
Potential Tear Biomarkers for the Diagnosis of Parkinson’s Disease—A Pilot Study

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Ophthalmological Examination
2.3. Neurological Examination
2.4. Tear-Sample Collection
2.5. Proteomics Analyses
2.6. Statistical Analyses
3. Results
3.1. Patients and Clinical Parameters
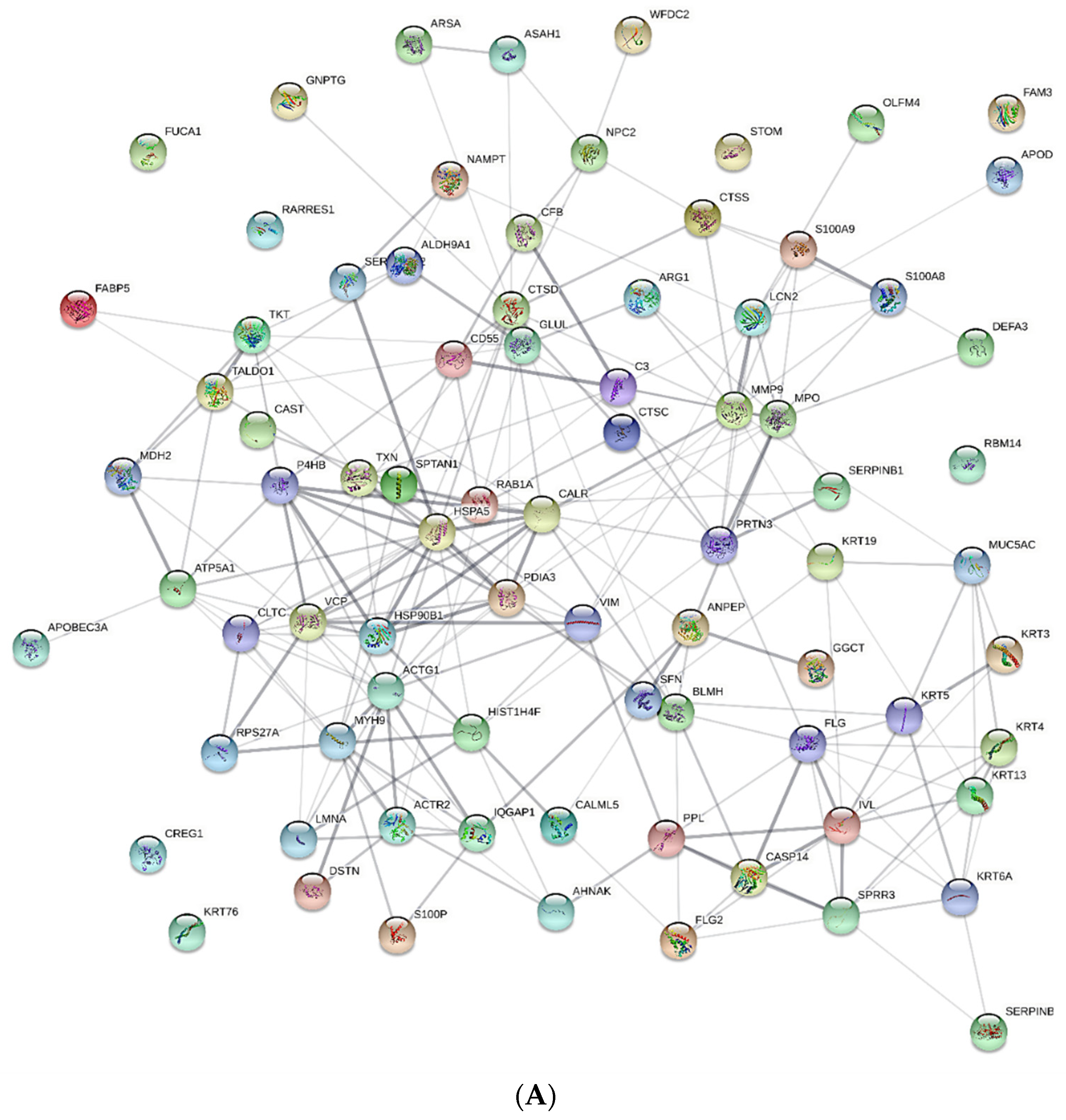
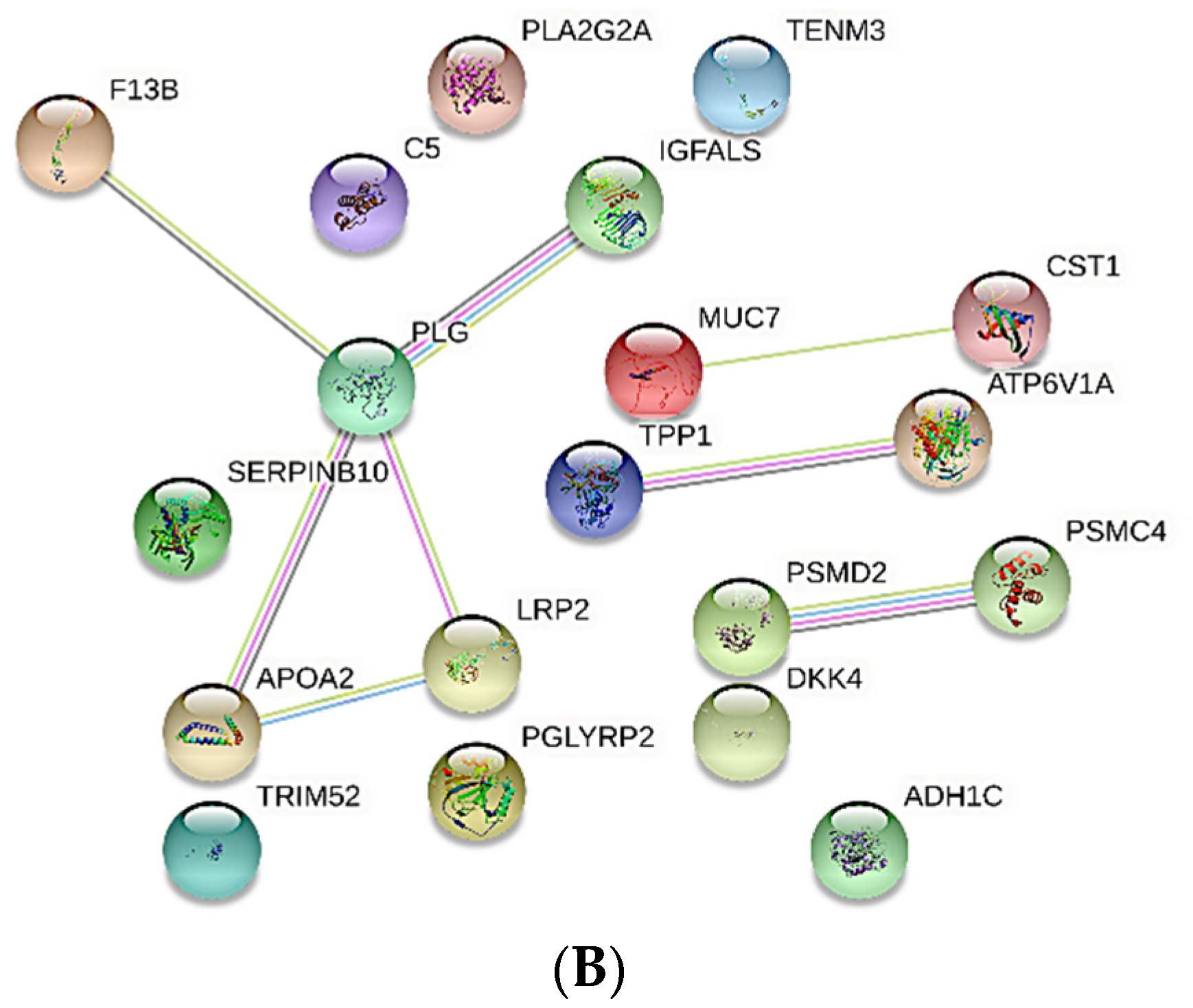
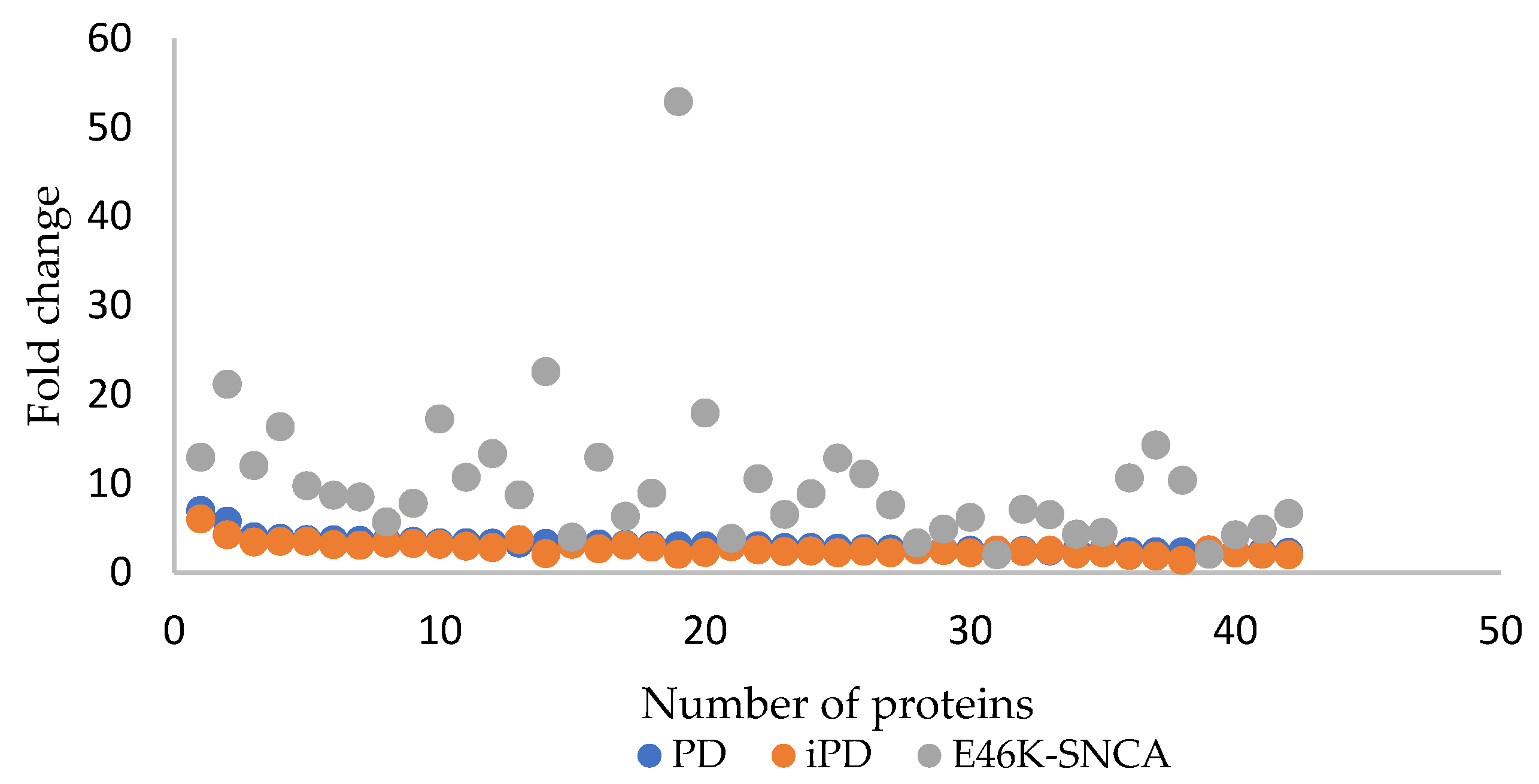
3.2. nLC MS/MS Data
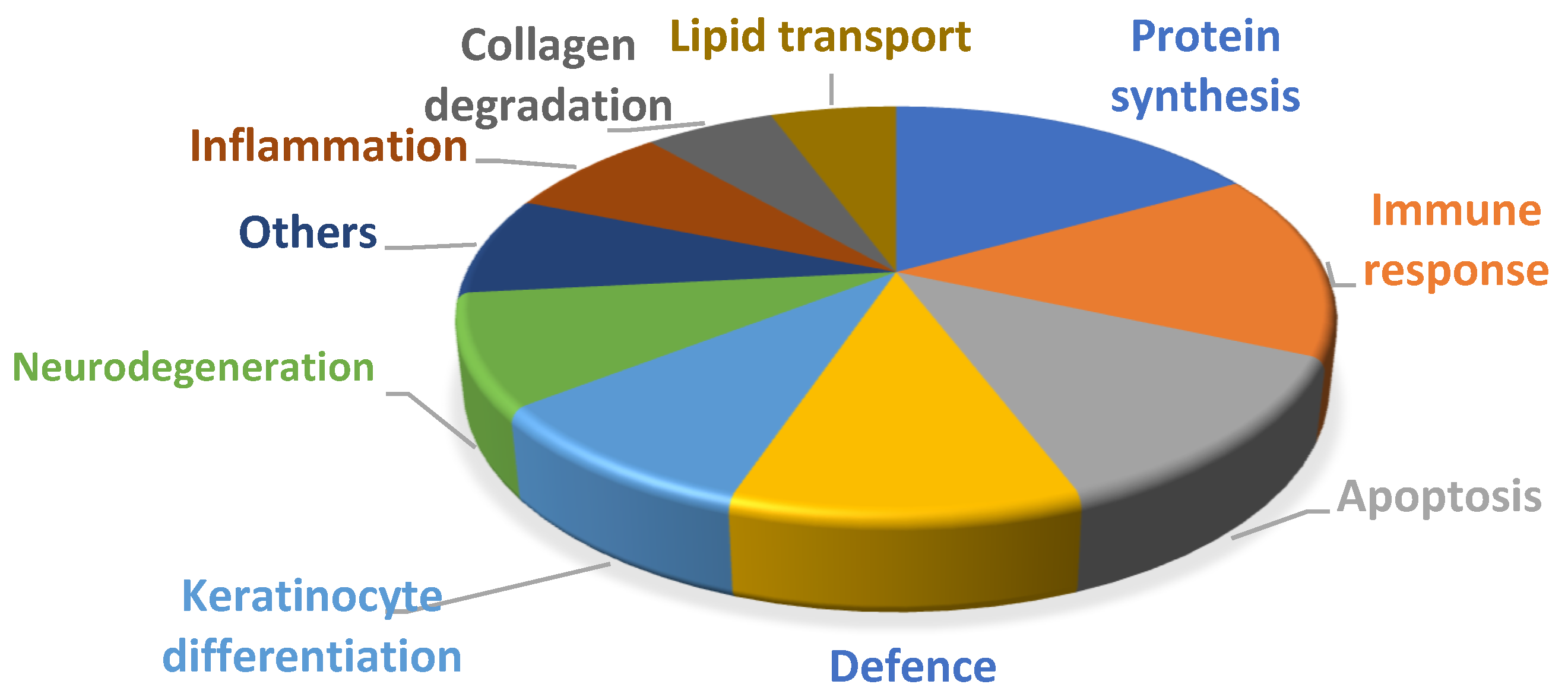
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- George, J.M. The synucleins. Genome Biol. 2001, 3, 1–6. [Google Scholar] [CrossRef]
- Love, S. Neuropathological investigation of dementia: A guide for neurologists. J. Neurol. Neurosurg. Psychiatry 2005, 76 (Suppl. 5), v8–v14. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Schaffer, A.A.; et al. Mapping of a Gene for Parkinson’s Disease to Chromosome 4q21-q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2003, 55, 164–173. [Google Scholar] [CrossRef]
- Archibald, N.K.; Clarke, M.P.; Mosimann, U.P.; Burn, D.J. Visual symptoms in Parkinson’s disease and Parkinson’s disease dementia. Mov. Disord. 2011, 26, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Safranow, K.; Nowacka, B.; Lubiński, W.; Honczarenko, K.; Potemkowski, A. Ophthalmological Features of Parkinson Disease. Med Sci. Monit. 2014, 20, 2243–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesnokova, N.B.; Pavlenko, T.A.; Ugrumov, M.V. Ophthalmic disorders as a manifestation of Parkinson’s disease. Zhurnal Nevrologii i Psikhiatrii Imeni SS Korsakova 2017, 117, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Murueta-Goyena, A.; Del Pino, R.; Galdós, M.; Arana, B.; Acera, M.; Carmona-Abellán, M.; Fernández-Valle, T.; Tijero, B.; Lucas-Jiménez, O.; Ojeda, N.; et al. Retinal Thickness Predicts the Risk of Cognitive Decline in Parkinson Disease. Ann. Neurol. 2020, 89, 165–176. [Google Scholar] [CrossRef]
