Selection, Characterization, and Optimization of DNA Aptamers against Challenging Marine Biotoxin Gymnodimine-A for Biosensing Application

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. GYM-A Aptamer Selection by Capture-SELEX
2.2. Cloning, Sequencing, Selection, and Affinity Identification of Candidate Aptamers
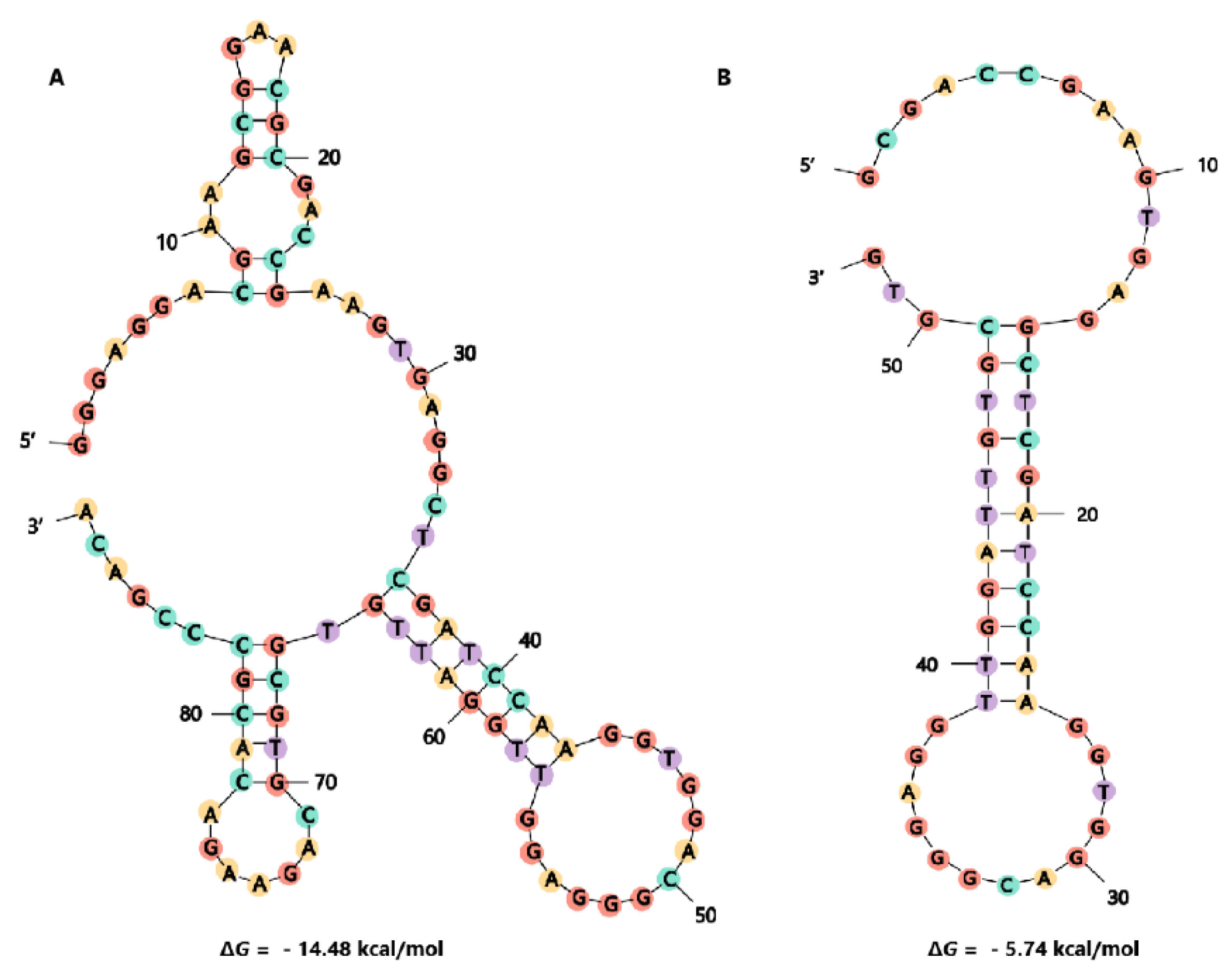
2.3. Truncation of Aptamer G48
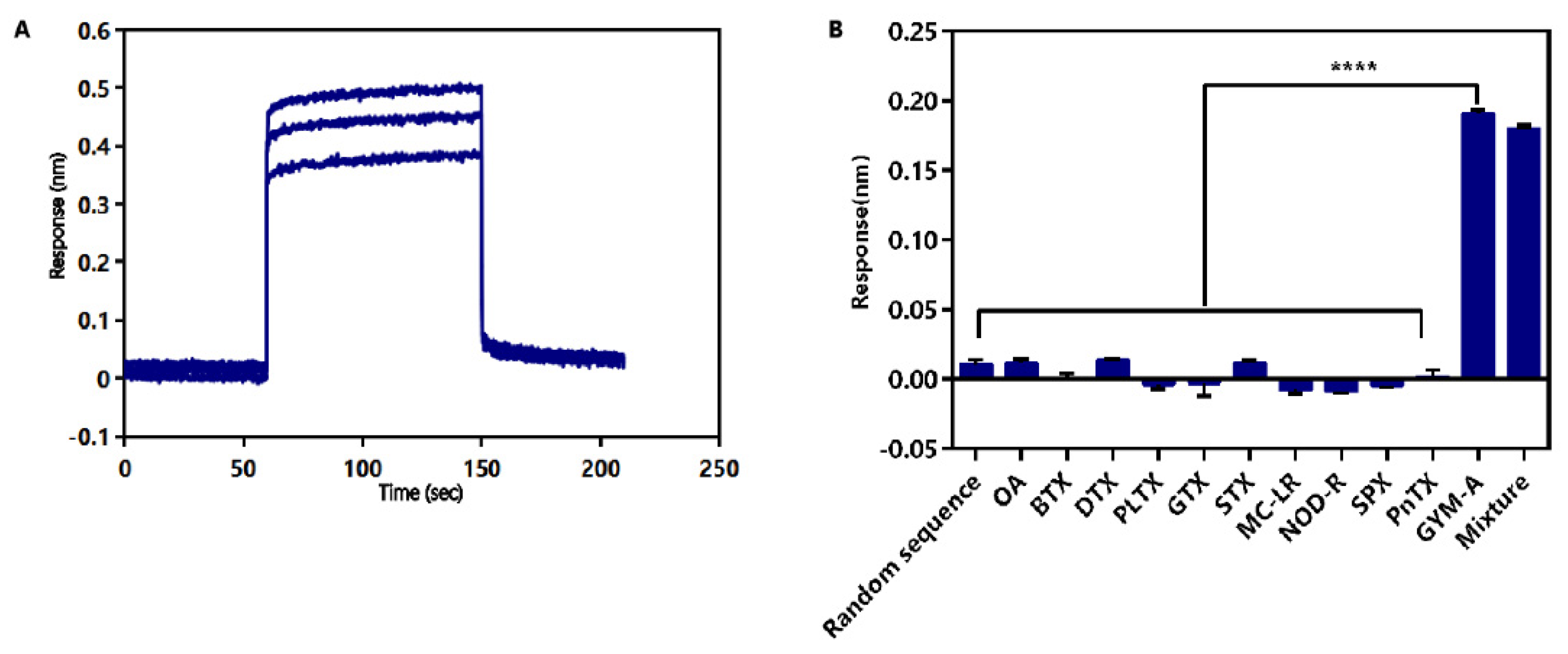
2.4. Identification of Affinity and Specificity of G48nop
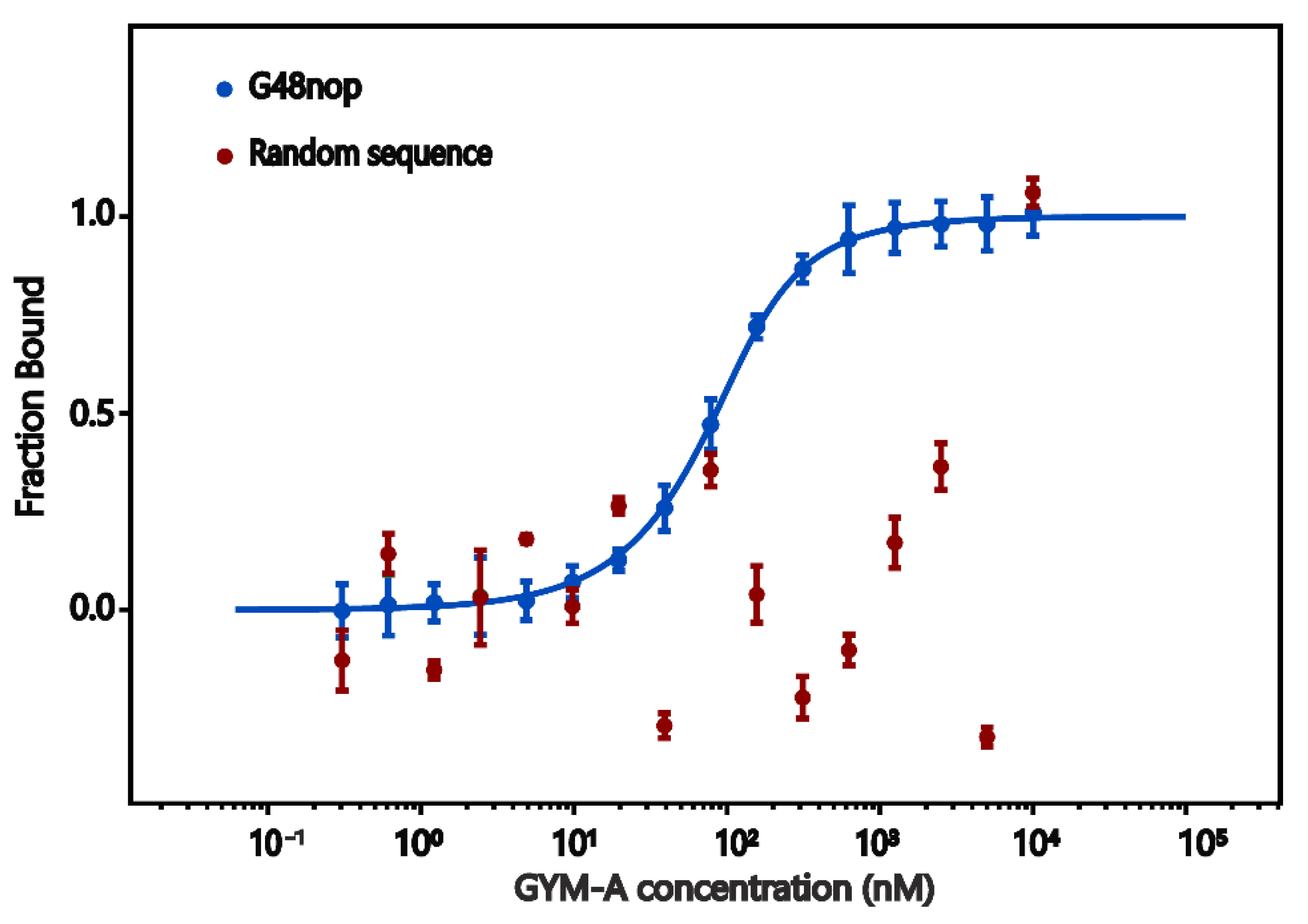
2.5. Microscale Thermophoresis (MST)
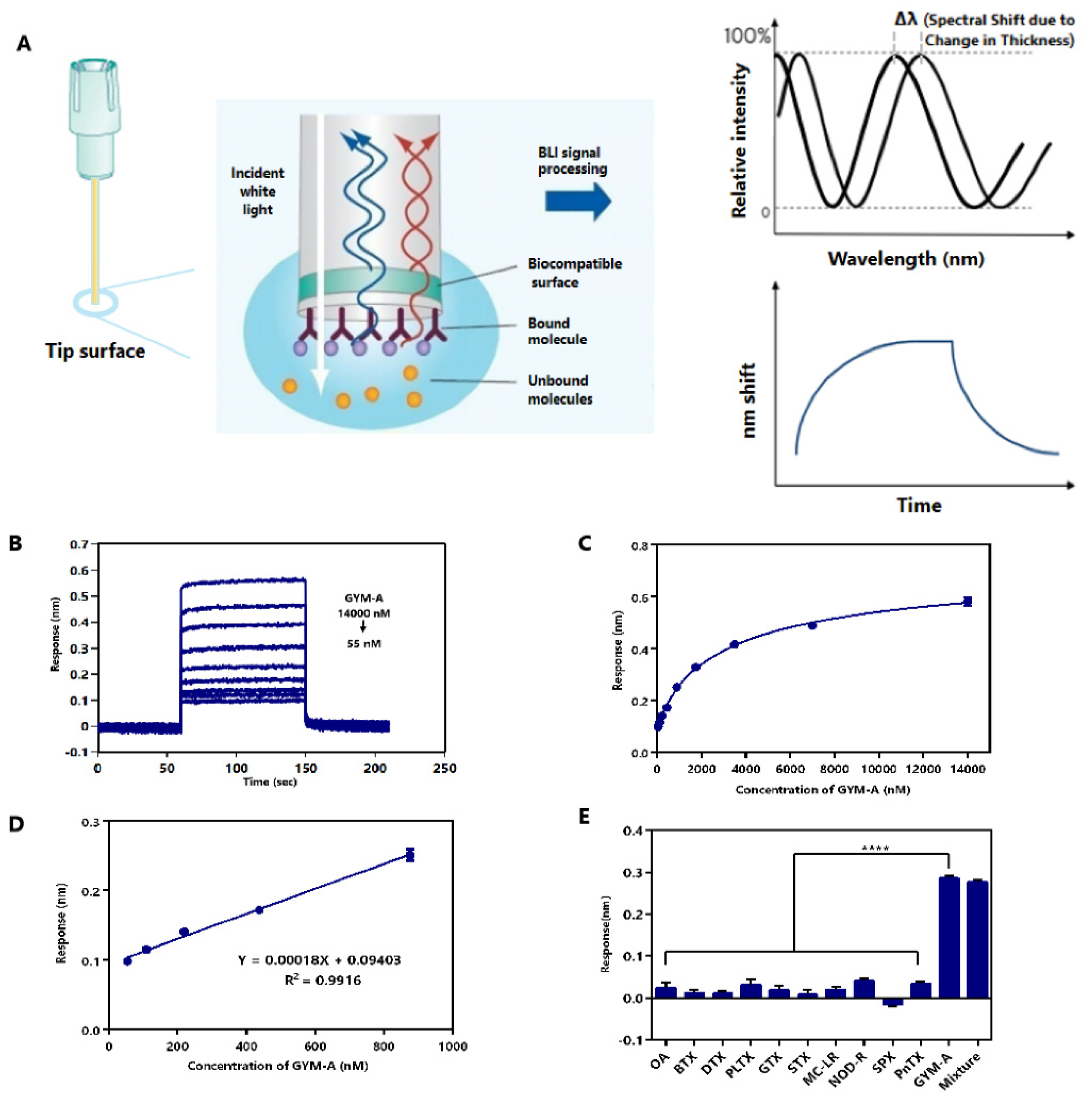
2.6. Label-Free BLI-Based Aptasenor for Detecting GYM-A
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Instruments
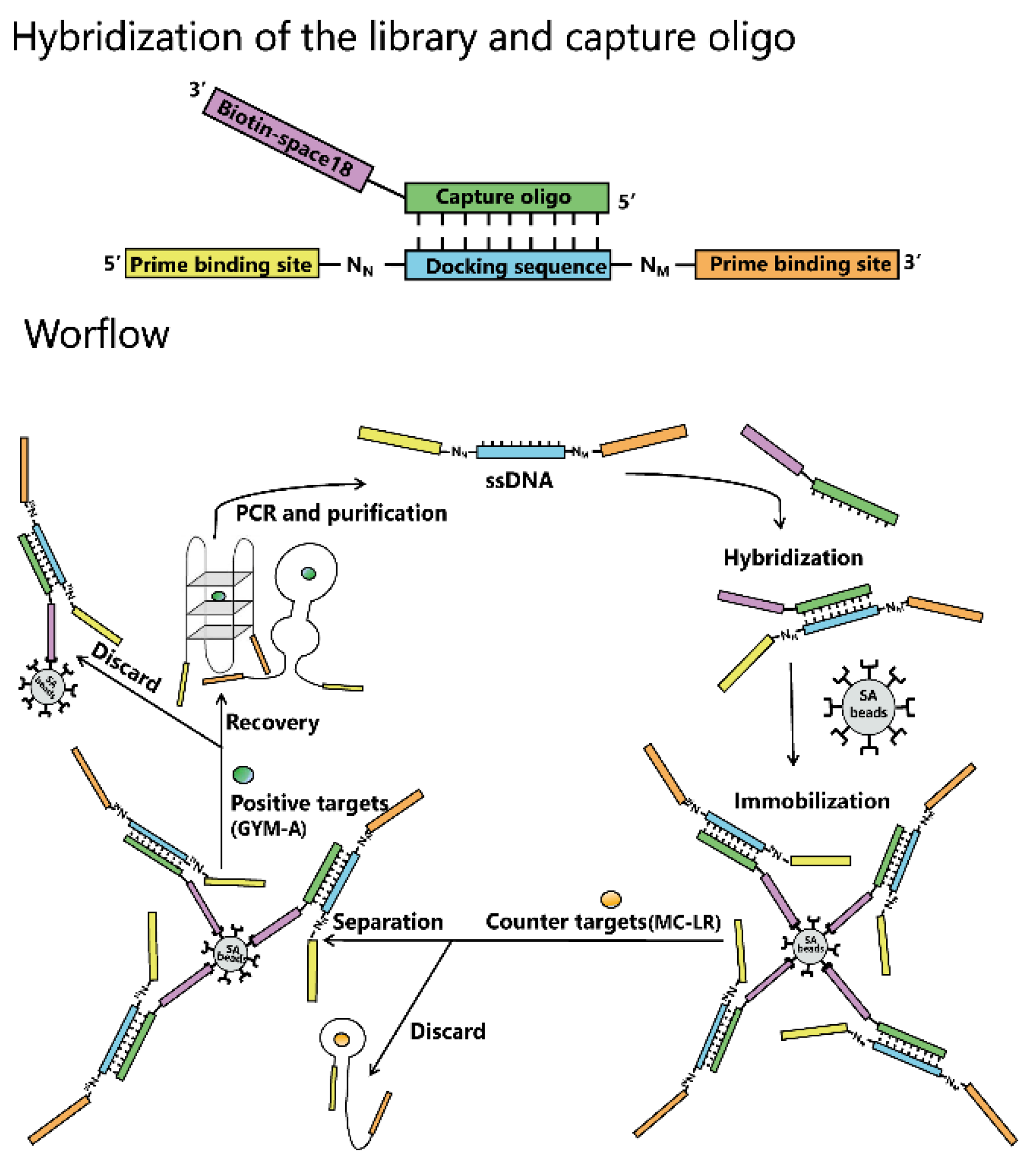
4.3. Capture-SELEX Library and Primers
4.4. Hybridization of the Library and Capture Oligo and Immobilization of Hybrid on Beads
4.5. Aptamer Selection by Capture-SELEX
4.6. Cloning, Sequencing, and Sequence Analysis
4.7. BLI Assays
4.8. MST Assays
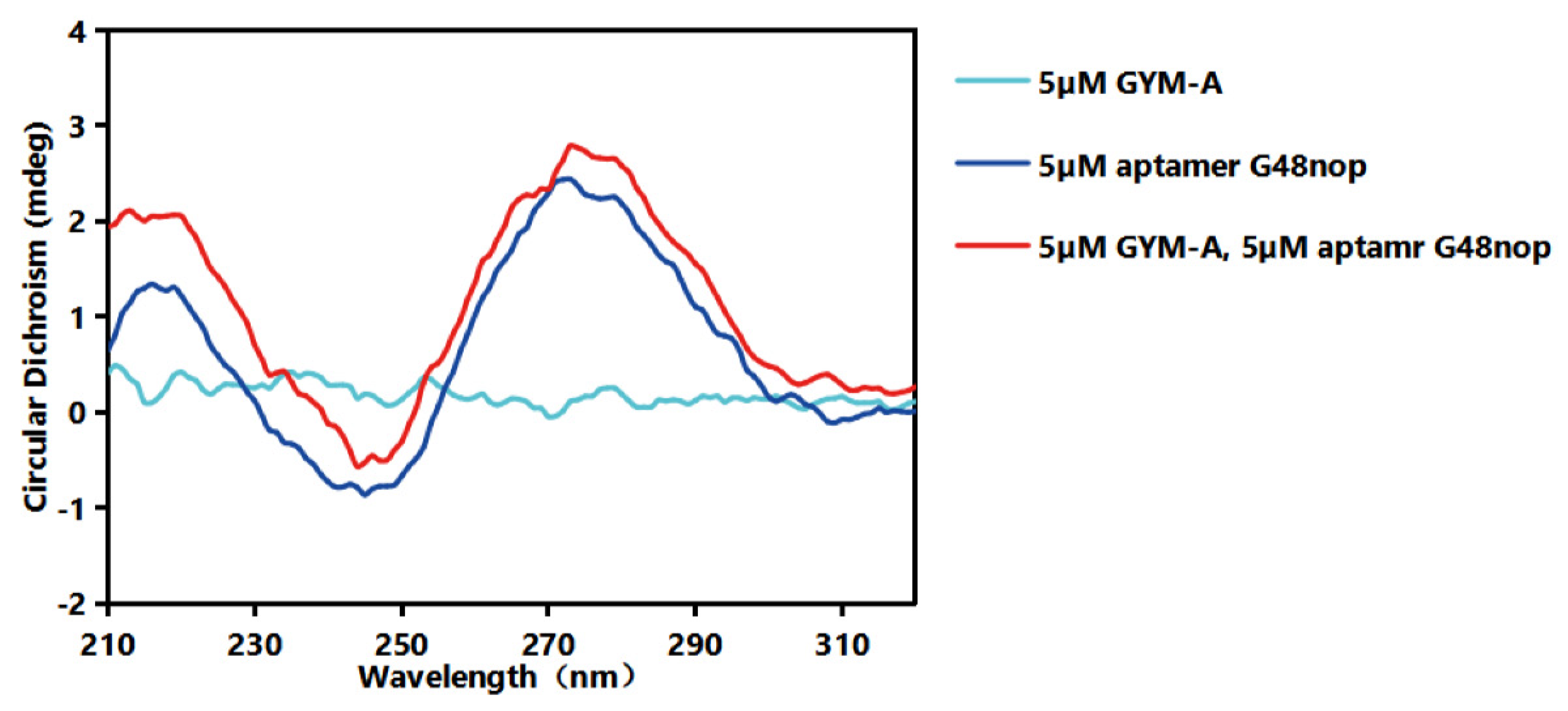
4.9. CD Assays
4.10. Treatment of Real Samples
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, E.; Yu, Q.; Eaglesham, G.; Connell, D.W.; McBroom, J.; Costanzo, S.; Shaw, G.R. Occurrence and seasonal variations of algal toxins in water, phytoplankton and shellfish from North Stradbroke Island, Queensland, Australia. Mar. Environ. Res. 2007, 64, 429–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, L.; Holland, P.; McNabb, P.; Beuzenberg, V.; Selwood, A.; Suzuki, T. Complex toxin profiles in phytoplankton and Greenshell mussels (Perna canaliculus), revealed by LC–MS/MS analysis. Toxicon 2002, 40, 1321–1330. [Google Scholar] [CrossRef]
- Stirling, D.J. Survey of historical New Zealand shellfish samples for accumulation of gymnodimine. N. Z. J. Mar. Freshw. Res. 2001, 35, 851–857. [Google Scholar] [CrossRef]
- Seki, T.; Satake, M.; Mackenzie, L.; Kaspar, H.F.; Yasumoto, T. Gymnodimine, a new marine toxin of unprecedented structure isolated from New Zealand oysters and the dinoflagellate, Gymnodinium sp. Tetrahedron Lett. 1995, 36, 7093–7096. [Google Scholar] [CrossRef]
- Salgado, P.; Riobó, P.; Rodríguez, F.; Franco, J.M.; Bravo, I. Differences in the toxin profiles of Alexandrium ostenfeldii (Dinophyceae) strains isolated from different geographic origins: Evidence of paralytic toxin, spirolide, and gymnodimine. Toxicon 2015, 103, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchiocchi, S.; Siracusa, M.; Campacci, D.; Ciriaci, M.; Dubbini, A.; Tavoloni, T.; Stramenga, A.; Gorbi, S.; Piersanti, A. Cyclic Imines (CIs) in Mussels from North-Central Adriatic Sea: First Evidence of Gymnodimine A in Italy. Toxins 2020, 12, 370. [Google Scholar] [CrossRef] [PubMed]
- Zurhelle, C.; Nieva, J.; Tillmann, U.; Harder, T.; Krock, B.; Tebben, J. Identification of novel gymnodimines and spirolides from the marine dinoflagellate Alexandrium ostenfeldii. Mar. Drugs 2018, 16, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujević, I.; Roje-Busatto, R.; Ezgeta-Balić, D. Comparison of amnesic, paralytic and lipophilic toxins profiles in cockle (Acanthocardia tuberculata) and smooth clam (Callista chione) from the central Adriatic Sea (Croatia). Toxicon 2019, 159, 32–37. [Google Scholar] [CrossRef]
- Kremp, A.; Tahvanainen, P.; Litaker, W.; Krock, B.; Suikkanen, S.; Leaw, C.P.; Tomas, C. Phylogenetic relationships, morphological variation, and toxin patterns in the Alexandrium ostenfeldii (Dinophyceae) complex: Implications for species boundaries and identities. J. Phycol. 2014, 50, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Mountfort, D.; Beuzenberg, V.; MacKenzie, L.; Rhodes, L. Enhancement of growth and gymnodimine production by the marine dinoflagellate, Karenia selliformis. Harmful Algae 2006, 5, 658–664. [Google Scholar] [CrossRef]
