DT389-YP7, a Recombinant Immunotoxin against Glypican-3 That Inhibits Hepatocellular Cancer Cells: An In Vitro Study

 , ,
, ,
Abstract
:1. Introduction
2. Results
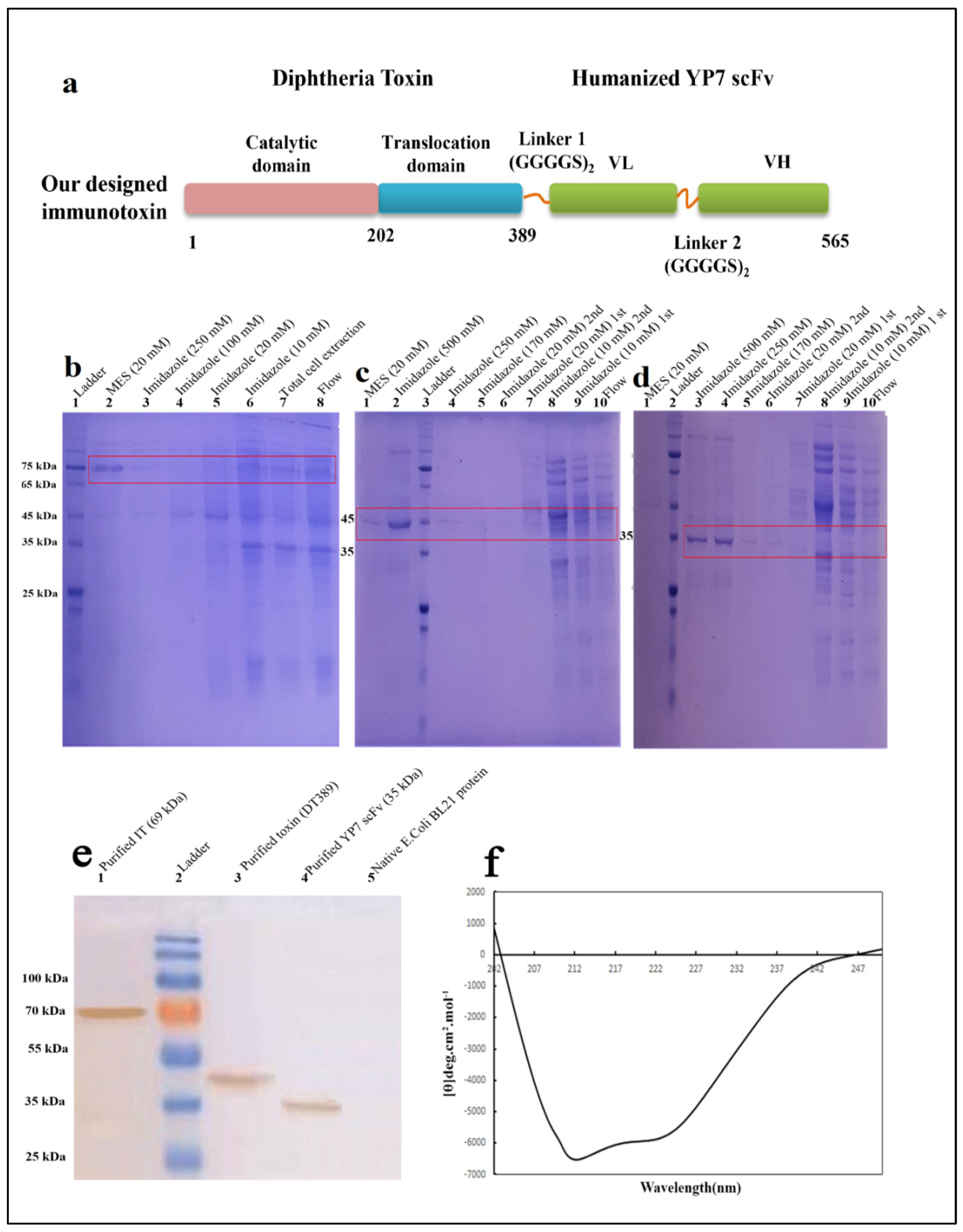
2.1. Construction Design
2.2. Purification and Validation of Proteins
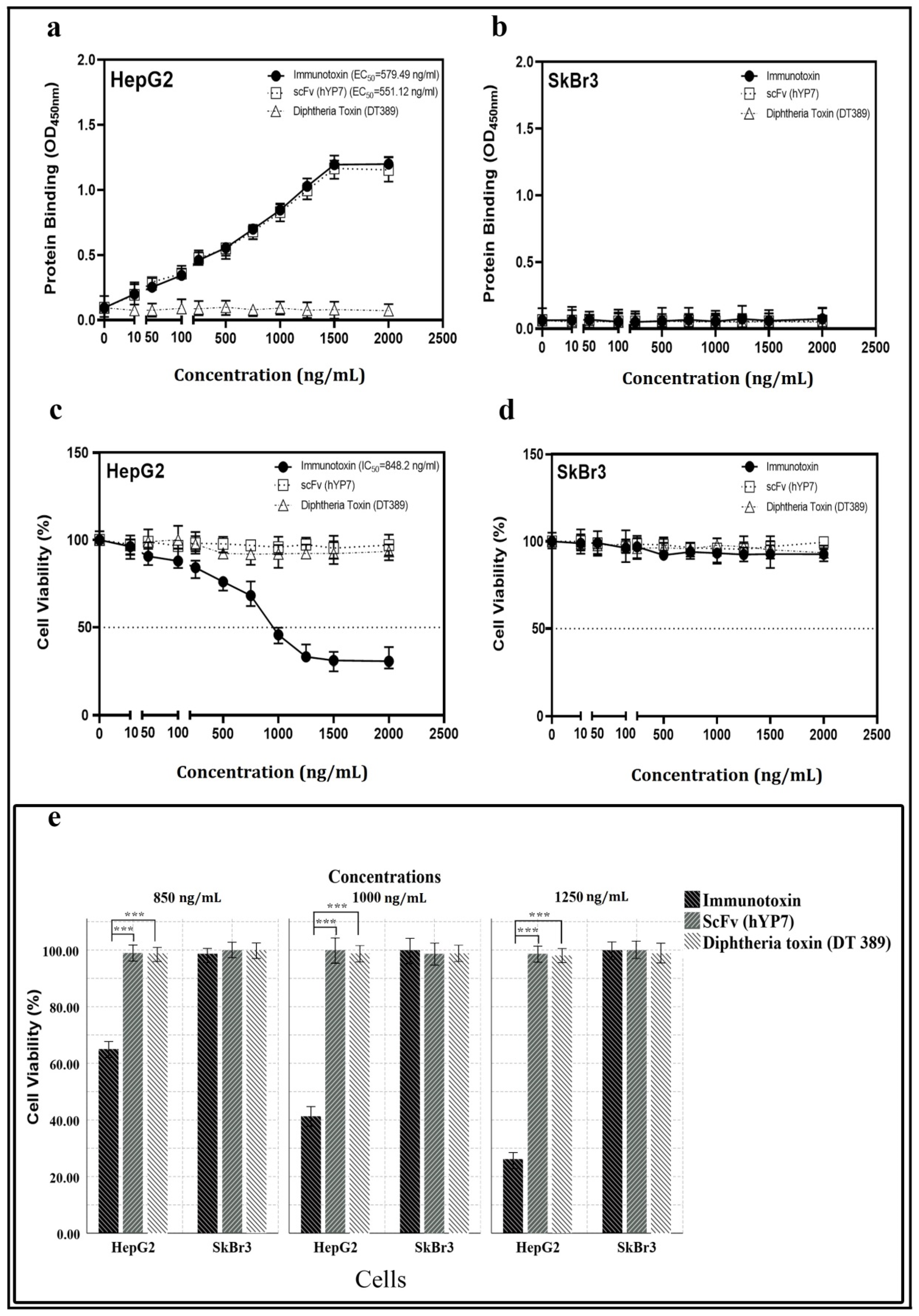
2.3. Binding Affinity and Bioactivity
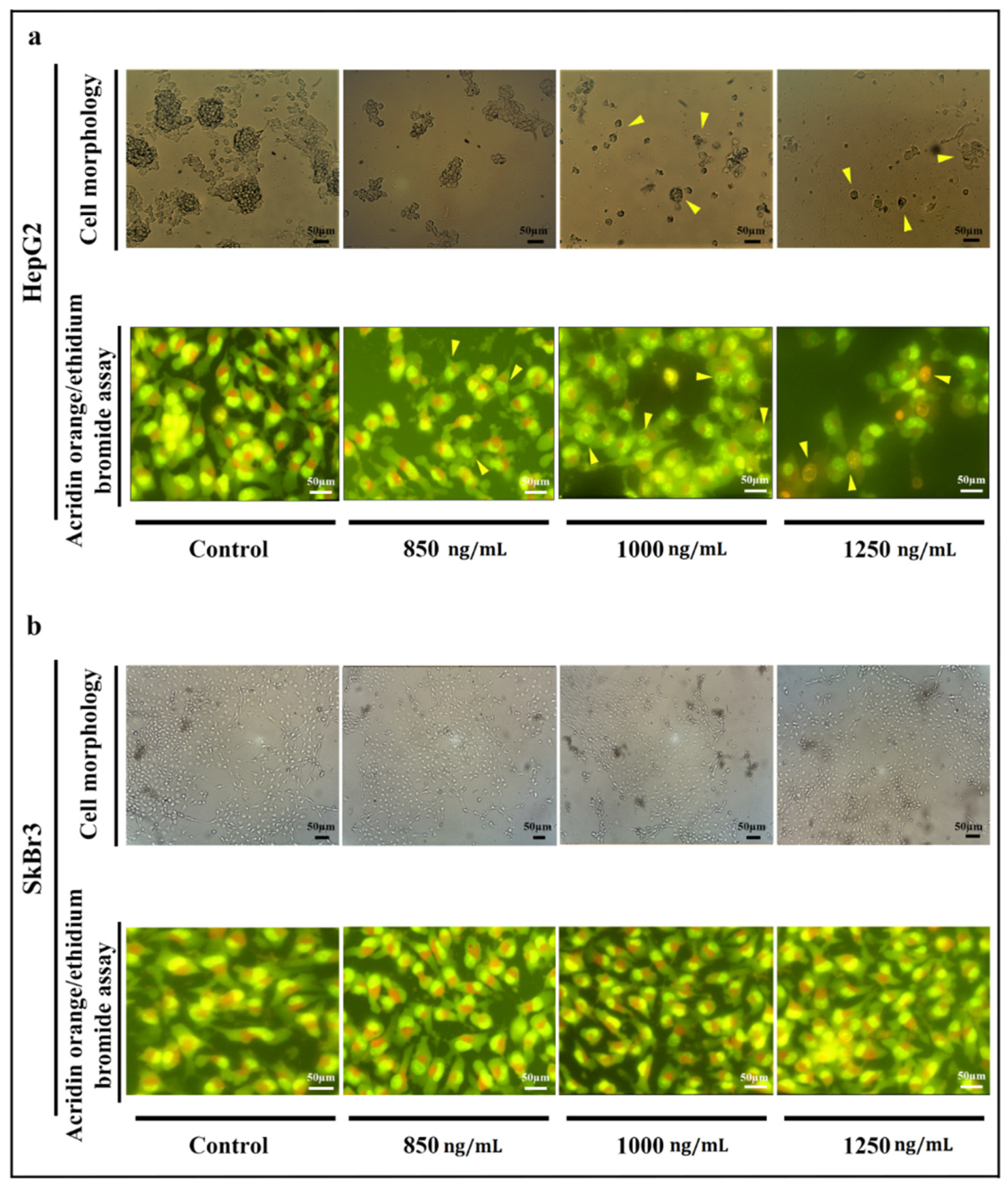
2.4. Cells’ Morphology
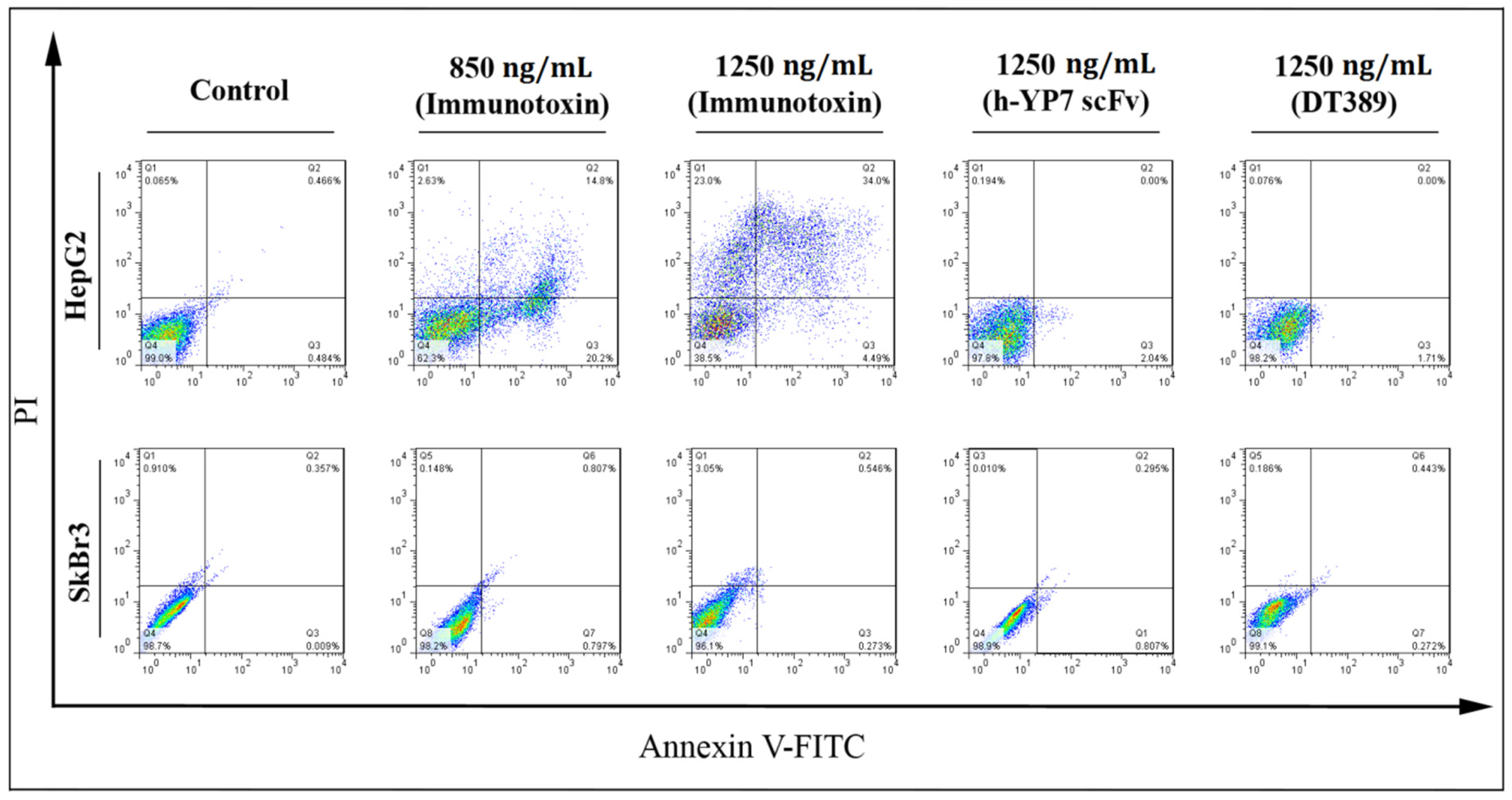
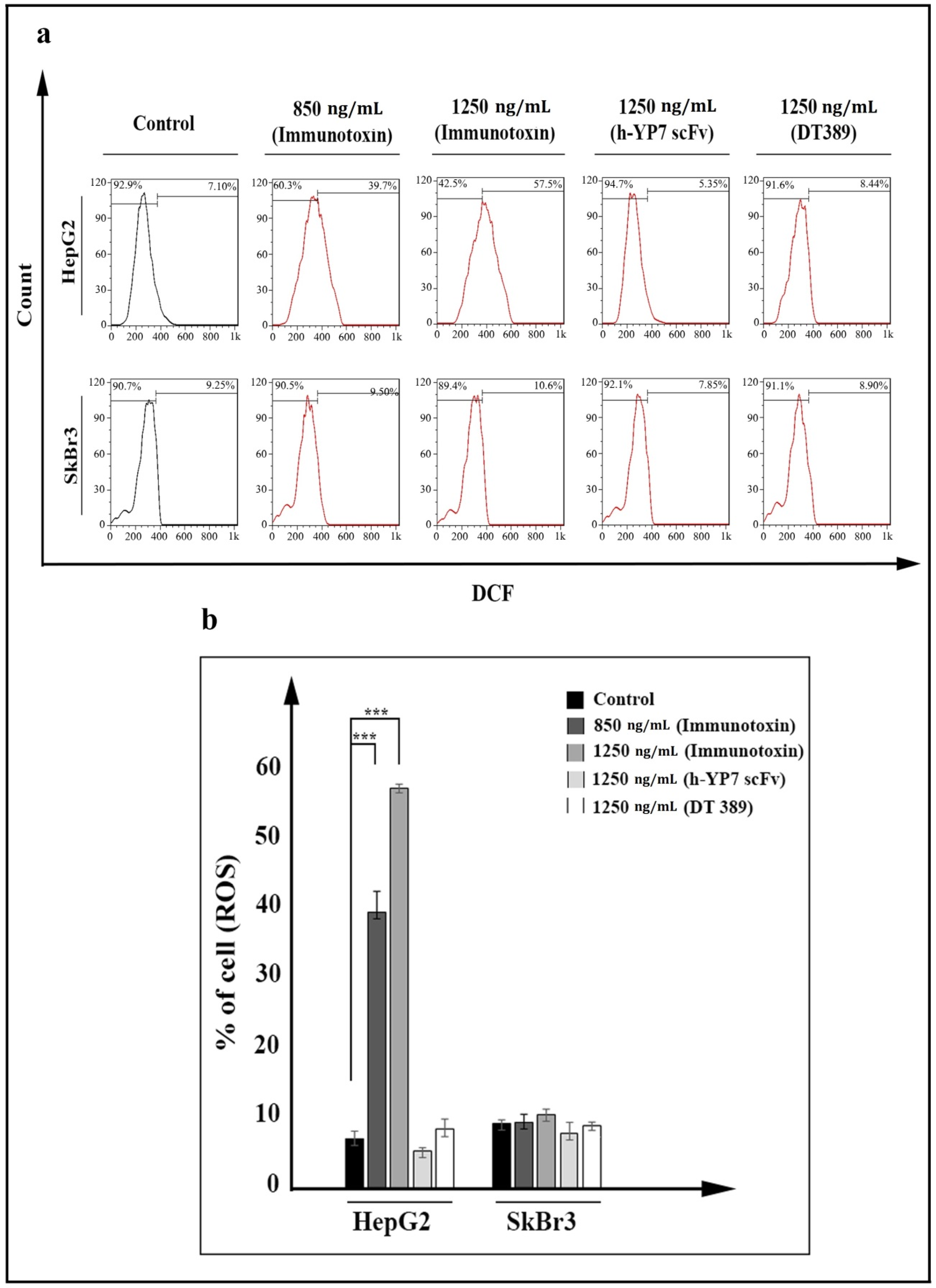
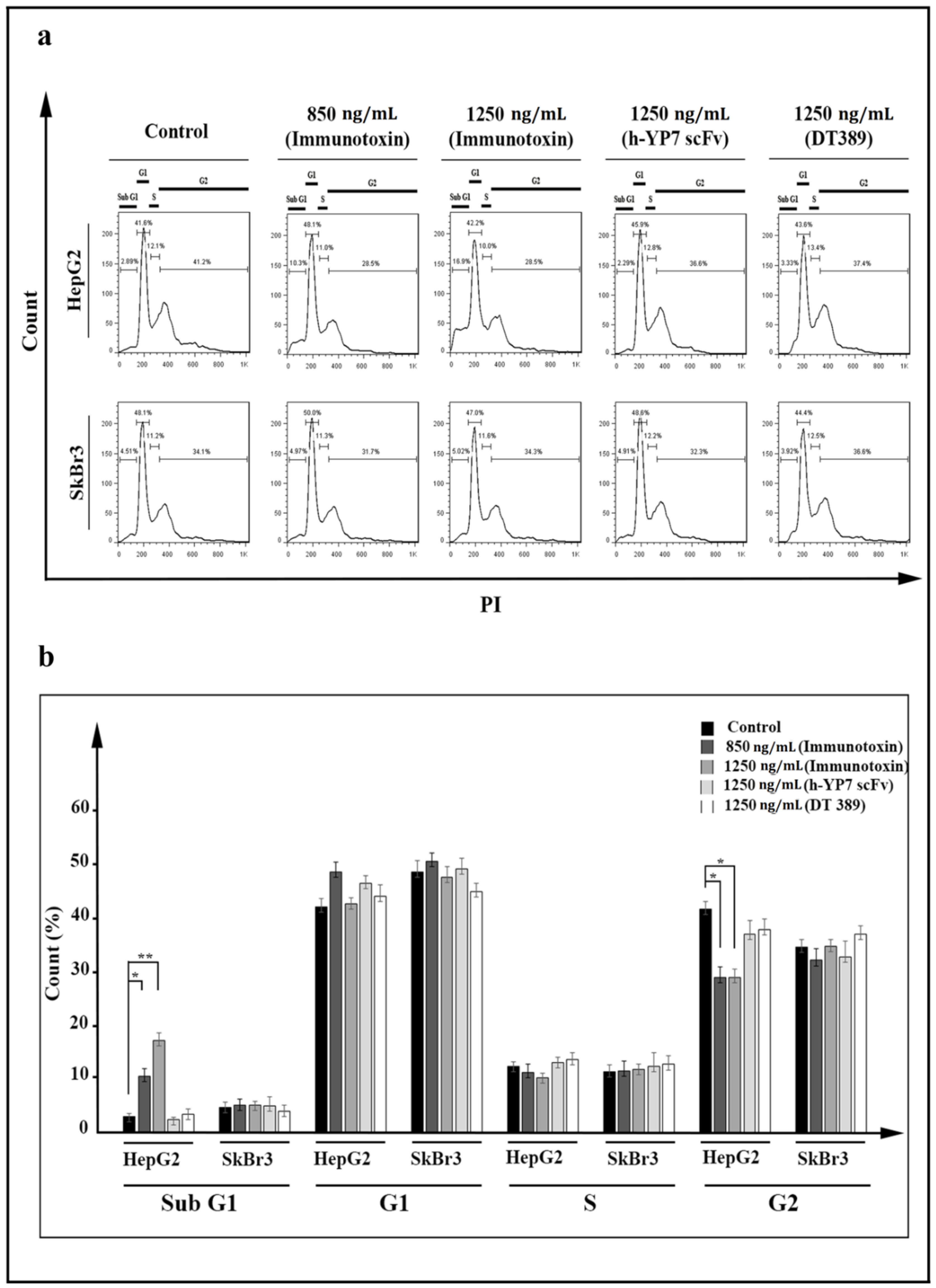
2.5. Apoptosis and Cell Cycle
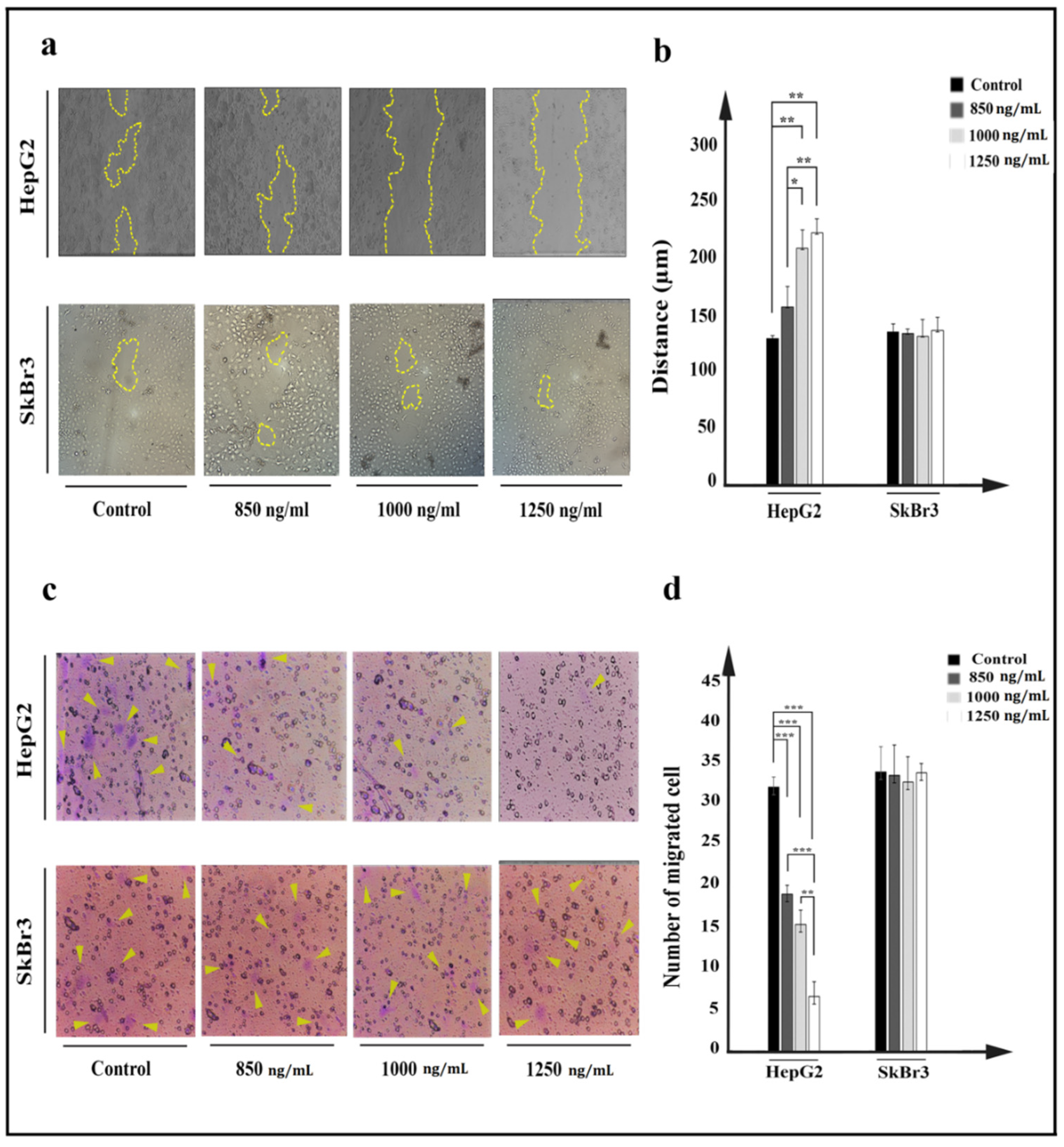
2.6. Cell Movement and Metastasis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Construction Design
5.2. Production, Purification, and Validation of Recombinant Protein by Affinity Chromatography and Western Blotting
5.3. Circular Dichroism Analysis
5.4. Cell Lines and Culture
5.5. Cytotoxic Effect of Immunotoxin
5.6. Cell Morphology Analysis
5.7. Cell Enzyme-Linked Immunosorbent Assay (Cell-ELISA)
5.8. Annexin V/PI Apoptosis Detection
5.9. Cell Cycle Investigation
5.10. Quantification of ROS in Cells
5.11. Cell Migration
5.12. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Haan, J.; Verheecke, M.; Van Calsteren, K.; Van Calster, B.; Shmakov, R.G.; Gziri, M.M.; Halaska, M.J.; Fruscio, R.; Lok, C.A.; Boere, I.A.; et al. Oncological management and obstetric and neonatal outcomes for women diagnosed with cancer during pregnancy: A 20-year international cohort study of 1170 patients. Lancet Oncol. 2018, 19, 337–346. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Sohda, M.; Kuwano, H. Current status and future prospects for esophageal cancer treatment. Ann. Thorac. Cardiovasc. Surg. 2017, 23, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Allahyari, H.; Heidari, S.; Ghamgosha, M.; Saffarian, P.; Amani, J. Immunotoxin: A new tool for cancer therapy. Tumor Biol. 2017, 39, 1010428317692226. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Alewine, C.; Pastan, I. New life for immunotoxin cancer therapy. Clin. Cancer Res. 2016, 22, 1055–1058. [Google Scholar] [CrossRef] [Green Version]
- Dandawate, P.R.; Subramaniam, D.; Jensen, R.A.; Anant, S. (Eds.) Targeting cancer stem cells and signaling pathways by phytochemicals: Novel approach for breast cancer therapy. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- El Hout, M.; Dos Santos, L.; Hamaï, A.; Mehrpour, M. (Eds.) A promising new approach to cancer therapy: Targeting iron metabolism in cancer stem cells. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Mohammadi, M.; Rezaie, E.; Sakhteman, A.; Zarei, N. A highly potential cleavable linker for tumor targeting antibody-chemokines. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Sohrabi, E.; Moslemi, M.; Rezaie, E.; Nafissi, N.; Khaledi, M.; Afkhami, H.; Fathi, J.; Zekri, A. The tissue expression of MCT3, MCT8, and MCT9 genes in women with breast cancer. Genes Genom. 2021, 43, 1065–1077. [Google Scholar] [CrossRef]