- Nakahara, T.; Mori, A.; Kurauchi, Y.; Sakamoto, K.; Ishii, K. Neurovascular Interactions in the Retina: Physiological and Pathological Roles. J. Pharmacol. Sci. 2013, 123, 79–84.3. [Google Scholar] [CrossRef] [Green Version]
- Lopatina, E.V.; Penniyaynen, V.A.; Tsyrline, V.A. Impact of norepinephrine and selective β1-adrenoceptor blockers on the growth of retinal tissue explants. Bull. Exp. Biol. Med. 2012, 153, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Bowd, C.; Zangwill, L.M.; Weinreb, R.N.; Girkin, C.A.; Fazio, M.A.; Liebmann, J.M.; Belghith, A. Racial Differences in Rate of Change of Spectral-Domain Optical Coherence Tomography–Measured Minimum Rim Width and Retinal Nerve Fiber Layer Thickness. Am. J. Ophthalmol. 2018, 196, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Winter, S.V.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448.3. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Boerger, M.; Funke, S.; Leha, A.; Roser, A.-E.; Wuestemann, A.-K.; Maass, F.; Bähr, M.; Grus, F.; Lingor, P. Proteomic analysis of tear fluid reveals disease-specific patterns in patients with Parkinson’s disease—A pilot study. Park. Relat. Disord. 2019, 63, 3–9. [Google Scholar] [CrossRef]
- Arlehamn, C.S.L.; Garretti, F.; Sulzer, D.; Sette, A. Roles for the adaptive immune system in Parkinson’s and Alzheimer’s diseases. Curr. Opin. Immunol. 2019, 59, 115–120. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Martin, T.A.; Mansel, R.E.; Jiang, W.G. Antagonistic effect of NK4 on HGF/SF induced changes in the transendothelial resistance (TER) and paracellular permeability of human vascular endothelial cells. J. Cell. Physiol. 2002, 192, 268–275. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix Metalloproteinase-2 and -9 Secreted by Leukemic Cells Increase the Permeability of Blood-Brain Barrier by Disrupting Tight Junction Proteins. PLoS ONE 2011, 6, e20599. [Google Scholar] [CrossRef]
- Gu, Y.; Zheng, G.-Q.; Xu, M.; Li, Y.; Chen, X.; Zhu, W.; Tong, Y.; Chung, S.K.; Liu, K.J.; Shen, J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J. Neurochem. 2011, 120, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Lorenzl, S.; Albers, D.S.; Narr, S.; Chirichigno, J.; Beal, M. Expression of MMP-2, MMP-9, and MMP-1 and Their Endogenous Counterregulators TIMP-1 and TIMP-2 in Postmortem Brain Tissue of Parkinson’s Disease. Exp. Neurol. 2002, 178, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Kim, E.-M.; Choi, J.H.; Son, H.J.; Ji, I.J.; Joh, T.H.; Chung, S.J.; Hwang, O. Matrix metalloproteinase-3 contributes to vulnerability of the nigral dopaminergic neurons. Neurochem. Int. 2010, 56, 161–167. [Google Scholar] [CrossRef]
- Acera, A.; Vecino, E.; Duran, J.A. Tear MMP-9 Levels as a Marker of Ocular Surface Inflammation in Conjunctivochalasis. Investig. Opthalmol. Vis. Sci. 2013, 54, 8285–8291. [Google Scholar] [CrossRef] [Green Version]
- Recalde, J.I.; Duran, J.A.; Rodriguez-Agirretxe, I.; Soria, J.; Sanchez-Tena, M.A.; Pereiro, X.; Suarez, T.; Acera, A. Changes in tear biomarker levels in keratoconus after corneal collagen crosslinking. Mol. Vis. 2019, 25, 12–21. [Google Scholar] [PubMed]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal Lipid