- Van de Waal, D.B.; Tillmann, U.; Martens, H.; Krock, B.; van Scheppingen, Y.; John, U. Characterization of multiple isolates from an Alexandrium ostenfeldii bloom in the Netherlands. Harmful Algae 2015, 49, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, K.M.; de Senerpont Domis, L.N.; Wohlrab, S.; Krock, B.; John, U.; van Scheppingen, Y.; van Donk, E.; Van de Waal, D.B. Combined physical, chemical and biological factors shape Alexandrium ostenfeldii blooms in the Netherlands. Harmful Algae 2017, 63, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Salas, M.F.; Rhodes, L.L.; Mackenzie, L.A.; Adamson, J.E. Gymnodinoid genera Karenia and Takayama (Dinophyceae) in New Zealand coastal waters. N. Z. J. Mar. Freshw. Res. 2005, 39, 135–139. [Google Scholar] [CrossRef]
- Holland, P.T.; Shi, F.; Satake, M.; Hamamoto, Y.; Ito, E.; Beuzenberg, V.; McNabb, P.; Munday, R.; Briggs, L.; Truman, P. Novel toxins produced by the dinoflagellate Karenia Brevisulcata. Harmful Algae 2012, 13, 47–57. [Google Scholar] [CrossRef]
- Stewart, M.; Blunt, J.W.; Munro, M.H.; Robinson, W.T.; Hannah, D.J. The absolute stereochemistry of the New Zealand shellfish toxin gymnodimine. Tetrahedron Lett. 1997, 38, 4889–4890. [Google Scholar] [CrossRef]
- Van Wagoner, R.M.; Misner, I.; Tomas, C.R.; Wright, J.L. Occurrence of 12-methylgymnodimine in a spirolide-producing dinoflagellate Alexandrium peruvianum and the biogenetic implications. Tetrahedron Lett. 2011, 52, 4243–4246. [Google Scholar] [CrossRef]
- Miles, C.O.; Wilkins, A.L.; Stirling, D.J.; MacKenzie, A.L. New analogue of gymnodimine from a Gymnodinium species. J. Agric. Food Chem. 2000, 48, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Strangman, W.; Anttila, M.; Tomas, C.; Wright, J.L. (5S)-5-[(4aR, 8aS, 9E, 11S, 13R, 14S, 16R, 17R, 19S)-11, 19-Dihydroxy-8, 10, 13, 16-tetramethyl-18-methylidene-3, 4, 5, 6, 8a, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21-hexadecahydro-2H-14, 17-epoxybenzo [2, 3] cyclohexadeca [1, 2-b] pyridine-7-yl]-3-methylfuran-2 (5H)-one (12-Methylgymnodimine B). Molbank 2016, 2016, M896. [Google Scholar]
- Miles, C.O.; Wilkins, A.L.; Stirling, D.J.; MacKenzie, A.L. Gymnodimine C, an isomer of gymnodimine B, from Karenia selliformis. J. Agric. Food Chem. 2003, 51, 4838–4840. [Google Scholar] [CrossRef]
- Martens, H.; Tillmann, U.; Harju, K.; Dell’Aversano, C.; Tartaglione, L.; Krock, B. Toxin variability estimations of 68 Alexandrium ostenfeldii (Dinophyceae) strains from The Netherlands reveal a novel abundant gymnodimine. Microorganisms 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharrat, R.; Servent, D.; Girard, E.; Ouanounou, G.; Amar, M.; Marrouchi, R.; Benoit, E.; Molgó, J. The marine phycotoxin gymnodimine targets muscular and neuronal nicotinic acetylcholine receptor subtypes with high affinity. J. Neurochem. 2008, 107, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Hauser, T.A.; Hepler, C.D.; Kombo, D.C.; Grinevich, V.P.; Kiser, M.N.; Hooker, D.N.; Zhang, J.; Mountfort, D.; Selwood, A.; Akireddy, S.R. Comparison of acetylcholine receptor interactions of the marine toxins, 13-desmethylspirolide C and gymnodimine. Neuropharmacology 2012, 62, 2239–2250. [Google Scholar] [CrossRef]
- Molgó, J.; Marchot, P.; Aráoz, R.; Benoit, E.; Iorga, B.I.; Zakarian, A.; Taylor, P.; Bourne, Y.; Servent, D. Cyclic imine toxins from dinoflagellates: A growing family of potent antagonists of the nicotinic acetylcholine receptors. J. Neurochem. 2017, 142, 41–51. [Google Scholar] [CrossRef]
