
Critical Thickness of Free-Standing Nanothin Films Made of Melted Polyethylene Chains via Molecular Dynamics

 , and
, and
Abstract
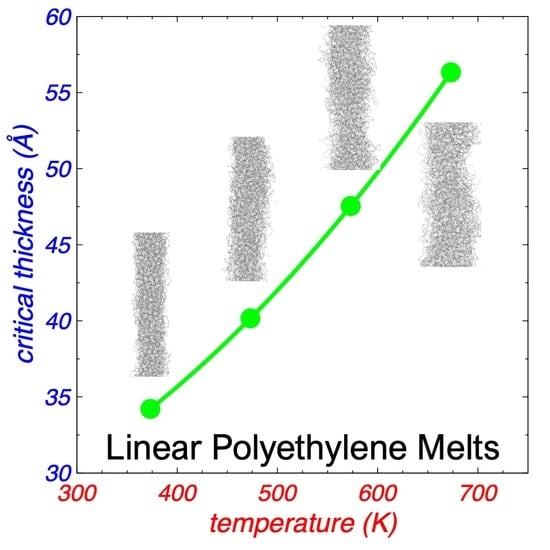
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methodology
3. Results
3.1. Density Profiles
3.2. Bulk Liquid Density
3.3. Radius of Gyration at the Interfaces
3.4. Critical Thickness
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robeson, L.M. Polymer membranes for gas separation. Curr. Opin. Solid State Mater. Sci. 1999, 4, 549–552. [Google Scholar] [CrossRef]
- Vinogradov, N.E.; Kagramanov, G.G. The development of polymer membranes and modules for air separation. J. Phys. Conf. Ser. 2016, 751, 12038. [Google Scholar] [CrossRef]
- Selyanchyn, R.; Fujikawa, S. Membrane thinning for efficient CO2 capture. Sci. Technol. Adv. Mater. 2017, 18, 816–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, S.A.; Mullhaupt, J.T.; Gareis, P.J. The effect of pressure on the permeation of gases and vapors through polyethylene. Usefulness of the corresponding states principle. AIChE J. 1969, 15, 64–73. [Google Scholar] [CrossRef]
- Wijmans, J.G.; Baker, R.W. The solution-diffusion model: A review. J. Memb. Sci. 1995, 107, 1–21. [Google Scholar] [CrossRef]
- Alqaheem, Y.; Alomair, A.; Vinoba, M.; Pérez, A. Polymeric Gas-Separation Membranes for Petroleum Refining. Int. J. Polym. Sci. 2017, 2017, 4250927. [Google Scholar] [CrossRef]
- Angarska, J.K.; Dimitrova, B.S.; Danov, K.D.; Kralchevsky, P.A.; Ananthapadmanabhan, K.P.; Lips, A. Detection of the Hydrophobic Surface Force in Foam Films by Measurements of the Critical Thickness of the Film Rupture. Langmuir 2004, 20, 1799–1806. [Google Scholar] [CrossRef]
- Rivera, J.L.; Douglas, J.F. Influence of film thickness on the stability of free-standing Lennard-Jones fluid films. J. Chem. Phys. 2019, 150, 144705. [Google Scholar] [CrossRef]
- Agboola, O.; Popoola, P.; Sadiku, R.; Sanni, S.E.; Babatunde, D.E.; Ayoola, A.; Abatan, O.G. Fabrication and Potential Applications of Nanoporous Membranes for Separation Processes BT—Environmental Nanotechnology. In Nanoscience in Food and Agriculture 2; Dasgupta, N., Ranjan, S., Lichtfouse, E., Mishra, B.N., Eds.; Springer: Cham, Switzerland, 2021; pp. 119–171. ISBN 978-3-030-73010-9. [Google Scholar]
- De Vries, A.J. Foam stability: Part IV. Kinetics and activation energy of film rupture. Recl. Trav. Chim. Pays-Bas 1958, 77, 383–399. [Google Scholar] [CrossRef]
- Vrij, A. Possible mechanism for the spontaneous rupture of thin, free liquid films. Discuss. Faraday Soc. 1966, 42, 23–33. [Google Scholar] [CrossRef]
- Ivanov, I.B.; Dimitrov, D.S. Hydrodynamics of thin liquid films. Colloid Polym. Sci. 1974, 252, 982–990. [Google Scholar] [CrossRef]
- Ivanov, I.B. Effect of surface mobility on the dynamic behavior of thin liquid films. Pure Appl. Chem. 1980, 52, 1241. [Google Scholar] [CrossRef]
- Rivera, J.L.; Douglas, J.F. Reducing uncertainty in simulation estimates of the surface tension through a two-scale finite-size analysis: Thicker is better. RSC Adv. 2019, 9, 35803–35812. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Jang, J.; Zin, W.-C. Thickness Dependence of the Glass Transition Temperature in Thin Polymer Films. Langmuir 2001, 17, 2703–2710. [Google Scholar] [CrossRef]
- Fryer, D.S.; Nealey, P.F.; de Pablo, J.J. Thermal Probe Measurements of the Glass Transition Temperature for Ultrathin Polymer Films as a Function of Thickness. Macromolecules 2000, 33, 6439–6447. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sanyal, M.K.; Geue, T.; Pietsch, U. Glass transition in ultrathin polymer films: A thermal expansion study. Phys. Rev. E 2005, 71, 41801. [Google Scholar] [CrossRef] [Green Version]