Peroxidation in Substantia Nigra Is Increased in Parkinson’s Disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Abbott, R.D.; Petrovitch, H.; Mailman, R.B.; Ross, G.W. Low LDL cholesterol and increased risk of Parkinson’s disease: Prospective results from Honolulu-Asia Aging Study. Mov. Disord. 2008, 23, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Alonso, A.; Guo, X.; Umbach, D.M.; Lichtenstein, M.L.; Ballantyne, C.M.; Mailman, R.; Mosley, T.H.; Chen, H. Statins, plasma cholesterol, and risk of Parkinson’s disease: A prospective study. Mov. Disord. 2015, 30, 552–559. [Google Scholar] [CrossRef]
- Ma, V.R.; Gurevich, T.; Giladi, N.; El-Ad, B.; Tsamir, J.; Hemo, B.; Peretz, C. Higher serum cholesterol and decreased Parkinson’s disease risk: A statin-free cohort study. Mov. Disord. 2018, 33, 1298–1305. [Google Scholar]
- Zhang, L.; Wang, X.; Wang, M.; Sterling, N.W.; Du, G.; Lewis, M.M.; Yao, T.; Mailman, R.B.; Li, R.; Huang, X. Circulating Cholesterol Levels May Link to the Factors Influencing Parkinson’s Risk. Front. Neurol. 2017, 8, 501. [Google Scholar] [CrossRef] [Green Version]
- Tamam, Y.; Tasdemir, N.; Yalman, M.; Tamam, B. Association of apolipoprotein E genotypes with prognosis in multiple sclerosis. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1122–1130. [Google Scholar]
- Ponsford, J.; McLaren, A.; Schönberger, M.; Burke, R.; Rudzki, D.; Olver, J.; Ponsford, M. The Association between Apolipoprotein E and Traumatic Brain Injury Severity and Functional Outcome in a Rehabilitation Sample. J. Neurotrauma 2011, 28, 1683–1692. [Google Scholar] [CrossRef]