- Bourne, Y.; Radić, Z.; Aráoz, R.; Talley, T.T.; Benoit, E.; Servent, D.; Taylor, P.; Molgó, J.; Marchot, P. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc. Natl. Acad. Sci. USA 2010, 107, 6076–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragunow, M.; Trzoss, M.; Brimble, M.A.; Cameron, R.; Beuzenberg, V.; Holland, P.; Mountfort, D. Investigations into the cellular actions of the shellfish toxin gymnodimine and analogues. Environ. Toxicol. Pharmacol. 2005, 20, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Estevez, P.; Castro, D.; Pequeño-Valtierra, A.; Giraldez, J.; Gago-Martinez, A. Emerging marine biotoxins in seafood from European coasts: Incidence and analytical challenges. Foods 2019, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, H. Joint FAO/WHO/IOC activities to provide scientific advice on marine biotoxins. Mar. Pollut. Bull. 2006, 52, 1735–1745. [Google Scholar] [CrossRef]
- Rodriguez, I.; Vieytes, M.R.; Alfonso, A. Analytical challenges for regulated marine toxins. Detection methods. Curr. Opin. Food Sci. 2017, 18, 29–36. [Google Scholar] [CrossRef]
- Davidson, K.; Baker, C.; Higgins, C.; Higman, W.; Swan, S.; Veszelovszki, A.; Turner, A.D. Potential threats posed by new or emerging marine biotoxins in UK waters and examination of detection methodologies used for their control: Cyclic imines. Mar. Drugs 2015, 13, 7087–7112. [Google Scholar] [CrossRef]
- Fonfría, E.S.; Vilariño, N.; Espiña, B.; Louzao, M.C.; Álvarez, M.; Molgó, J.; Aráoz, R.; Botana, L.M. Feasibility of gymnodimine and 13-desmethyl C spirolide detection by fluorescence polarization using a receptor-based assay in shellfish matrixes. Anal. Chim. Acta 2010, 657, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Aráoz, R.; Ramos, S.; Pelissier, F.; Guérineau, V.; Benoit, E.; Vilariño, N.; Botana, L.M.; Zakarian, A.; Molgó, J. Coupling the Torpedo microplate-receptor binding assay with mass spectrometry to detect cyclic imine neurotoxins. Anal. Chem. 2012, 84, 10445–10453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visciano, P.; Schirone, M.; Berti, M.; Milandri, A.; Tofalo, R.; Suzzi, G. Marine biotoxins: Occurrence, toxicity, regulatory limits and reference methods. Front. Microbiol. 2016, 7, 1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garthwaite, I.; Ross, K.M.; Miles, C.O.; Briggs, L.R.; Towers, N.R.; Borrell, T.; Busby, P. Integrated enzyme-linked immunosorbent assay screening system for amnesic, neurotoxic, diarrhetic, and paralytic shellfish poisoning toxins found in New Zealand. J. AOAC Int. 2001, 84, 1643–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munday, R.; Towers, N.R.; Mackenzie, L.; Beuzenberg, V.; Holland, P.T.; Miles, C.O. Acute toxicity of gymnodimine to mice. Toxicon 2004, 44, 173–178. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation 2019/627 of 15 March 2019 laying down uniform practical arrangements for the performance of official controls on products of animal origin intended for human consumption. Off. J. Eur. Union L 2019, 131, 51–100. [Google Scholar]
- Varriale, F.; Tartaglione, L.; Cinti, S.; Milandri, A.; Dell’Aversano, C. Development of a data dependent acquisition-based approach for the identification of unknown fast-acting toxins and their ester metabolites. Talanta 2020, 224, 121842. [Google Scholar] [CrossRef]
- Oller-Ruiz, A.; Campillo, N.; Hernández-Córdoba, M.; Gilabert, J.; Viñas, P. Monitoring Lipophilic Toxins in Seawater Using Dispersive Liquid-Liquid Microextraction and Liquid Chromatography with Triple Quadrupole Mass Spectrometry. Toxins 2021, 13, 57. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, R.; Kong, F.; Li, C.; Dai, L.; Chen, Z.; Zhou, M. Lipophilic marine toxins discovered in the Bohai Sea using high performance liquid chromatography coupled with tandem mass spectrometry. Chemosphere 2017, 183, 380–388. [Google Scholar] [CrossRef]
