
A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle


Department of Chemistry and Biochemistry, University of Windsor, 401 Sunset Ave., Windsor, ON N9B 3P4, Canada
*
Authors to whom correspondence should be addressed.
Symmetry 2021, 13(4), 627; https://doi.org/10.3390/sym13040627
Submission received: 20 March 2021
/
Revised: 2 April 2021
/
Accepted: 5 April 2021
/
Published: 9 April 2021
(This article belongs to the Special Issue Recent Advances in Structural and Synthetic Supramolecular Systems)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Anions are important hydrogen bond acceptors in a range of biological, chemical, environmental and medical molecular recognition processes. These interactions have been exploited for the design and synthesis of ditopic resorcinarenes as the hydrogen bond strength can be tuned through the modification of the substituent at the 2-position. However, many potentially useful compounds, especially those incorporating electron-withdrawing functionalities, have not been prepared due to the challenge of their synthesis: their incorporation slows resorcinarene formation that is accessed by electrophilic aromatic substitution. As part of our broader campaign to employ resorcinarenes as selective recognition elements, we need access to these specialized materials. In this article, we report a straightforward synthetic pathway for obtaining a 2-(carboxymethyl)-resorcinarene, and resorcinarene esters in general. We discuss the unusual conformation it adopts and propose that this arises from the electron-withdrawing nature of the ester substituents that renders them better hydrogen bond acceptors than the phenols, ensuring that each of them acts as a donor only. Density Functional Theory (DFT) calculations show that this conformation arises as a consequence of the unusual configurational isomerism of this compound and interruption of the archetypal hydrogen bonding by the ester functionality.
1. Introduction
Resorcinarenes are (usually) bowl-shaped macrocyclic compounds stabilized by a circular network of intramolecular O···H−O hydrogen bonds [1,2]. These compounds represent a unique family of host compounds which have been extensively studied in supramolecular host–guest chemistry because they display several sites for non-covalent interactions, excellent pKa tunability and an electron-rich bowl-shaped cavity in the C4v symmetric conformation, among a myriad of other interesting properties [2,3,4]. Their cavities can accommodate a wide range of guest molecules through non-covalent interactions including (but not limited to) hydrogen bonding, halogen bonding, cation···π, C−H···π as well as π···π interactions depending on both the size and charge distribution of the respective guest molecules and the functionalization of the resorcinarene.4 In