- Amouyel, P.; French Research Group on Epidemiology of Human Spongiform Encephalopathies; Vidal, O.; Laplanche, J.-L.; Launay, J. The apolipoprotein E alleles as major susceptibility factors for Creutzfeldt-Jakob disease. Lancet 1994, 344, 1315–1318. [Google Scholar] [CrossRef]
- Koob, A.O.; Ubhi, K.; Paulsson, J.F.; Kelly, J.; Rockenstein, E.; Mante, M.; Adame, A.; Masliah, E. Lovastatin ameliorates α-synuclein accumulation and oxidation in transgenic mouse models of α-synucleinopathies. Exp. Neurol. 2010, 221, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Apolipoprotein E-Containing Lipoproteins Protect Neurons from Apoptosis via a Signaling Pathway Involving Low-Density Lipoprotein Receptor-Related Protein-1. J. Neurosci. 2007, 27, 1933–1941. [Google Scholar] [CrossRef]
- Navarro, A.; Mendez, E.; Diaz, C.; del Valle, E.; Martinez-Pinilla, E.; Ordonez, C.; Tolivia, J. Lifelong expression of apolipoprotein D in the human brainstem: Correlation with reduced age-related neurodegeneration. PLoS ONE 2013, 8, e77852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clin. Lipidol. 2010, 5, 555–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, E.; Anastasopoulos, D.; Konitsiotis, S.; Lavedan, C.; Polymeropoulos, M.H. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson’s disease. Hum. Genet. 1998, 103, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef]
- Datta, I.; Ganapathy, K.; Razdan, R.; Bhonde, R. Location and Number of Astrocytes Determine Dopaminergic Neuron Survival and Function Under 6-OHDA Stress Mediated Through Differential BDNF Release. Mol. Neurobiol. 2017, 55, 5505–5525. [Google Scholar] [CrossRef]