- Raftery, M.; Schmidt, J.; Clark, D. Specificity of α-bungarotoxin binding to Torpedo californica electroplax. Arch. Biochem. Biophys. 1972, 152, 882–886. [Google Scholar] [CrossRef]
- Heidmann, T.; Changeux, J.P. Fast kinetic studies on the interaction of a fluorescent agonist with the membrane-bound acetylcholine receptor from Torpedo Marmorata. Eur. J. Biochem. 1979, 94, 255–279. [Google Scholar] [CrossRef]
- Rodríguez, L.P.; Vilariño, N.; Molgó, J.; Aráoz, R.; Louzao, M.C.; Taylor, P.; Talley, T.; Botana, L.M. Development of a solid-phase receptor-based assay for the detection of cyclic imines using a microsphere-flow cytometry system. Anal. Chem. 2013, 85, 2340–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Liu, T.; Zhang, W.; Zhu, M.; Liu, Z.; Kong, Y.; Liu, S. Marine Toxins Detection by Biosensors Based on Aptamers. Toxins 2019, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent advances in aptamer discovery and applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citartan, M.; Tang, T.-H. Recent developments of aptasensors expedient for point-of-care (POC) diagnostics. Talanta 2019, 199, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.G.; Carrillo, M.P.; Phillips, T.; Andrews, C.J. A novel screening method for competitive FRET-aptamers applied to E. coli assay development. J. Fluoresc. 2010, 20, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.Y.; Pestilli, F.; Rokem, A. Deconvolution of high dimensional mixtures via boosting, with application to diffusion-weighted MRI of human brain. Adv. Neural Inf. Processing Syst. 2014, 27, 1–9. [Google Scholar]
- Burke, D.H.; Hoffman, D.C.; Brown, A.; Hansen, M.; Pardi, A.; Gold, L. RNA aptamers to the peptidyl transferase inhibitor chloramphenicol. Chem. Biol. 1997, 4, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Kim, Y.S.; Niazi, J.H.; Gu, M.B. Electrochemical aptasensor for tetracycline detection. Bioprocess Biosyst. Eng. 2010, 33, 31–37. [Google Scholar] [CrossRef]
- Mufhandu, H.T.; Gray, E.S.; Madiga, M.C.; Tumba, N.; Alexandre, K.B.; Khoza, T.; Wibmer, C.K.; Moore, P.L.; Morris, L.; Khati, M. UCLA1, a synthetic derivative of a gp120 RNA aptamer, inhibits entry of human immunodeficiency virus type 1 subtype C. J. Virol. 2012, 86, 4989–4999. [Google Scholar] [CrossRef] [Green Version]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood J. Am. Soc. Hematol. 2014, 123, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson III, W.E.; Haque, F.; Xu, R.; Guo, P. Systemic delivery of anti-miRNA for suppression of triple negative breast cancer utilizing RNA nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef]
- Gijs, M.; Aerts, A.; Impens, N.; Baatout, S.; Luxen, A. Aptamers as radiopharmaceuticals for nuclear imaging and therapy. Nucl. Med. Biol. 2016, 43, 253–271. [Google Scholar] [CrossRef]
- Handy, S.M.; Yakes, B.J.; DeGrasse, J.A.; Campbell, K.; Elliott, C.T.; Kanyuck, K.M.; DeGrasse, S.L. First report of the use of a saxitoxin–protein conjugate to develop a DNA aptamer to a small molecule toxin. Toxicon 2013, 61, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, S.; Ng, A.; Siaj, M.; Tavares, A.C.; Zourob, M. Selection and identification of DNA aptamers against okadaic acid for biosensing application. Anal. Chem. 2013, 85, 11794–11801. [Google Scholar] [CrossRef]
