]
- Tran, D.; Carpentier, J.L.; Sawano, F.; Gorden, P.; Orci, L. Ligands internalized through coated or noncoated invaginations follow a common intracellular pathway. Proc. Natl. Acad. Sci. USA 1987, 84, 7956–7961. [Google Scholar]
- Singh, R.D.; Puri, V.; Valiyaveettil, J.T.; Marks, D.L.; Bittman, R.; Pagano, R.E. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol. Biol. Cell 2003, 14, 3254–3265. [Google Scholar]
- Hansen, G.H.; Dalskov, S.M.; Rasmussen, C.R.; Immerdal, L.; Niels-Christiansen, L.L.; Danielsen, E.M. Cholera toxin entry into pig enterocytes occurs via lipid raft- and clathrin-dependent mechanism. Biochemistry 2005, 44, 873–882. [Google Scholar] [PubMed]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar] [CrossRef]
- Amessou, M.; Fradagrada, A.; Falguieres, T.; Lord, J.M.; Smith, D.C.; Roberts, L.M.; Lamaze, C.; Johannes, L. Syntaxin 16 and syntaxin 5 are required for efficient retrograde transport of several exogenous and endogenous cargo proteins. J. Cell Sci. 2007, 120, 1457–1468. [Google Scholar] [CrossRef]
- Ganley, I.G.; Espinosa, E.; Pfeffer, S.R. A syntaxin 10-SNARE complex distinguishes two distinct transport routes from endosomes to the trans-Golgi in human cells. J. Cell Biol. 2008, 180, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Del Nery, E.; Miserey-Lenkei, S.; Falguieres, T.; Nizak, C.; Johannes, L.; Perez, F.; Goud, B. Rab6A and Rab6A' GTPases play non-overlapping roles in membrane trafficking. Traffic 2006, 7, 394–407. [Google Scholar]
- Lu, L.; Tai, G.; Hong, W. Autoantigen Golgin-97, an effector of Arl1 GTPase, participates in traffic from the endosome to the trans-golgi network. Mol. Biol. Cell 2004, 15, 4426–4443. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Spilsberg, B.; Lauvrak, S.U.; Torgersen, M.L.; Iversen, T.G.; van Deurs, B. Pathways followed by protein toxins into cells. Int. J. Med. Microbiol 2004, 293, 483–490. [Google Scholar] [CrossRef]
- Popoff, V.; Mardones, G.A.; Bai, S.K.; Chambon, V.; Tenza, D.; Burgos, P.V.; Shi, A.; Benaroch, P.; Urbe, S.; Lamaze, C.; Grant, B.D.; Raposo, G.; Johannes, L. Analysis of articulation between clathrin and retromer in retrograde sorting on early endosomes. Traffic 2009, 10, 1868–1880. [Google Scholar]
- Popoff, V.; Mardones, G.A.; Tenza, D.; Rojas, R.; Lamaze, C.; Bonifacino, J.S.; Raposo, G.; Johannes, L. The retromer complex and clathrin define an early endosomal retrograde exit site. J. Cell Sci. 2007, 120, 2022–2031. [Google Scholar]
- Nambiar, M.P.; Oda, T.; Chen, C.; Kuwazuru, Y.; Wu, H.C. Involvement of the Golgi region in the intracellular trafficking of cholera toxin. J. Cell Physiol. 1993, 154, 222–228. [Google Scholar] [CrossRef]
- Lencer, W.I.; Constable, C.; Moe, S.; Jobling, M.; Webb, H.M.; Ruston, S.; Madara, J.L.; Hirst, T.; Holmes, R. Targeting of cholera toxin and E. coli heat labile toxin in polarized epithelia: role of C-terminal KDEL. J. Cell Biol. 1995, 131, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Mekalanos, J.J.; Collier, R.J.; Romig, W.R. Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J. Biol. Chem. 1979, 254, 5855–5861. [Google Scholar] [PubMed]
- Forster, M.L.; Sivick, K.; Park, Y.N.; Arvan, P.; Lencer, W.I.; Tsai, B. Protein disulfide isomerase-like proteins play opposing roles during retrotranslocation. J. Cell Biol. 2006, 173, 853–859. [Google Scholar]
- Goins, B.; Freire, E. Thermal stability and intersubunit interactions of cholera toxin in solution and in association with its cell-surface receptor ganglioside GM1. Biochem. 1988, 27, 2046–2052. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Leddy, J.J.; Mantsch, H.H. Structure, stability, and receptor interaction of cholera toxin as studied by Fourier-transform infrared spectroscopy. Biochemistry 1990, 29, 8106–8111. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.H.; Scaglione, P.; Taylor, M.; Nemec, K.N.; Tuthill, S.; Moe, D.; Holmes, R.K.; Tatulian, S.A.; Teter, K. Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 2007, 374, 1114–1128. [Google Scholar] [CrossRef]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Tsai, B.; Rapoport, T. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nature Rev. Cell Biol. 2002, 3, 246–255. [Google Scholar]
- Ampapathi, R.S.; Creath, A.L.; Lou, D.I.; Craft, J.W., Jr.; Blanke, S.R.; Legge, G.B. Order-Disorder-Order Transitions Mediate the Activation of Cholera Toxin. J. Mol. Biol. 2008. [Google Scholar]
- Tsai, B.; Rapoport, T. Unfolded cholera toxin is transferred to the ER membrane and released from protein disulfide isomerase upon oxidation by Ero1. J. Cell Biol. 2002, 159, 207–215. [Google Scholar] [CrossRef]
- Nishikawa, S.; Brodsky, J.L.; Nakatsukasa, K. Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J. Biol. Chem. 2005, 137, 551–555. [Google Scholar]
- Brodsky, J.L.; McCracken, A.A. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 1999, 10, 507–513. [Google Scholar]