- Inoue, R.; Kanaya, T.; Miyazaki, T.; Nishida, K.; Tsukushi, I.; Shibata, K. Glass transition and thermal expansivity of polystyrene thin films. Mater. Sci. Eng. A 2006, 442, 367–370. [Google Scholar] [CrossRef]
- Wang, Y.; Rafailovich, M.; Sokolov, J.; Gersappe, D.; Araki, T.; Zou, Y.; Kilcoyne, A.D.L.; Ade, H.; Marom, G.; Lustiger, A. Substrate Effect on the Melting Temperature of Thin Polyethylene Films. Phys. Rev. Lett. 2006, 96, 28303. [Google Scholar] [CrossRef]
- Mohammadi, H.; Vincent, M.; Marand, H. Investigating the equilibrium melting temperature of linear polyethylene using the non-linear Hoffman-Weeks approach. Polymer 2018, 146, 344–360. [Google Scholar] [CrossRef]
- Rivera, J.L.; Molina-Rodríguez, L.; Ramos-Estrada, M.; Navarro-Santos, P.; Lima, E. Interfacial properties of the ionic liquid [bmim][triflate] over a wide range of temperatures. RSC Adv. 2018, 8, 10115–10123. [Google Scholar] [CrossRef] [Green Version]
- Arroyo-Valdez, J.A.; Viramontes-Gamboa, G.; Guerra-Gonzalez, R.; Ramos-Estrada, M.; Lima, E.; Rivera, J.L. Cation folding and the thermal stability limit of the ionic liquid [BMIM+][BF4−] under total vacuum. RSC Adv. 2021, 11, 12951–12960. [Google Scholar] [CrossRef]
- McKechnie, D.; Cree, J.; Wadkin-Snaith, D.; Johnston, K. Glass transition temperature of a polymer thin film: Statistical and fitting uncertainties. Polymer 2020, 195, 122433. [Google Scholar] [CrossRef]
- Reiter, G.; Kindl, P. Positron lifetime investigations on linear polyethylene compared to branched polyethylene. Phys. Status Solidi 1990, 118, 161–168. [Google Scholar] [CrossRef]
- Sustaita-Rodríguez, J.M.; Medellín-Rodríguez, F.J.; Olvera-Mendez, D.C.; Gimenez, A.J.; Luna-Barcenas, G. Thermal Stability and Early Degradation Mechanisms of High-Density Polyethylene, Polyamide 6 (Nylon 6), and Polyethylene Terephthalate. Polym. Eng. Sci. 2019, 59, 2016–2023. [Google Scholar] [CrossRef]
- Zong, R.; Wang, Z.; Liu, N.; Hu, Y.; Liao, G. Thermal degradation kinetics of polyethylene and silane-crosslinked polyethylene. J. Appl. Polym. Sci. 2005, 98, 1172–1179. [Google Scholar] [CrossRef]
- Noseé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Trokhymchuk, A.; Alejandre, J. Computer simulations of liquid/vapor interface in Lennard-Jones fluids: Some questions and answers. J. Chem. Phys. 1999, 111, 8510–8523. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D.; Swenson, C.J. Optimized intermolecular potential functions for liquid hydrocarbons. J. Am. Chem. Soc. 1984, 106, 6638–6646. [Google Scholar] [CrossRef]
- Hagita, K.; Fujiwara, S.; Iwaoka, N. Structure formation of a quenched single polyethylene chain with different force fields in united atom molecular dynamics simulations. AIP Adv. 2018, 8, 115108. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Locker, C.R.; in’t Veld, P.J.; Rutledge, G.C. Effect of Short Chain Branching on the Interlamellar Structure of Semicrystalline Polyethylene. Macromolecules 2017, 50, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.Z.; Nishimura, R.; Yasuoka, K.; Masubuchi, Y. Molecular Dynamics Simulations for Resolving Scaling Laws of Polyethylene Melts. Polymers 2017, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Moyassari, A.; Gkourmpis, T.; Hedenqvist, M.S.; Gedde, U.W. Molecular dynamics simulation of linear polyethylene blends: Effect of molar mass bimodality on topological characteristics and mechanical behavior. Polymer 2019, 161, 139–150. [Google Scholar] [CrossRef]
- Ramos, J.; Vega, J.F.; Martínez-Salazar, J. Molecular Dynamics Simulations for the Description of Experimental Molecular Conformation, Melt Dynamics, and Phase Transitions in Polyethylene. Macromolecules 2015, 48, 5016–5027. [Google Scholar] [CrossRef]