addition to their structural role enforcing the upper rim of the macrocycle, the hydroxyl groups at the 1 and 3 positions on the aromatic subunits can participate extensively in hydrogen bonding with hydrogen bond-accepting guest molecules [5,6,7,8,9]. As a direct result of these hydrogen bonded supramolecular networks, resorcinarenes have been extensively exploited as appropriate hosts to accommodate a myriad of guests ranging from alcohols [10,11,12,13,14] to sugars [15,16,17,18], steroids [19,20,21] and even heterocyclic five- and six-membered ring compounds as guest molecules [22,23,24,25,26].
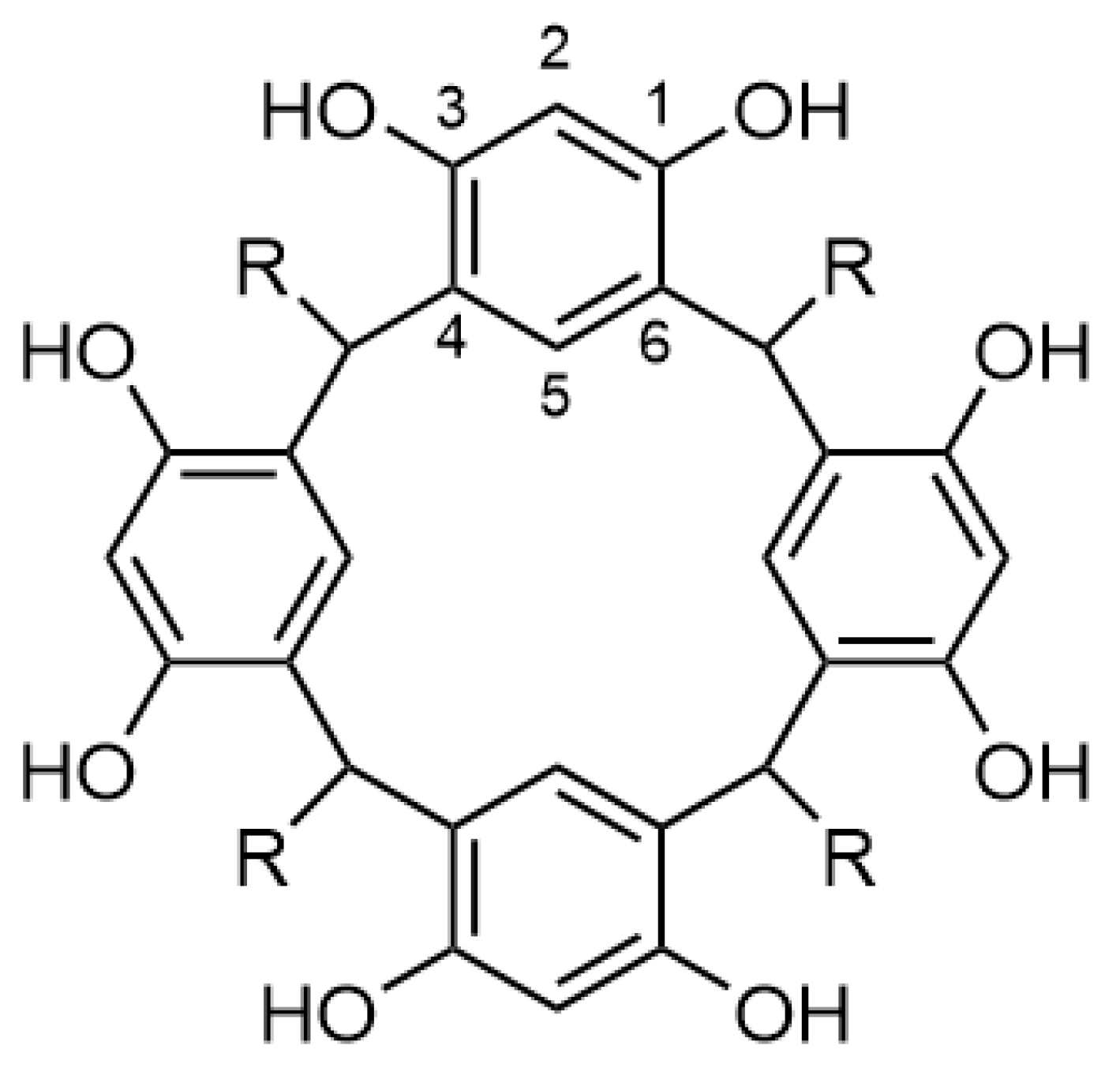
On resorcinarenes themselves, reaction at C2 (see Figure 1 for numbering) is selective over C4 and C6 positions, as these are blocked by the lower rim linkages of the resorcinarene ring. The hydrogen bonded network of hydroxyl groups enhances the acidity of the phenol while increasing π-basicity inside the cavity [27]. Attenuation or cleavage of the O−H bonds, exo to the upper rim, by bases results in increased electron density on the oxygen, effectively strengthening the hydrogen bonding [26,28,29,30].
Functionalization of resorcinarenes at the 2-position tunes the relative acidity of the phenolic hydrogens, allowing for selective reactions with certain guests. Deprotonation of the phenolic hydrogens with amine bases creates protonated ammonium cations which form interesting supramolecular complexes with the anionic resorcinarenes. These assemblies may have enhanced crystallinity that can then be studied both in the solid state and solution state by single crystal X-ray diffraction and 1H NMR, respectively, as well as in the gas phase by mass spectrometry. The challenge is to access a wide enough variety of resorcinarenes to take advantage of these potential specific interactions. As part of our campaign to access a greater variety of these molecules, we wish to report the synthesis of a simple ester resorcinarene, and its very un-resorcinarene like conformation.
2. Results and Discussion
A resorcin[4]arene with an ester functionality in the 2-position has not been reported; this moiety would act as an electron-withdrawing functionality that would increase the acidity of the phenols.
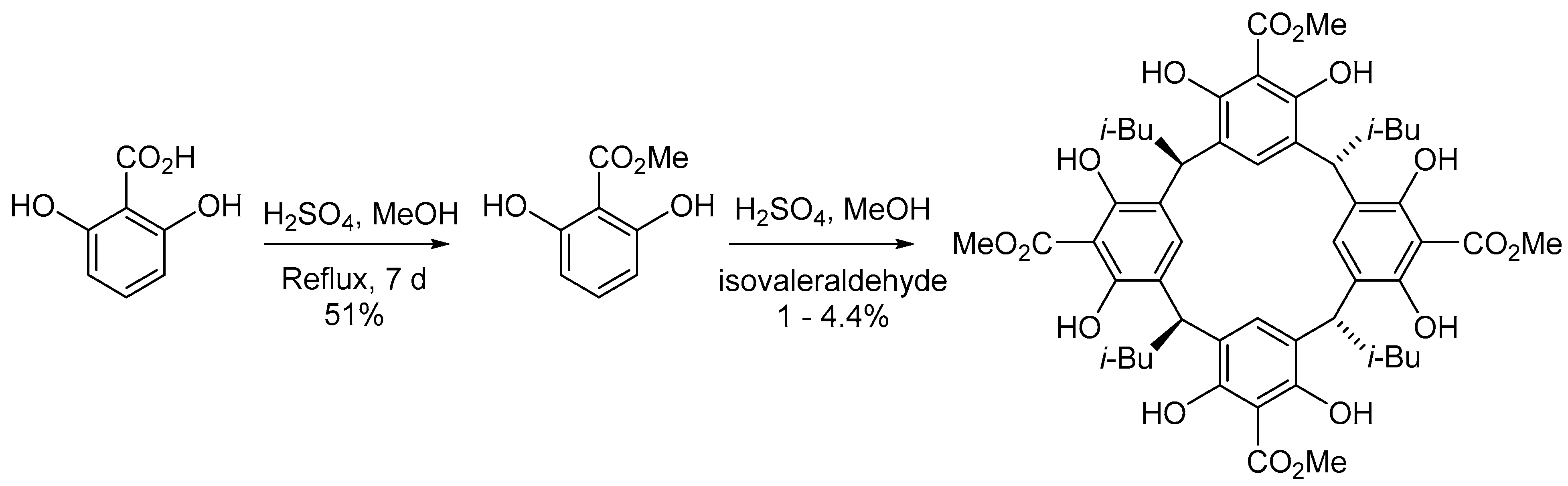
The formation of resorcinarene macrocycles as crystalline solids with high melting points through the acid-catalyzed condensation of resorcinol (or functionalized resorcinols) with aldehydes is well established [1]. Högberg was one of the first to discover the synthesis of resorcinarenes using formaldehyde and resorcinol in acidic conditions [31]. This approach works extremely well for simple 2-haloresorcinarenes and we have found success employing it for other functionalities, so it was the starting point for our synthesis [32].