- Partanen, S.; Haapanen, A.; Kielar, C.; Pontikis, C.; Alexander, N.; Inkinen, T.; Saftig, P.; Gillingwater, T.H.; Cooper, J.D.; Tyynela, J. Synaptic changes in the thalamocortical system of cathepsin D-deficient mice: A model of human congenital neuronal ceroid-lipofuscinosis. J. Neuropathol. Exp. Neurol. 2008, 67, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Siintola, E.; Partanen, S.; Stromme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.E.; Tyynela, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, V.; Lindfors, M.; Ng, J.; Paetau, A.; Swinton, E.; Kolodziej, P.; Boston, H.; Saftig, P.; Woulfe, J.; Feany, M.B.; et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2009, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Thomas, L.M.; Watkins, S.C.; Franchi, L.; Nunez, G.; Salter, R.D. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J. Leukoc. Biol. 2009, 86, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Padiath, Q.S.; Fu, Y.H. Autosomal dominant leukodystrophy caused by lamin B1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol. 2010, 98, 337–357. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables, Units | iPD | E46K-SNCA | Control |
|---|---|---|---|
| n | 24 | 3 | 27 |
| Age, years | 60.4 (9.21) | 45 (14.73) | 49.69 (12.17) |
| Gender (F/M) (%) | 38/62 | 0/100 | 54/46 |
| Disease duration, years | 9.36 (6.66) | 3.33 (3.51) | N/A |
| Hoehn and Yahr score | 2 (0.82) | 2 (1) | N/A |
| UPDRS score (%) | 32.39 (9.03) | 16.56 (11.12) | N/A |
| Part I UPDRS score | 17.19 (12.6) | 6.25 (0.0) | N/A |
| Part II UPDRS score | 27.52 (10.07) | 11.54 (8.38) | N/A |
| Part III UPDRS score | 45.13 (15.09) | 25.00 (16.7) | N/A |
| Part IV UPDRS score | 16.30 (9.46) | 10.14 (9.05) | N/A |
| Blepharitis (%) | 40.74 | 0 | 0 |
| Entry Name | Protein Name | p-Value | Fold |
|---|---|---|---|
| OLFM4 | Olfactomedin-4 | 7.08 × 10−5 | 4.25 |
| SPB3 | Serpin B3 | 5.29 × 10−4 | 3.99 |