- Gao, S.; Zheng, X.; Hu, B.; Sun, M.; Wu, J.; Jiao, B.; Wang, L. Enzyme-linked, aptamer-based, competitive biolayer interferometry biosensor for palytoxin. Biosens. Bioelectron. 2016, 952, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, B.; Zheng, X.; Cao, Y.; Liu, D.; Sun, M.; Jiao, B.; Wang, L. Gonyautoxin 1/4 aptamers with high-affinity and high-specificity: From efficient selection to aptasensor application. Biosens. Bioelectron. 2016, 79, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Eissa, S.; Siaj, M.; Zourob, M. Aptamer-based competitive electrochemical biosensor for brevetoxin-2. Biosens. Bioelectron. 2015, 69, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liang, L.; Zhou, S.; Xie, W.; He, S.; Wang, Y.; Chaker, T.; Tong, S.; Wang, D. Label-free sensitive detection of Microcystin-LR via aptamer-conjugated gold nanoparticles based on solid-state nanopores. Langmuir 2018, 34, 14825–14833. [Google Scholar]
- Caglayan, M.O.; Stünda, Z. Saxitoxin aptasensor based on attenuated internal reflection ellipsometry for seafood. Toxicon 2020, 187. [Google Scholar] [CrossRef]
- Yang, K.-A.; Chun, H.; Zhang, Y.; Pecic, S.; Nakatsuka, N.; Andrews, A.M.; Worgall, T.S.; Stojanovic, M.N. High-affinity nucleic-acid-based receptors for steroids. ACS Chem. Biol. 2017, 12, 3103–3112. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Luo, Y.; Alkhamis, O.; Canoura, J.; Yu, B.; Xiao, Y. Isolation of natural DNA aptamers for challenging small-molecule targets, cannabinoids. Anal. Chem. 2021, 93, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Nutiu, R.; Li, Y. In vitro selection of structure-switching signaling aptamers. Angew. Chem. Int. Ed. 2005, 44, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.A.; Barbu, M.; Halim, M.; Pallavi, P.; Kim, B.; Kolpashchikov, D.M.; Pecic, S.; Taylor, S.; Worgall, T.S.; Stojanovic, M.N. Recognition and sensing of low-epitope targets via ternary complexes with oligonucleotides and synthetic receptors. Nat. Chem. 2014, 6, 1003–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinemann, C.; Von Fritsch, U.F.; Rudolph, S.; Strehlitz, B. Generation and characterization of quinolone-specific DNA aptamers suitable for water monitoring. Biosens. Bioelectron. 2016, 77, 1039–1047. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Ouyang, S.; Hu, B.; Zhou, R.; Liu, D.; Peng, D.; Li, Z.; Li, Z.; Jiao, B.; Wang, L. Rapid and sensitive detection of nodularin-R in water by a label-free BLI aptasensor. Analyst 2018, 143, 4316–4322. [Google Scholar] [CrossRef]
- Li, Z.; Hu, B.; Zhou, R.; Zhang, X.; Wang, R.; Gao, Y.; Sun, M.; Jiao, B.; Wang, L. Selection and application of aptamers with high-affinity and high-specificity against dinophysistoxin-1. RSC Adv. 2020, 10, 8181–8189. [Google Scholar] [CrossRef]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef]
- Edwards, S.L.; Poongavanam, V.; Kanwar, J.R.; Roy, K.; Hillman, K.M.; Prasad, N.; Leth-Larsen, R.; Petersen, M.; Marušič, M.; Plavec, J. Targeting VEGF with LNA-stabilized G-rich oligonucleotide for efficient breast cancer inhibition. Chem. Commun. 2015, 51, 9499–9502. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Zhang, B.; He, Y.; Wang, J.; Wang, S. A rapid fluorometric method for determination of aflatoxin B1 in plant-derived food by using a thioflavin T-based aptasensor. Microchim. Acta 2019, 186, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Carter, Z.; Kataky, R. A G-quadruplex aptamer based impedimetric sensor for free lysine and arginine. Sens. Actuators B Chem. 2017, 243, 904–909. [Google Scholar] [CrossRef]