To obtain 2-substituted resorcinarenes, functionalization can take place either before or after cyclization. As macrocycle formation blocks the 4 and 6 positions, the post-cyclization strategy can have advantages in terms of regioselectivity, although as four functional group transformations must occur in every step, incomplete substitution can lead to complex mixtures, difficult purification and low yields. Pre-cyclization methods, by contrast, introduce regioselectivity issues, but the use of a purified monomer ensures uniform substitution in the macrocycle. In this case, we pursued a pre-cyclization derivatization protocol because of the ready availability of a suitable precursor; the monomer unit was readily obtainable via a slow Fischer esterification of commercially available 2,6-dihydroxybenzoic acid using sulfuric acid in methanol. Following removal of the solvent in vacuo, the residue was dissolved in dichloromethane and washed with saturated sodium bicarbonate, which removed any unreacted starting material along with the sulfuric acid catalyst. Pure methyl 2,6-dihydroxy benzoate was obtained as a pinkish solid in 51% yield (Scheme 1).
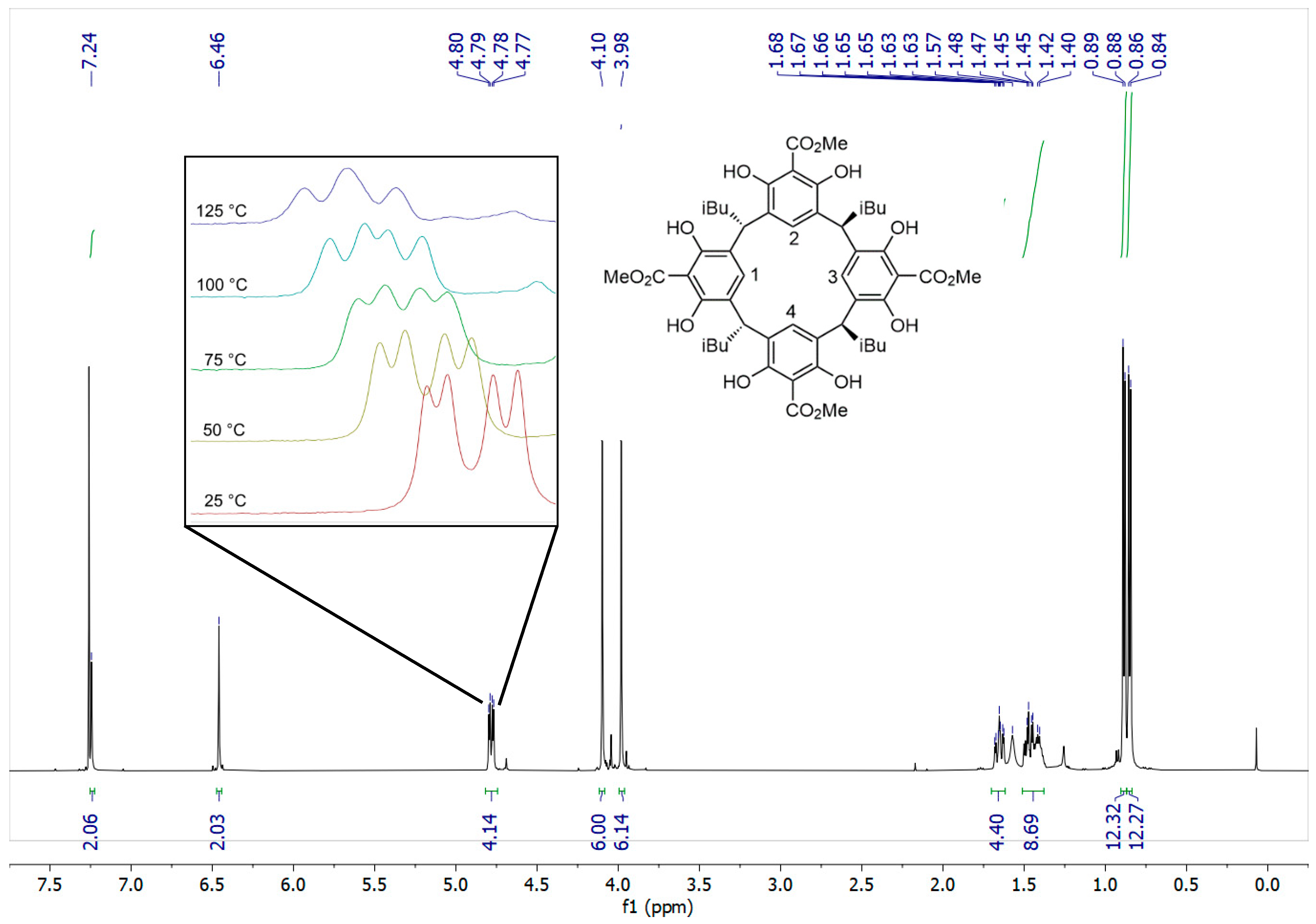
With the functionalized resorcinol in hand, several approaches toward macrocyclization were attempted using isovaleraldehyde, as the tetra isobutyl resorcinarenes are typically highly crystalline in our experience. After some exploration of conditions (see SI for discussion), we were able to effect the desired macrocyclization by employing concentrated sulfuric acid in methanol, providing the desired resorcinarene as a white solid in a poor 1–4.4% yield over repeated trials, with a great majority of the lost mass balance attributed to the formation of polymer and oligomer (Scheme 1). Curiously the NMR spectrum was not as we expected and initially gave us grave concern (Figure 2). Generally, resorcinarenes are, as we have emphasized, found in a C4v symmetric bowl-shaped conformation. In this form, the protons on each of the subunits are magnetically equivalent to congeners on the others. Consequently, one only observes a single aromatic signal, a single benzylic signal, and a single set of peaks for the lower rim alkyl chain. This is not what we found. Instead, our spectrum was consistent with a pair of isomers. We spent a significant amount of time attempting to separate these isomers but the apparent mixture behaves as a single compound by TLC, and HPLC. Seeking clarification on this issue, we attempted to recrystallize the material, but this also did not change the ratio of the signals or enrich our sample in either compound. However, it did provide us with material of sufficient quality for X-ray analysis.