- Vembar, S.S.; Brodsky, J.L. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar]
- Pilon, M.; Schekman, R.; Romisch, K. Sec61p mediates export of a misfolded secretory protein from the nedoplasmic reticulum to the cytososl for degradation. EMBO J. 1997, 16, 4540–4548. [Google Scholar] [CrossRef]
- Ploegh, H.L. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 2007, 448, 435–438. [Google Scholar]
- Denic, V.; Quan, E.M.; Weissman, J.S. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 2006, 126, 349–359. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Wojcikiewicz, R.J. Substrate-specific mediators of ER associated degradation (ERAD). Curr. Opin. Cell Biol. 2009, 21, 516–521. [Google Scholar] [CrossRef]
- Oda, Y.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Nagata, K.; Mori, K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006, 172, 383–393. [Google Scholar] [CrossRef]
- Okuda-Shimizu, Y.; Hendershot, L.M. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell 2007, 28, 544–554. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: the long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membran. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T. A. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticumul membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar]
- Chen, B.; Mariano, J.; Tsai, Y.C.; Chan, A.H.; Cohen, M.; Weissman, A.M. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 341–346. [Google Scholar]
- Hirsch, C.; Gauss, R.; Horn, S.C.; Neuber, O.; Sommer, T. The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [Google Scholar]
- Horn, S.C.; Hanna, J.; Hirsch, C.; Volkwein, C.; Schutz, A.; Heinemann, U.; Sommer, T.; Jarosch, E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol. Cell 2009, 36, 782–793. [Google Scholar] [CrossRef]
- Schulze, A.; Standera, S.; Buerger, E.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Koning, F.; Kloetzel, P.M.; Seeger, M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005, 354, 1021–1027. [Google Scholar]
- Rodighiero, C.; Tsai, B.; Rapoport, T.A.; Lencer, W.I. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002, 3, 1222–1227. [Google Scholar]
- Wernick, N.L.; De Luca, H.; Kam, W.R.; Lencer, W.I. N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retro-translocation from ER to cytosol. J. Biol. Chem. 1990, 265, 20723–20726. [Google Scholar]
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 1786, 153–177.
- Flierman, D.; Ye, Y.; Dai, M.; Chau, V.; Rapoport, T.A. Polyubiquitin serves as a recognition signal, rather than a ratcheting molecule, during retrotranslocation of proteins across the endoplasmic reticulum membrane. J. Biol. Chem. 2003, 278, 34774–34782. [Google Scholar] [PubMed]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef]
- Werner, E.D.; Brodsky, J.L.; McCracken, A.A. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA 1996, 93, 13797–13801. [Google Scholar]
- Lee, R.J.; Liu, C.W.; Harty, C.; McCracken, A.A.; Latterich, M.; Romisch, K.; DeMartino, G.N.; Thomas, P.J.; Brodsky, J.L. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J. 2004, 23, 2206–2215. [Google Scholar]
- McConnell, E.; Lass, A.; Wojcik, C. Ufd1-Npl4 is a negative regulator of cholera toxin retrotranslocation. Biochem. Biophys. Res. Commun. 2007, 355, 1087–1090. [Google Scholar] [CrossRef]
- Kothe, M.; Ye, Y.; Wagner, J.S.; De Luca, H.E.; Kern, E.; Rapoport, T.A.; Lencer, W.I. Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J. Biol. Chem. 2005, 280, 28127–28132. [Google Scholar]
- Abujarour, R.J.; Dalal, S.; Hanson, P.I.; Draper, R.K. p97 is in a complex with cholera toxin and influences the transport of cholera toxin and related toxins to the cytoplasm. J. Biol. Chem. 2005, 280, 15865–15871. [Google Scholar]
- Lencer, W.I.; Tsai, B. The intracellular voyage of cholera toxin: going retro. Trends Biochem. Sci. 2003, 28, 639–645. [Google Scholar] [CrossRef]
- Teter, K.; Allyn, R.L.; Jobling, M.G.; Holmes, R.K. Transfer of the cholera toxin A1 polypeptide from the endoplasmic reticulum to the cytosol is a rapid process facilitated by the endoplasmic reticulum-associated degradation pathway. Infect Immun. 2002, 70, 6166–6171. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wernick, N.L.B.; Chinnapen, D.J.-F.; Cho, J.A.; Lencer, W.I. Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum. Toxins 2010, 2, 310-325. https://doi.org/10.3390/toxins2030310
Wernick NLB, Chinnapen DJ-F, Cho JA, Lencer WI. Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum. Toxins. 2010; 2(3):310-325. https://doi.org/10.3390/toxins2030310
Chicago/Turabian StyleWernick, Naomi L. B., Daniel J.-F. Chinnapen, Jin Ah Cho, and Wayne I. Lencer. 2010. "Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum" Toxins 2, no. 3: 310-325. https://doi.org/10.3390/toxins2030310