- Juárez-Guerra, F.M.; Rivera, J.L.; Zúñiga-Moreno, A.; Galicia-Luna, L.A.; Rico, J.L.; Lara, J. Molecular modeling of thiophene in the vapor-liquid equilibrium. Sep. Sci. Technol. 2006, 41, 261–281. [Google Scholar] [CrossRef]
- Rivera, J.L.J.L.; Nicanor-Guzman, H.; Guerra-Gonzalez, R. The Intramolecular Pressure and the Extension of the Critical Point’s Influence Zone on the Order Parameter. Adv. Condens. Matter Phys. 2015, 2015, 258601. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Sun, C.; Bai, B.; Gatapova, E.Y.; Kabov, O.A. Ionic hydration-induced evolution of decane–water interfacial tension. Phys. Chem. Chem. Phys. 2017, 19, 14606–14614. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, N.; Narayanan Nair, A.K.; Che Ruslan, M.F.A.; Sun, S. Bulk and interfacial properties of decane in the presence of carbon dioxide, methane, and their mixture. Sci. Rep. 2019, 9, 19784. [Google Scholar] [CrossRef] [Green Version]
- Katiyar, P.; Singh, J.K. The effect of ionisation of silica nanoparticles on their binding to nonionic surfactants in oil–water system: An atomistic molecular dynamic study. Mol. Phys. 2018, 116, 2022–2031. [Google Scholar] [CrossRef]
- Sato, Y.; Hashiguchi, H.; Inohara, K.; Takishima, S.; Masuoka, H. PVT properties of polyethylene copolymer melts. Fluid Phase Equilib. 2007, 257, 124–130. [Google Scholar] [CrossRef]
- Zhang, W.; Larson, R.G. A metastable nematic precursor accelerates polyethylene oligomer crystallization as determined by atomistic simulations and self-consistent field theory. J. Chem. Phys. 2019, 150, 244903. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Z.; Hao, T.; Ye, X.; Ne, Y. Temperature Dependence of Structural Properties and Chain Configurational Study: A Molecular Dynamics Simulation of Polyethylene Chains. Macromol. Theory Simul. 2015, 24, 335–343. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jang, K.-L.; Ahn, K.; Yoon, T.; Lee, T.-I.; Kim, T.-S. Thermal expansion behavior of thin films expanding freely on water surface. Sci. Rep. 2019, 9, 7071. [Google Scholar] [CrossRef] [Green Version]
- Hanakata, P.Z.; Douglas, J.F.; Starr, F.W. Local variation of fragility and glass transition temperature of ultra-thin supported polymer films. J. Chem. Phys. 2012, 137, 244901. [Google Scholar] [CrossRef] [Green Version]
- Sheludko, A. Thin liquid films. Adv. Colloid Interface Sci. 1967, 1, 391–464. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Jain, R.K. Spontaneous rupture of thin liquid films. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1974, 70, 132–147. [Google Scholar] [CrossRef]
- Morariu, M.D.; Schäffer, E.; Steiner, U. Capillary instabilities by fluctuation induced forces. Eur. Phys. J. E 2003, 12, 375–381. [Google Scholar] [CrossRef]
- Reiter, G.; Sharma, A.; Casoli, A.; David, M.-O.; Khanna, R.; Auroy, P. Thin Film Instability Induced by Long-Range Forces. Langmuir 1999, 15, 2551–2558. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Mijangos, J.A.; Lima, E.; Guerra-González, R.; Ramírez-Zavaleta, F.I.; Rivera, J.L. Critical Thickness of Free-Standing Nanothin Films Made of Melted Polyethylene Chains via Molecular Dynamics. Polymers 2021, 13, 3515. https://doi.org/10.3390/polym13203515
González-Mijangos JA, Lima E, Guerra-González R, Ramírez-Zavaleta FI, Rivera JL. Critical Thickness of Free-Standing Nanothin Films Made of Melted Polyethylene Chains via Molecular Dynamics. Polymers. 2021; 13(20):3515. https://doi.org/10.3390/polym13203515
Chicago/Turabian StyleGonzález-Mijangos, José Antonio, Enrique Lima, Roberto Guerra-González, Fernando Iguazú Ramírez-Zavaleta, and José Luis Rivera. 2021. "Critical Thickness of Free-Standing Nanothin Films Made of Melted Polyethylene Chains via Molecular Dynamics" Polymers 13, no. 20: 3515. https://doi.org/10.3390/polym13203515