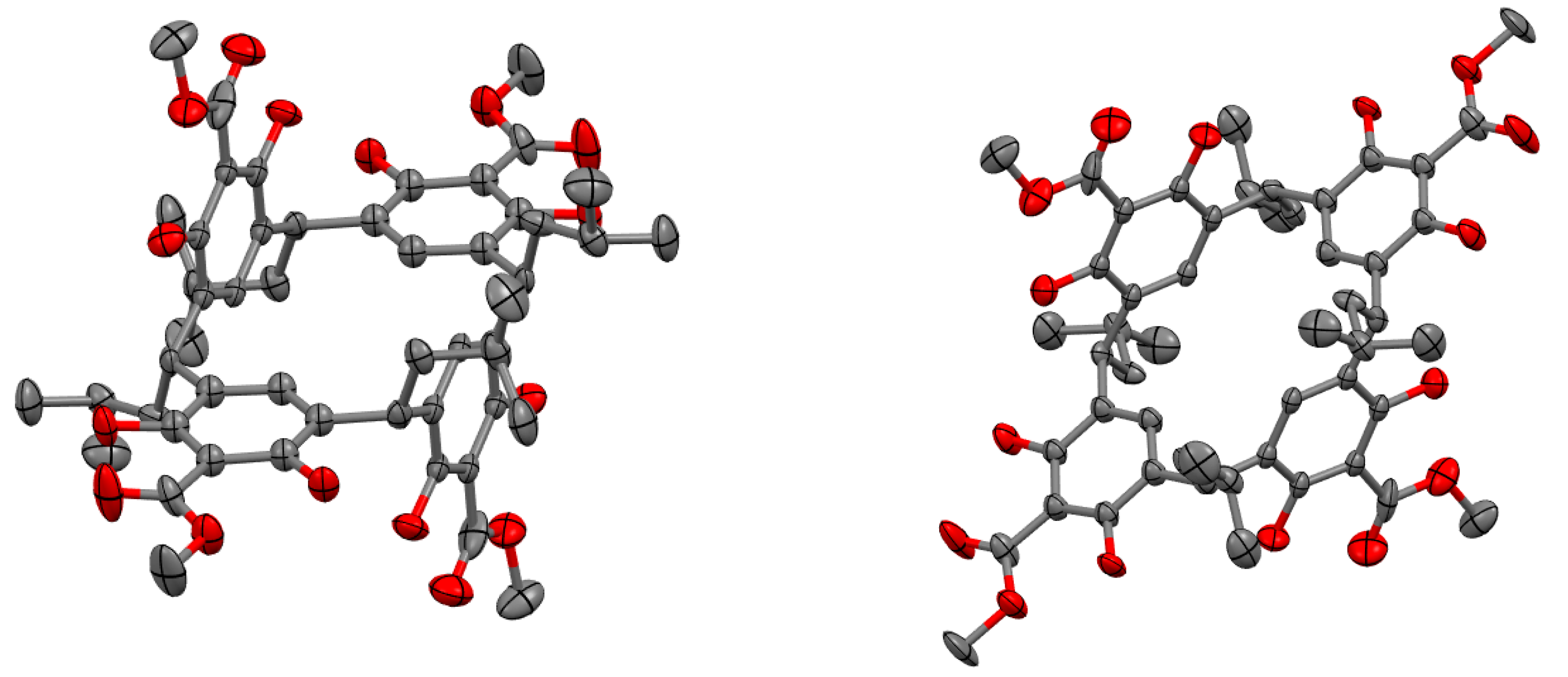
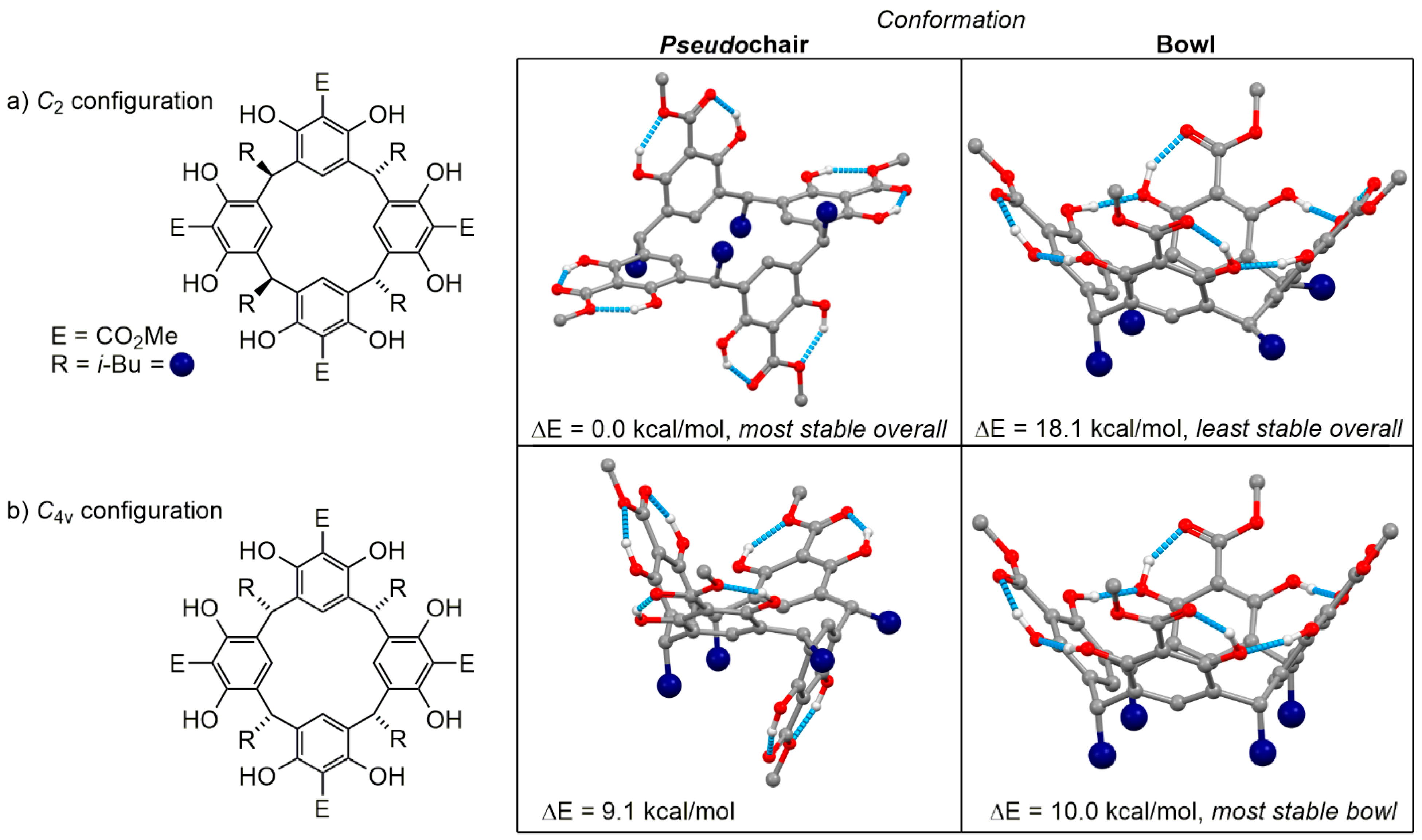
Crystals suitable for single-crystal X-ray diffraction could be obtained from the white powder by slow evaporation of a chloroform solution (Figure 3). The crystal structure of the obtained compound revealed two unusual features. Firstly, the configuration of the isobutyl groups around the lower rim of the resorcinarene is reminiscent of C2 symmetry (this can be seen in the 2D representation in Figure 4a), in contrast to the more commonly observed C4v isomer. Secondly, in the majority of resorcinarene crystal structures, the observed conformation is the archetypal bowl shape. This crystal instead exhibited a “chair” conformer with a pseudo-C2 rotation axis where two of the resorcinol subunits (2 and 4, see Figure 2 inset for numbering) are coplanar with one another, while the other two (1 and 3) sit orthogonal to the plane and antiperiplanar to one another. This result also clearly contextualizes the doubling of the resonances in the NMR spectra: this conformation is not an artifact of crystallization but appears to persist in solution and not rapidly interconvert or “flip” the pseudochair, in which case we would observe a single set of peaks as the average of the two chemical environments. The 1H NMR spectrum can now be understood in terms of this conformational preference, where the two 6H singlets at 4.10 and 3.98 ppm correspond to the methyl esters in two different environments, with the two 2H singlets at 7.24 and 6.46 ppm corresponding to the two aryl C−H environments. We were particularly surprised by the chemical shift of the singlet at 6.46 pm, which is unusually shielded for an aromatic C−H bond para to an electron-withdrawing group; in the methyl 2,6-dihydroxybenzoate starting material, the peak for the para C−H is at 7.30 ppm. After careful consideration of the crystal structure, we noticed the close proximity of the C5 atoms of aromatic rings 1 and 3 to the ArC−H hydrogens of rings 2 and 4; we thus propose that the ring currents of the 1 and 3 π-systems shield the ring 2 and 4 C−Hs. Another unusual feature of the 1H NMR spectrum is a peak at 4.78 ppm split into a doublet of doublets. This is curious as the dibenzylic protons are expected to split into a triplet by the CH2 of the isobutyl group. We hypothesize that the isobutyl groups were unable to rotate significantly in this conformation and, because of this, the adjacent methylene protons are in fact magnetically nonequivalent, leading to the observed doublet of doublets.