| SPRR3 | Small proline-rich protein 3 | 1.77 × 10−2 | 3.31 |
| MMP9 | Matrix metalloproteinase-9 | 7.33 × 10−3 | 3.08 |
| CASPE | Caspase-14 | 4.88 × 10−2 | 2.67 |
| PRTN3 | Myeloblastin | 1.76 × 10−2 | 2.63 |
| GGCT | Gamma-glutamylcyclotransferase | 2.56 × 10−2 | 2.40 |
| CATD | Cathepsin D | 1.24 × 10−3 | 2.31 |
| CALL5 | Calmodulin-like protein 5 | 3.48 × 10−2 | 1.91 |
| TIG1 | Retinoic acid receptor responder protein 1 | 3.87 × 10−2 | 1.90 |
| MDHM | Malate dehydrogenase mitochondrial | 8.59 × 10−3 | 1.66 |
| NGAL | Neutrophil gelatinase-associated lipocalin | 3.51 × 10−2 | 1.54 |
| NQO1 | NAD(P)H dehydrogenase (quinone) 1 | 1.92 × 10−2 | 0.49 |
| PLST | Plastin-3 | 1.15 × 10−2 | 0.40 |
| MYH14 | Myosin-14 | 4.80 × 10−2 | 0.37 |
| AMPL | Cytosol aminopeptidase | 3.78 × 10−2 | 0.36 |
| AK1A1 | Alcohol dehydrogenase (NADP(+)) | 3.53 × 10−2 | 0.35 |
| ADH1G | Alcohol dehydrogenase 1C | 2.64 × 10−3 | 0.35 |
| PRDX5 | Peroxiredoxin-5 mitochondrial | 4.71 × 10−2 | 0.35 |
| RINI | Ribonuclease inhibitor | 2.21 × 10−2 | 0.34 |
| VATA | V-type proton ATPase catalytic subunit A | 1.49 × 10−4 | 0.25 |
| Entry Name | Protein Name | Peptides | Unique Peptides | p-Value | Fold Change | AUC % |
|---|---|---|---|---|---|---|
| LMNA | Prelamin-A/C | 25 | 24 | 4.00 × 10−2 | 2.25 | 67.1 |
| CATD | Cathepsin D | 19 | 19 | 1.48 × 10−3 | 1.82 | 75.4 |
| ASAH1 | Acid ceramidase | 6 | 6 | 2.98 × 10−3 | 1.80 | 72.3 |
| TERA | Transitional endoplasmic reticulum ATPase | 18 | 18 | 1.99 × 10−2 | 1.60 | 65 |
| DYHC1 | Cytoplasmic dynein 1 heavy chain 1 | 9 | 9 | 7.50 × 10−3 | 1.32 | 70.2 |
| TPP1 | Tripeptidyl-peptidase 1 | 11 | 11 | 4.13 × 10−2 | 0.64 | 69 |
| Unique Variable Used in Model | Estimate | p-Value |