- Hudson, J.S.; Brooks, S.C.; Graves, D.E. Interactions of actinomycin D with human telomeric G-quadruplex DNA. Biochemistry 2009, 48, 4440–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorlíčková, M.; Kejnovská, I.; Bednářová, K.; Renčiuk, D.; Kypr, J. Circular dichroism spectroscopy of DNA: From duplexes to quadruplexes. Chirality 2012, 24, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Vorlíčková, M.; Kejnovská, I.; Sagi, J.; Renčiuk, D.; Bednářová, K.; Motlová, J.; Kypr, J. Circular dichroism and guanine quadruplexes. Methods 2012, 57, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B. Review on Marine Biotoxins Toxicity and Detection Methods. In Proceedings of the 2020 10th International Conference on Biomedical Engineering and Technology; Association for Computing Machinery: New York, NY, USA, 2020; pp. 28–33. [Google Scholar]
- Rundberget, T.; Aasen, J.A.B.; Selwood, A.I.; Miles, C.O. Pinnatoxins and spirolides in Norwegian blue mussels and seawater. Toxicon 2011, 58, 700–711. [Google Scholar] [CrossRef]
- Mcnabb, P.S.; Mccoubrey, D.J.; Rhodes, L.; Smith, K.; Holland, P.T. New perspectives on biotoxin detection in Rangaunu Harbour, New Zealand arising from the discovery of pinnatoxins. Harmful Algae 2012, 13, 34–39. [Google Scholar] [CrossRef]
- Wu, H.; Yao, J.; Guo, M.; Tan, Z.; Zhou, D.; Zhai, Y. Distribution of marine lipophilic toxins in shellfish products collected from the Chinese market. Mar. Drugs 2015, 13, 4281–4295. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.; Jo, H.; Kim, S.-H.; Kang, G.-J. Exposure assessment to paralytic shellfish toxins through the shellfish consumption in Korea. Food Res. Int. 2018, 108, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entzian, C.; Schubert, T. Studying small molecule–aptamer interactions using MicroScale Thermophoresis (MST). Methods 2016, 97, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Lee, J.E. Measuring protein-protein and protein-nucleic acid interactions by biolayer interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.; Witte, K.; Wartchow, C.; Choo, S.; Yao, D.; Persson, H.; Wei, J.; Li, P.; Heidecker, B.; Ma, W. Label-free detection of biomolecular interactions using bioLayer interferometry for kinetic characterization. Comb. Chem. High Throughput Screen. 2009, 12, 791–800. [Google Scholar] [CrossRef]
- Chandran, S.; Singh, R. Comparison of various international guidelines for analytical method validation. Die Pharm. -Int. J. Pharm. Sci. 2007, 62, 4–14. [Google Scholar]
- Shrivastava, A.; Gupta, V.B. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Shechtman, O. The coefficient of variation as an index of measurement reliability. In Methods of Clinical Epidemiology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 39–49. [Google Scholar]
- Marrouchi, R.; Dziri, F.; Belayouni, N.; Hamza, A.; Benoit, E.; Molgó, J.; Kharrat, R. Quantitative determination of gymnodimine-A by high performance liquid chromatography in contaminated clams from Tunisia coastline. Mar. Biotechnol. 2010, 12, 579–585. [Google Scholar] [CrossRef]
- Krock, B.; Pitcher, G.C.; Ntuli, J.; Cembella, A.D. Confirmed identification of Gymnodimine in Oysters from the west coast of South Africa by Liquid Chromatography-Tandem Mass Spectrometry. Afr. J. Mar. Sci. 2009, 31, 113–118. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain. Scientific Opinion on marine biotoxins in shellfish-Cyclic imines (spirolides, gymnodimines, pinnatoxins and pteriatoxins). EFSA J. 2010, 8, 1628. [Google Scholar]
- Vignon, A.; Flaget, A.; Michelas, M.; Djeghdir, M.; Defrancq, E.; Coche-Guerente, L.; Spinelli, N.; Van der Heyden, A.; Dejeu, J. Direct detection of low-molecular-weight compounds in 2D and 3D aptasensors by biolayer interferometry. ACS Sens. 2020, 5, 2326–2330. [Google Scholar] [CrossRef]
- Kumaraswamy, S.; Tobias, R. Label-free kinetic analysis of an antibody-antigen interaction using biolayer interferometry. In Protein-Protein Interactions; Springer: Berlin/Heidelberg, Germany, 2015; pp. 165–182. [Google Scholar]