To further investigate the solution phase conformation, a variable-temperature (VT) NMR experiment was performed. The high-temperature NMR was necessary to determine whether this species would, in fact, interconvert to the bowl conformation if enough heat was applied, or if the chair conformation “flips” fast enough so that only an average signal is recorded. The experiment was done in increments of 25 °C up to a maximum temperature of 150 °C, at which point substantial decomposition was observed; however, up to 125 °C, there was no change in the two ester methyl peaks at 4.10 and 3.98 ppm, suggesting that the conformation of the ring remains fixed in the pseudochair conformation, where rings 1 and 3 are related by a C2 rotation axis (through the C2/C5 atoms of rings 2 and 4) and 2 and 4 are related by a mirror plane that bisects rings 1 and 3. Unlike the methyl ester peaks, there was a change observed in the dibenzylic peak at 4.78 ppm (see Figure 2, inset); at 125 °C, the doublet of doublets had converged to a triplet, suggesting the isobutyl groups were able to rotate fast enough for an averaged coupling constant to be observed on the NMR timescale. We are currently developing a model to explain this unusual conformation and stereoisomeric product, and are also preparing additional electron-poor members that might show similar behavior. However, we speculate that without the phenols working together to form the hydrogen bond network and template the forming resorcinarene, the typical C4v conformation might not be favored.
To investigate the unusual conformational preference of the resorcinarenes, DFT calculations were performed at the ωB97XD/6-311G(d,p) level of theory in the gas phase and using the polarized continuum solvation model (PCM) to consider solvent effects. Geometric optimizations of the “chair” conformation and a theoretical “bowl” geometry were performed. The initial geometry for the “chair” conformer was obtained from the solid-state molecular structure, whereas the “bowl” conformer was based on the solid-state molecular structure of known resorcinarenes. The energies and structures of the solvent-corrected optimized conformations are provided in Figure 4a; the optimized structures (as mol2 files), gas phase energies and all thermodynamic parameters can be found in the Supplementary Information. These calculations showed that the classic resorcinarene bowl conformation was disfavored by 18.1 kcal/mol using the solvent correction (15.1 kcal/mol in the gas phase), an enormous preference for the observed conformer. This is consistent with our VT NMR data, where only signals corresponding to the pseudochair ring conformation were observed until the material decomposed at 150 °C. This large preference has two possible contributing factors: first, the ester functional groups are Lewis basic and therefore have the ability to act as hydrogen bond acceptors to the phenolic hydrogen bond donors when coplanar to the benzene ring. Second, most resorcinarenes are found as the C4v configurational isomer; the steric hinderance of the isobutyl groups in the isomer obtained in this case may also impact the conformational preference of the macrocycle.
To investigate the impact of these factors, chair and bowl structures of a C4v resorcinarene were also calculated (Figure 4b). The chair conformer is still preferred in the solvated structures, although the preference is much reduced compared to the C2 isomer (0.9 kcal/mol using the chloroform solvent correction). Surprisingly, the C4v bowl is slightly favored (by 1.6 kcal/mol) in the gas phase calculations. These energy differences are so small as to be considered within the error of DFT methods, and we conclude that for the C4v isomer there is no preference between the two conformations. This shows that the configuration at the carbons bridging the resorcinol subunits can have a significant effect on the conformational preference of the macrocycle; the steric effects of the C4v configuration thus work to reinforce the upper rim, whereas in the C2 configuration, the steric effects work to pull it apart. The upper rim of a resorcinarene bowl comprises a hydrogen bond network; we see here that this is interrupted by the presence of the esters as hydrogen bond acceptors. The esters in the crystal structure are all coplanar with the benzene rings; this maximizes the delocalization of electron density from the electron-rich ring into the carbonyl of the ester, which enhances its Lewis basicity. This likely has a synergistic effect with the hydrogen bond donor phenols, which will hold the ester coplanar. We can therefore conclude that the ester acting as a hydrogen bond acceptor has the most significant effect upon the conformational preference of this macrocycle. Investigations into the generality of this phenomenon are underway in our laboratory.
3. Conclusions
We have successfully synthesized a novel 2-methyl ester resorcin[4]arene under simple acid-catalyzed conditions, if in poor yield, and the structure and solid-state conformation were determined by single crystal X-ray diffraction and NMR spectra. Computational investigations of this system revealed a significant preference for the observed pseudochair conformation and shed light upon the interplay of configurational and hydrogen bonding effects that are in operation in resorcinarene structures. Our studies in this area will further investigate the conformational preference of this and related systems, and methods to rationalize and control this aspect of supramolecular architecture will be developed.
Supplementary Materials
The followings are available online at https://www.mdpi.com/article/10.3390/sym13040627/s1, The structure of 2-(methylcarboxyl)resorcin[4]arene has been deposited in the Cambridge Crystallographic Databank (#2070167). The geometry-optimized structures (as mol2 files), complete methods and materials are found in the accompanying Supplementary Information.
Author Contributions
Conceptualization, J.F.T.; Funding acquisition, J.F.T.; Investigation, M.R.R., J.J.H.; Methodology, M.R.R., J.J.H., F.S.P.; Visualization and crystal analysis, F.S.P.; Project administration, J.F.T.; Supervision, J.F.T., J.J.H.; Writing original draft, M.R.R.; Writing—review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
Natural Sciences and Engineering Research Council of Canada Discovery Grants Program (NSERC, 2018-06338 to JFT) the Canadian Foundation for Innovation and Ontario Research Fund (37425), and MITACS and Odd Society Spirits (IT13760, salary to FP)
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data relevant to this investigation can be found in the accompanying Supplementary Information. The crystallographic data have also been deposited in the Cambridge Crystallographic Databank (#2070167).
Acknowledgments
The authors would like to thank the Natural Sciences and Engineering Research Council of Canada Discovery Grants Program (NSERC, 2018-06338 to JFT) the Canadian Foundation for Innovation and Ontario Research Fund (37425), and MITACS and Odd Society Spirits (IT13760, salary to FP) for support for this work. The authors would like to thank Lara Watanabe, University of Windsor, for acquiring the crystallographic data. The authors would like to thank A. Dina Dilinaer, University of Windsor, for providing the artwork for table of contents graphic.