- Sohrabi, E.; Rezaie, E.; Heiat, M.; Sefidi-Heris, Y. An Integrated Data Analysis of mRNA, miRNA and Signaling Pathways in Pancreatic Cancer. Biochem. Genet. 2021, 59, 1326–1358. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, B.V.; Centeno, G.; Pascuccelli, H.; Ward, F.; Peters, M.G.; Puricelli, L.; Bal de Kier Joffé, E. Expression pattern of glypican-3-GPC3-during human embryonic and fetal development. Histol. Histopathol. 2008, 23, 1333–1340. [Google Scholar] [PubMed]
- Ligato, S.; Mandich, D.; Cartun, R.W. Utility of glypican-3 in differentiating hepatocellular carcinoma from other primary and metastatic lesions in FNA of the liver: An immunocytochemical study. Mod. Pathol. 2008, 21, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Kim, H.; Feng, M.; Phung, Y.; Xavier, C.P.; Rubin, J.S.; Ho, M. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology 2014, 60, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Tang, Z.; Zhang, Y.-F.; Feng, M.; Qian, M.; Dimitrov, D.S.; Ho, M. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat. Commun. 2015, 6, 6536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Gao, W.; Wang, R.; Chen, W.; Man, Y.G.; Figg, W.D.; Wang, X.W.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, E1083–E1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.K.; Chua, M.-S.; He, J.; Samuel, K.S. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-β2. Neoplasia 2011, 13, 735-IN25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounajjed, T.; Zhang, L.; Wu, T.T. Glypican-3 expression in gastrointestinal and pancreatic epithelial neoplasms. Hum. Pathol. 