|---|---|---|
| Sex | 1.163 | 0.072 |
| Age | 0.088 | 0.00 * |
| LMNA | 0.351 | 0.055 |
| CATD | 1.426 | 0.010 * |
| ASAH1 | 0.990 | 0.020 * |
| TERA | 0.801 | 0.040 * |
| DYHC1 | 1.197 | 0.085 |
| TPP1 | −0.527 | 0.147 |
| Time with disease | 9.483 | 0.995 |
| UPDRS score | 488.104 | 0.995 |
| Part I UPDRS score | 311.136 | 0.994 |
| Part II UPDRS score | 964.522 | 0.995 |
| Part III UPDRS score | 313.201 | 0.995 |
| Part IV UPDRS score | 398.234 | 0.995 |
| HY Score | 20.013 | 0.995 |
| Multivariate Model Variables | Estimate | p-Value |
|---|---|---|
| (Intercept) | −50.576 | 0.053 |
| Age | 0.166 | 0.040 * |
| LMNA | 0.495 | 0.181 |
| CATD | 1.670 | 0.040 * |
| ASAH1 | 0.141 | 0.912 |
| TERA | 0.470 | 0.655 |
| DYHC1 | 0.798 | 0.602 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acera, A.; Gómez-Esteban, J.C.; Murueta-Goyena, A.; Galdos, M.; Azkargorta, M.; Elortza, F.; Ruzafa, N.; Ibarrondo, O.; Pereiro, X.; Vecino, E. Potential Tear Biomarkers for the Diagnosis of Parkinson’s Disease—A Pilot Study. Proteomes 2022, 10, 4. https://doi.org/10.3390/proteomes10010004
Acera A, Gómez-Esteban JC, Murueta-Goyena A, Galdos M, Azkargorta M, Elortza F, Ruzafa N, Ibarrondo O, Pereiro X, Vecino E. Potential Tear Biomarkers for the Diagnosis of Parkinson’s Disease—A Pilot Study. Proteomes. 2022; 10(1):4. https://doi.org/10.3390/proteomes10010004
Chicago/Turabian StyleAcera, Arantxa, Juan Carlos Gómez-Esteban, Ane Murueta-Goyena, Marta Galdos, Mikel Azkargorta, Felix Elortza, Noelia Ruzafa, Oliver Ibarrondo, Xandra Pereiro, and Elena Vecino. 2022. "Potential Tear Biomarkers for the Diagnosis of Parkinson’s Disease—A Pilot Study" Proteomes 10, no. 1: 4. https://doi.org/10.3390/proteomes10010004