Conflicts of Interest
The authors claim no conflicts of interest.
References
- Sliwa, W.; Kozlowski, C. Calixarenes and Resorcinarenes; Wiley: Weinheim, Germany, 2009. [Google Scholar]
- Timmerman, P.; Verboom, W.; Reinhoudt, D.N. Resorcinarenes. Tetrahedron 1996, 52, 2663–2704. [Google Scholar] [CrossRef] [Green Version]
- Böhmer, V. Calixarenes, macrocycles with (almost) unlimited possibilities. Angew. Chem. Int. Ed. 1995, 34, 713–745. [Google Scholar] [CrossRef]
- Rissanen, K. Very large container molecules. Angew. Chem. Int. Ed. 2005, 44, 3652–3654. [Google Scholar] [CrossRef]
- Atwood, J.L.; Szumna, A. Anion-sealed single-molecule capsules. Chem. Commun. 2003, 8, 940–941. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J.; Jerga, A. Organization of the interior of molecular capsules by hydrogen bonding. Proc. Natl. Acad. Sci. USA 2002, 99, 4837–4841. [Google Scholar] [CrossRef] [Green Version]
- MacGillivray, L.; Atwood, J. Unique guest inclusion within multi-component, extended-cavity resorcin[4]arenes. Chem. Commun. 1999, 2, 181–182. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Kogej, M.; Åhman, A.; Rissanen, K.; Schalley, C.A. Flying capsules: Mass spectrometric detection of pyrogallarene and resorcinarene hexamers. Angew. Chem. Int. Ed. 2006, 45, 5214–5218. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Paulus, E.F.; Rissanen, K.; Kolehmainen, E.; Böhmer, V. Resorcarenes in the boat conformation as building blocks for hydrogen-bonded assemblies including two ammonium cations. Chem. Eur. J. 2001, 7, 1944–1951. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Weimann, D.P.; Kaufmann, L.; Schalley, C.A.; Rissanen, K. Ion-pair recognition of tetramethylammonium salts by halogenated resorcinarenes. Chem. Eur. J. 2012, 18, 5552–5557. [Google Scholar] [CrossRef] [PubMed]
- Avram, L.; Cohen, Y.; Rebek, J., Jr. Recent advances in hydrogen-bonded hexameric encapsulation complexes. Chem. Commun. 2011, 47, 5368–5375. [Google Scholar] [CrossRef] [PubMed]
- Koshets, I.A.; Kazantseva, Z.I.; Belyaev, A.E.; Kalchenko, V.I. Sensitivity of resorcinarene films towards aliphatic alcohols. Sens. Actuators B 2009, 140, 104–108. [Google Scholar] [CrossRef]
- Ugono, O.; Holman, K.T. An achiral form of the hexameric resorcin[4]arene capsule sustained by hydrogen bonding with alcohols. Chem. Commun. 2006, 20, 2144–2146. [Google Scholar] [CrossRef] [PubMed]
- Fox, O.D.; Leung, J.F.Y.; Hunter, J.M.; Dalley, N.K.; Harrison, R.G. Metal-assembled cobalt(II) resorc[4]arene-based cage molecules that reversibly capture organic molecules from water and act as NMR shift reagents. Inorg. Chem 2000, 39, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kalenius, E.; Kekäläinen, T.; Neitola, R.; Beyeh, K.; Rissanen, K.; Vainiotalo, P. Size- and structure-selective noncovalent recognition of saccharides by tetraethyl and tetraphenyl resorcinarenes in the gas phase. Chem. Eur. J. 2008, 14, 5220–5228. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Johnson, R.J.; Escobedo, J.O.; Beck, P.A.; Kim, K.K.; St. Luce, N.N.; Davis, C.J.; Lewis, P.T.; Fronczek, F.R.; Melancon, B.J.; et al. Chromophore formation in resorcinarene solutions and the visual detection of mono- and oligosaccharides. J. Am. Chem. Soc. 2002, 124, 5000–5009. [Google Scholar] [CrossRef] [Green Version]
- Evan-Salem, T.; Baruch, I.; Avram, L.; Cohen, Y.; Palmer, L.C.; Rebek, J. Resorcinarenes are hexameric capsules in solution. Proc. Natl. Acad. Sci. USA 2006, 103, 12296. [Google Scholar] [CrossRef] [Green Version]
- Rhlalou, T.; Ferhat, M.; Frouji, M.A.; Langevin, D.; Métayer, M.; Verchère, J.F. Facilitated transport of sugars by a resorcinarene through a supported liquid membrane. J. Membr. Sci. 2000, 168, 63–73. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Rebek, J.J. Hydrogen-bonded capsules in polar, protic solvents. Chem. Commun. 2001, 22, 2374–2375. [Google Scholar] [CrossRef]
- Faull, J.D.; Gupta, V.K. Chemical selectivity of self-assembled monolayers of calix[4]resorcinarene. Thin Solid Films 2003, 440, 129–137. [Google Scholar] [CrossRef]
- Faull, J.D.; Gupta, V.K. Impact of host structure on guest−host recognition at self-assembled surfaces of tetrathiol and tetrasulfide derivatives of calix[4]resorcinarene. Langmuir 2002, 18, 6584–6592. [Google Scholar] [CrossRef]
- Puttreddy, R.; Beyeh, N.K.; Jurček, P.; Turunen, L.; Trant, J.F.; Ras, R.H.A.; Rissanen, K. Host-guest complexes of C-propyl-2-bromoresorcinarene with aromatic N-oxides. Supramol. Chem. 2018, 30, 445–454. [Google Scholar] [CrossRef]