2013, 44, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Moek, K.L.; Fehrmann, R.S.; van der Vegt, B.; de Vries, E.G.; de Groot, D.J. Glypican 3 overexpression across a broad spectrum of tumor types discovered with functional genomic mRNA profiling of a large cancer database. Am. J. Pathol. 2018, 188, 1973–1981. [Google Scholar] [CrossRef]
- Keshtvarz, M.; Salimian, J.; Yaseri, M.; Bathaie, S.Z.; Rezaie, E.; Aliramezani, A.; Norouzbabaei, Z.; Amani, J.; Douraghi, M. Bioinformatic prediction and experimental validation of a PE38-based recombinant immunotoxin targeting the Fn14 receptor in cancer cells. Immunotherapy 2017, 9, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, E.; Amani, J.; Pour, A.B.; Hosseini, H.M. A new scfv-based recombinant immunotoxin against EPHA2-overexpressing breast cancer cells; High in vitro anti-cancer potency. Eur. J. Pharmacol. 2020, 870, 172912. [Google Scholar] [CrossRef]
- Rezaie, E.; Mohammadi, M.; Sakhteman, A.; Bemani, P.; Ahrari, S. Application of molecular dynamics simulations to design a dual-purpose oligopeptide linker sequence for fusion proteins. J. Mol. Modeling 2018, 24, 313. [Google Scholar] [CrossRef]
- Rezaie, E.; Nekoie, H.; Miri, A.; Oulad, G.; Ahmadi, A.; Saadati, M.; Bozorgmehr, M.; Ebrahimi, M.; Salimian, J. Different frequencies of memory B-cells induced by tetanus, botulinum, and heat-labile toxin binding domains. Microb. Pathog. 2019, 127, 225–232. [Google Scholar] [CrossRef]