- Rodríguez, L.P.; Vilarino, N.; Molgó, J.; Aráoz, R.; Antelo, A.; Vieytes, M.R.; Botana, L.M. Solid-phase receptor-based assay for the detection of cyclic imines by chemiluminescence, fluorescence, or colorimetry. Anal. Chem. 2011, 83, 5857–5863. [Google Scholar] [CrossRef] [PubMed]
- Aráoz, R.; Barnes, P.; Séchet, V.; Delepierre, M.; Zinn-Justin, S.; Molgó, J.; Zakarian, A.; Hess, P.; Servent, D. Cyclic imine toxins survey in coastal european shellfish samples: Bioaccumulation and mode of action of 28-O-palmitoyl ester of pinnatoxin-G. first report of portimine-A bioaccumulation. Harmful Algae 2020, 98, 101887. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Gong, L.; Baibado, J.T.; Dong, W.; Wang, Y.; Dai, Z.; Cheung, H.Y. Graphene based pipette tip solid phase extraction of marine toxins in shellfish muscle followed by UPLC–MS/MS analysis. Talanta 2013, 116, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Yao, X.; Wang, L.; Li, J. Solid-phase extraction-based ultra-sensitive detection of four lipophilic marine biotoxins in bivalves by high-performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. Sci. 2015, 53, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan-long, W.; Jun-hui, C.; Zhao-yong, L.; Shuai, W.; Xiao-ling, Z.; Xiao-ru, W. Analysis of typical cyclic imine toxins by atmospheric pressure chemical ionization-mass spectrometry. J. Chin. Mass Spectrom. Soc. 2015, 36, 551. [Google Scholar]
- Zhang, Y.; Chen, D.; Hong, Z.; Zhou, S.; Zhao, Y. Polymeric ion exchange material based dispersive micro solid-phase extraction of lipophilic marine toxins in seawater followed by the Q Exactive mass spectrometer analysis using a scheduled high resolution parallel reaction monitoring. Microchem. J. 2018, 138, 526–532. [Google Scholar] [CrossRef]
- Mattarozzi, M.; Cavazza, A.; Calfapietra, A.; Cangini, M.; Pigozzi, S.; Bianchi, F.; Careri, M. Analytical screening of marine algal toxins for seafood safety assessment in a protected Mediterranean shallow water environment. Food Addit. Contam. Part A 2019, 36, 612–624. [Google Scholar] [CrossRef]
- Fang, L.; Qiu, F.; Yu, X. Determination of lipophilic marine biotoxins in shellfish by online turbulent flow chromatography coupled to liquid chromatography–tandem mass spectrometry. Chromatographia 2019, 82, 1321–1331. [Google Scholar] [CrossRef]
- O’Neill, A.; Morrell, N.; Turner, A.D.; Maskrey, B.H. Method performance verification for the combined detection and quantitation of the marine neurotoxins cyclic imines and brevetoxin shellfish metabolites in mussels (Mytilus edulis) and oysters (Crassostrea gigas) by UHPLC-MS/MS. J. Chromatogr. B 2021, 1179, 122864. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; He, X.; Hao, S.; Wang, Y.; Zheng, X.; Wang, B. Simple determination of six groups of lipophilic marine algal toxins in seawater by automated on-line solid phase extraction coupled to liquid chromatography-tandem mass spectrometry. Chemosphere 2021, 262, 128374. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, B.; Zhao, Q.; Wang, Z.; Liu, W.; Zhang, J.; Zhou, Y.; Sun, Q.; Huang, H.; Huang, X. Shellfish contamination with lipophilic toxins and dietary exposure assessments from consumption of shellfish products in Shenzhen, China. Ecotoxicol. Environ. Saf. 2021, 221, 112446. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zheng, X.; Wu, J. A biolayer interferometry-based competitive biosensor for rapid and sensitive detection of saxitoxin. Sens. Actuators B Chem. 2017, 246, 169–174. [Google Scholar] [CrossRef]
- Mukherjee, M.; Sistla, S.; Veerabhadraiah, S.R.; Bettadaiah, B.; Thakur, M.; Bhatt, P. DNA aptamer selection and detection of marine biotoxin 20 Methyl Spirolide, G. Food Chem. 2021, 363, 130332. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of DNA Aptamers | Family | Sequence (5′-3′) | KD (nM) |