- Puttreddy, R.; Beyeh, N.K.; Taimoory, S.M.; Meister, D.; Trant, J.F.; Rissanen, K. Host–guest complexes of conformationally flexible C-hexyl-2-bromoresorcinarene and aromatic N-oxides: Solid-state, solution and computational studies. Beilstein J. Org. Chem. 2018, 14, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puttreddy, R.; Beyeh, N.K.; Ras, R.H.A.; Trant, J.; Rissanen, K. Endo-/exo- and halogen bonded complexes of conformationally rigid C-ethyl-2-bromoresorcinarene and aromatic N-oxides. CrystEngComm 2017, 19, 4312–4320. [Google Scholar] [CrossRef] [Green Version]
- Nissinen, M.; Rissanen, K. Crystal engineering studies of the complexes of ethyl resorcinarene with aromatic nitrogen heterocycles. Supramol. Chem. 2003, 15, 581–590. [Google Scholar] [CrossRef]
- Nissinen, M.; Wegelius, E.; Falábu, D.; Rissanen, K. Melamine induced conformational change of ethyl resorcinarene in solid state. CrystEngComm 2000, 2, 151–153. [Google Scholar] [CrossRef]
- Taimoory, S.M.; Twum, K.; Dashti, M.; Pan, F.; Lahtinen, M.; Rissanen, K.; Puttreddy, R.; Trant, J.F.; Beyeh, N.K. Bringing a molecular plus one: Synergistic binding creates guest-mediated three-component complexes. J. Org. Chem. 2020, 85, 5884–5894. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-L.; Gembicky, M.; Messerschmidt, M.; Dominiak, P.M.; Coppens, P. Effect of the environment on molecular properties: Synthesis, structure, and photoluminescence of Cu(I) bis(2,9-dimethyl-1,10-phenanthroline) nanoclusters in eight different supramolecular frameworks. Inorg. Chem. 2006, 45, 9281–9289. [Google Scholar] [CrossRef]
- Murayama, K. Resorcin[4]arene dimer linked by eight water molecules and incorporating a tetraethylammonium ion: Guest-driven capsule formation via cation–π interactions. Chem. Commun. 1998, 5, 607–608. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J.; Hardie, M.J.; Lygris, E.; Raston, C.L.; Webb, H.R. Inclusion complexes of 18-crown-6 and (Na+⊂[2.2.2]cryptand) in [C-methylcalix[4]resorcinarene-Hn], n⊕=⊕0, 1. CrystEngComm 2001, 3, 41–43. [Google Scholar] [CrossRef]
- Högberg, A.G.S. Two stereoisomeric macrocyclic resorcinol-acetaldehyde condensation products. J. Org. Chem. 1980, 45, 4498–4500. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Pan, F.; Rissanen, K. A halogen-bonded dimeric resorcinarene capsule. Angew. Chem. Int. Ed. 2015, 54, 7303–7307. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A generic resorcinarene illustrating the numbering convention.

Scheme 1.
Synthesis of the resorcin[4]arene from 2,6-dihydroxybenzoic acid.

Figure 2.
1H NMR spectrum in CDCl3 showing the features of the C2 configuration in solution. Inset zoom shows convergence of dd peak to triplet in the VT experiment from 25 °C to 125 °C, in DMSO-d6.
Figure 2.
1H NMR spectrum in CDCl3 showing the features of the C2 configuration in solution. Inset zoom shows convergence of dd peak to triplet in the VT experiment from 25 °C to 125 °C, in DMSO-d6.

Figure 3.
Front and side view of the crystal structure of 2-(methylcarboxyl)resorcin[4]arene with ellipsoids drawn at 50% probability. H-atoms omitted for clarity. Deposition #2070167 in the CCDC.
Figure 3.
Front and side view of the crystal structure of 2-(methylcarboxyl)resorcin[4]arene with ellipsoids drawn at 50% probability. H-atoms omitted for clarity. Deposition #2070167 in the CCDC.

Figure 4.
Computed energies of (a) the obtained C2 isomer and (b) expected C4v isomer. All C−H bonds have been omitted and the isobutyl groups are represented by navy spheres for clarity. Hydrogen bonds have been highlighted in blue. Note: the configuration refers purely to the relative arrangement of the isobutyl groups.
Figure 4.
Computed energies of (a) the obtained C2 isomer and (b) expected C4v isomer. All C−H bonds have been omitted and the isobutyl groups are represented by navy spheres for clarity. Hydrogen bonds have been highlighted in blue. Note: the configuration refers purely to the relative arrangement of the isobutyl groups.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Reynolds, M.R.; Pick, F.S.; Hayward, J.J.; Trant, J.F. A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle. Symmetry 2021, 13, 627. https://doi.org/10.3390/sym13040627
AMA Style
Reynolds MR, Pick FS, Hayward JJ, Trant JF. A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle. Symmetry. 2021; 13(4):627. https://doi.org/10.3390/sym13040627
Chicago/Turabian StyleReynolds, Michael R., Fraser S. Pick, John J. Hayward, and John F. Trant. 2021. "A Concise Synthesis of a Methyl Ester 2-Resorcinarene: A Chair-Conformation Macrocycle" Symmetry 13, no. 4: 627. https://doi.org/10.3390/sym13040627
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.