- Keshtvarz, M.; Salimian, J.; Amani, J.; Douraghi, M.; Rezaie, E. In silico analysis of STX2a-PE15-P4A8 chimeric protein as a novel immunotoxin for cancer therapy. Silico Pharmacol. 2021, 9, 19. [Google Scholar] [CrossRef]
- Rezaie, E.; Pour, A.B.; Amani, J.; Hosseini, H.M. Bioinformatics Predictions, Expression, Purification and Structural Analysis of the PE38KDEL-scfv Immunotoxin against EPHA2 Receptor. Int. J. Pept. Res. Ther. 2020, 26, 979–996. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Fleming, B.D.; Ho, M. Glypican-3 targeting immunotoxins for the treatment of liver cancer. Toxins 2016, 8, 274. [Google Scholar] [CrossRef]
- Wang, C.; Gao, W.; Feng, M.; Pastan, I.; Ho, M. Construction of an immunotoxin, HN3-mPE24, targeting glypican-3 for liver cancer therapy. Oncotarget 2017, 8, 32450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-F.; Ho, M. Humanization of high-affinity antibodies targeting glypican-3 in hepatocellular carcinoma. Sci. Rep. 2016, 6, 33878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiat, M.; Hashemi Yeganeh, H.; Alavian, S.M.; Rezaie, E. Immunotoxins Immunotherapy against Hepatocellular Carcinoma: A Promising Prospect. Toxins 2021, 13, 719. [Google Scholar] [CrossRef]
- Ogawa, K.; Tanaka, S.; Matsumura, S.; Murakata, A.; Ban, D.; Ochiai, T.; Irie, T.; Kudo, A.; Nakamura, N.; Tanabe, M.; et al. EpCAM-Targeted Therapy for Human Hepatocellular Carcinoma. Ann. Surg. Oncol. 2014, 21, 1314–1322. [Google Scholar] [CrossRef]