|---|---|---|---|
| G48 | A | GGGAGGACGAAGCGGAACGCGACCGAAGTGAGGCTCGATCCAAGGTGGACGGGAGGTTGGATTGTGCGTGCAGAAGACACGCCCGACA | 288 |
| G13 | B | GGGAGGACGAAGCGGAACCCATGGGGTGTGAGGCTCGATCAGGAGGTAAAAGCAGCGCGTCGTCAGTGTGCAGAAGACACGCCCGACA | 168,000 |
| G16 | C | GGGAGGACGAAGCGGAACCACCTTGAGATGAGGCTCGATCACCGGTGGGTATAGTGGTCGCGATGTATACCAGAAGACACGCCCGACA | 495 |
| G79 | D | GGGAGGACGAAGCGGAACGGGGGTGGGTTGAGGCTCGATCATGATAGCGAATTGGACACGATTCGTGTACCAGAAGACACGCCCGACA | 341 |
| G29 | E | GGGAGGACGAAGCGGAACCGTAGGAAAGTGAGGCTCGATCTACCCTTGGATCGATTTGTCAGGTGGGGACCAGAAGACACGCCCGACA | 308 |
| G24 | F | GGGAGGACGAAGCGGAACGGGCGGGATTTGAGGCTCGATCCGACTACGGATACGGCTTGCTTGTATCCGCCAGAAGACACGCCCGACA | 859 |
| Name of DNA Aptamers | Sequence (5′–3′) | KD (nM) |
|---|---|---|
| G48nop | GCGACCGAAGTGAGGCTCGATCCAAGGTGGACGGGAGGTTGGATTGTGCGTG | 95.30 |
| G48nors | TGAGGCTCGATCCAAGGTGGACGGGAGGTTGGATTGTGCGTG | 354.00 |
| G48norsj | CGATCCAAGGTGGACGGGAGGTTGGATTG | 501.00 |
| Samples | Spiked GYM-A (nM) | Recovery Rate (%) | RSD (%) |
|---|---|---|---|
| Shellfish | 875 | 96.65 | 2.28 |
| 1750 | 104.98 | 0.91 | |
| 3500 | 106.06 | 0.55 | |
| Water | 875 | 105.10 | 1.09 |
| 1750 | 109.67 | 1.28 | |
| 3500 | 99.63 | 1.87 |
| Analytical Techniques | LOD | LOQ | Linear Range | Recovery Rate | Reference |
|---|---|---|---|---|---|
| Fluorescence Polarization | 80 nM | — | — | 82.8–98.4% | [30] |
| Microplate-receptor binding assay | 2 nM | 20 nM | — | — | [31] |
| Chemiluminescence | 154 ± 64.3 nM | — | — | — | [95] |
| HPLC–UV | 5 ng/mL | 8 ng/g | 0.005–1 µg/mL | 96% | [90] |
| HPLC–MS/MS | 0.06 ng/mL | 0.2 ng/mL | 0.90–42.6 ng/mL | 92.1 ± 2.1% | [38] |
| UPLC–MS/MS | 100 pM or 0.10 pg | 1 nM or 1.0 pg | 1pM–1µM | — | [96] |
| Pipette tip solid-phase extraction UPLC–MS/MS | 0.1 µg/kg | 0.2 µg/kg | 0.5–100 ng/mL | 86–97.92% | [97] |
| SPE-HPLC–MS/MS | 0.052 µg/kg | 0.16 µg/kg | 0.5–16.0 ng/mL | 71% | [98] |
| IT-APCI-MS/MS | 1.00 pg | — | — | — | [99] |
| DMSPE-LC–HRMS/MS | 0.03 ng/L | 0.1 ng/L | 0.01–2 µg/L | — | [100] |
| LC–MS/MS multiresidue method | 11 µg/kg | 20 µg/kg | — | 81–107% | [101] |
| Online TFC-LC–MS/MS | 0.5 µg/kg | 1.5 µg/kg | 2.5–200 µg/kg | 92.7–116.0% | [102] |
| LC–HRMS | 0.6 ng/mL (µg/kg) | 1.2 ng/mL (µg/kg) | — | — | [36] |
| UHPLC–MS/MS | Mussel 0.11 µg/kg, P oyster 0.07 µg/kg | Mussel 0.38 µg/kg, P oyster 0.25 µg/kg | 0.4–40 µg/kg | Mussel 84–88%, P oyster 88–90% | [103] |
| On-line SPE-LC–MS/MS | 0.003 ng/L | 0.007 ng/L | 0.06–31.25 ng/L | 90–90.5% | [104] |
| DLLME-LC–QqQ-MS/MS | 0.7 ng/L | 2.3 ng/L | 2.5–1000 ng/L | 98–112% | [37] |
| LC–ESI-MS/MS | 1 μg/kg | 3 μg/kg | 0.5–20 ng/mL | — | [105] |
| BLI | 6.21 nM | 20.72 nM | 55–875 nM | 97–110% | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Gao, Y.; Deng, B.; Hu, B.; Zhao, L.; Guo, H.; Yang, C.; Ma, Z.; Sun, M.; Jiao, B.; et al. Selection, Characterization, and Optimization of DNA Aptamers against Challenging Marine Biotoxin Gymnodimine-A for Biosensing Application. Toxins 2022, 14, 195. https://doi.org/10.3390/toxins14030195
Zhang X, Gao Y, Deng B, Hu B, Zhao L, Guo H, Yang C, Ma Z, Sun M, Jiao B, et al. Selection, Characterization, and Optimization of DNA Aptamers against Challenging Marine Biotoxin Gymnodimine-A for Biosensing Application. Toxins. 2022; 14(3):195. https://doi.org/10.3390/toxins14030195
Chicago/Turabian StyleZhang, Xiaojuan, Yun Gao, Bowen Deng, Bo Hu, Luming Zhao, Han Guo, Chengfang Yang, Zhenxia Ma, Mingjuan Sun, Binghua Jiao, and et al. 2022. "Selection, Characterization, and Optimization of DNA Aptamers against Challenging Marine Biotoxin Gymnodimine-A for Biosensing Application" Toxins 14, no. 3: 195. https://doi.org/10.3390/toxins14030195