- Liu, Z.; Feng, Z.; Zhu, X.; Xu, W.; Zhu, J.; Zhang, X.; Fan, Z.; Ji, G. Construction, expression, and characterization of an anti-tumor immunotoxin containing the human anti-c-Met single-chain antibody and PE38KDEL. Immunol. Lett. 2013, 149, 30–40. [Google Scholar] [CrossRef]
- Lv, M.; Qiu, F.; Li, T.; Sun, Y.; Zhang, C.; Zhu, P.; Qi, X.; Wan, J.; Yang, K.; Zhang, K. Construction, expression, and characterization of a recombinant immunotoxin targeting EpCAM. Mediat. Inflamm. 2015, 2015, 460264. [Google Scholar] [CrossRef]
- Marvig, R.L.; Sommer, L.M.M.; Molin, S.; Johansen, H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef] [Green Version]
- Majtnerová, P.; Roušar, T. An overview of apoptosis assays detecting DNA fragmentation. Mol. Biol. Rep. 2018, 45, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Oelsner, M.; Kreitman, R.J.; Salvatore, G.; Wang, Q.-C.; Pastan, I.; Peschel, C.; Licht, T. Induction of caspase-dependent programmed cell death in B-cell chronic lymphocytic leukemia by anti-CD22 immunotoxins. Blood 2004, 103, 2718–2726. [Google Scholar] [CrossRef]
- Keppler-Hafkemeyer, A.; Brinkmann, U.; Pastan, I. Role of caspases in immunotoxin-induced apoptosis of cancer cells. Biochemistry 1998, 37, 16934–16942. [Google Scholar] [CrossRef]
- Mathew, M.; Zaineb, K.; Verma, R.S. GM-CSF-DFF40: A novel humanized immunotoxin induces apoptosis in acute myeloid leukemia cells. Apoptosis 2013, 18, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tian, Z.; Ding, Y.; Li, X.; Zhang, Z.; Yang, L.; Zhao, F.; Ren, F.; Guo, R. EGFR-targeted immunotoxin exerts antitumor effects on esophageal cancers by increasing ROS accumulation and inducing apoptosis via inhibition of the Nrf2-Keap1 pathway. J. Immunol. Res. 2018, 2018, 1090287. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-J.; Zhu, X.-Z. Reactive oxygen species-induced apoptosis in PC12 cells and protective effect of bilobalide. J. Pharmacol. Exp. Ther. 2000, 293, 982–988. [Google Scholar] [PubMed]
- Ghetie, M.-A.; Picker, L.J.; Richardson, J.A.; Tucker, K.; Uhr, J.W.; Vitetta, E.S. Anti-CD19 inhibits the growth of human B-cell tumor lines in vitro and of Daudi cells in SCID mice by inducing cell cycle arrest. Blood 1994, 83, 1329–1336. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.F.; Wood, S.J.; Roy, R.; Reiser, J.; Kuzel, T.M.; Shafikhani, S.H. Pseudomonas aeruginosa ExoT induces G1 cell cycle arrest in melanoma cells. Cell. Microbiol. 2021, 23, e13339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cai, Y.; Dai, X.; Wu, J.; Lan, Y.; Zhang, H.; Lu, M.; Liu, J.; Xie, J. Novel EGFR-bispecific recombinant immunotoxin based on cucurmosin shows potent anti-tumor efficiency in vitro. Oncol. Rep. 2021, 45, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Haldar, S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhou, Y.; Casanova, E.; Chai, M.; Kiss, E.; Gröne, H.-J.; Schütz, G.; Grummt, I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 2005, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Stamenkovic, I. Metastatic cancer cell. Annu. Rev. Pathol. 2008, 3, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Bruell, D.; Stöcker, M.; Huhn, M.; Redding, N.; Küpper, M.; Schumacher, P.; Paetz, A.; Bruns, C.J.; Haisma, H.J.; Fischer, R.; et al. The recombinant anti-EGF receptor immunotoxin 425(scFv)-ETA’ suppresses growth of a highly metastatic pancreatic carcinoma cell line. Int. J. Oncol. 2003, 23, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Bayat, S.; Zeinoddini, M.; Azizi, A.; Khalili, M.A.N. Co-solvents effects on the stability of recombinant immunotoxin denileukin diftitox: Structure and function assessment. Iran. J. Sci. Technol. Trans. A Sci. 2019, 43, 2091–2097. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Fusion Protein | Alpha Helix | Extended Strand | Random Coil |
|---|---|---|---|
| DT289-(GGGGS)2-YP7 | 40.23 | 29.81 | 29.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashemi Yeganeh, H.; Heiat, M.; Kieliszek, M.; Alavian, S.M.; Rezaie, E. DT389-YP7, a Recombinant Immunotoxin against Glypican-3 That Inhibits Hepatocellular Cancer Cells: An In Vitro Study. Toxins 2021, 13, 749. https://doi.org/10.3390/toxins13110749
Hashemi Yeganeh H, Heiat M, Kieliszek M, Alavian SM, Rezaie E. DT389-YP7, a Recombinant Immunotoxin against Glypican-3 That Inhibits Hepatocellular Cancer Cells: An In Vitro Study. Toxins. 2021; 13(11):749. https://doi.org/10.3390/toxins13110749
Chicago/Turabian StyleHashemi Yeganeh, Hamid, Mohammad Heiat, Marek Kieliszek, Seyed Moayed Alavian, and Ehsan Rezaie. 2021. "DT389-YP7, a Recombinant Immunotoxin against Glypican-3 That Inhibits Hepatocellular Cancer Cells: An In Vitro Study" Toxins 13, no. 11: 749. https://doi.org/10.3390/toxins13110749