
Improving Recycled Poly(lactic Acid) Biopolymer Properties by Chain Extension Using Block Copolymers Synthesized by Nitroxide-Mediated Polymerization (NMP)


 and
and
Abstract
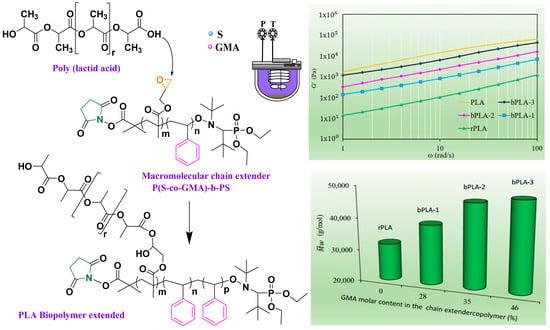
:
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Polymers
2.2. Synthesis of PLA
2.3. Synthesis of Poly(S-co-GMA) Copolymers by NMP
Chain Extension Experiments
2.4. Characterization of P(S-co-GMA) and P(S-co-GMA)-b-PS Copolymers
2.5. Extrusion Processing
2.6. Intrinsic Viscosity, Viscosity Average Molecular Weight and Melt Flow Rate Measurements
2.7. Rheological Analyses
2.8. Thermal Analyses
3. Results and Discussion
3.1. Synthesis and Characterization of Reactive Copolymers
3.2. Reactive Processing of rPLA/P(S-co-GMA)-b-PS Blends
3.3. Intrinsic Viscosity (IV) Measurements
3.4. Thermal Analyses
3.5. Effect of the Incorporation of Chain Extenders on rPLA Rheological Behavior
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castro-Aguirre, E.; Iñiguez-Franco, F.; Samsudin, H.; Fang, X.; Auras, R. Poly (lactic acid)—Mass production, processing, industrial applications, and end of life. Adv. Drug Deliv. Rev. 2016, 107, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Balla, E.; Daniilidis, V.; Karlioti, G.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Vlachopoulos, A.; Koumentakou, I.; Bikiaris, D.N. Poly (lactic Acid): A Versatile Biobased Polymer for the Future with Multifunctional Properties—From Monomer Synthesis, Polymerization Techniques and Molecular Weight Increase to PLA Applications. Polymers 2021, 13, 1822. [Google Scholar] [CrossRef]
- Beltrán, F.R.; Barrio, I.; Lorenzo, V.; del Río, B.; Martínez Urreaga, J.; de la Orden, M.U. Valorization of poly (lactic acid) wastes via mechanical recycling: Improvement of the properties of the recycled polymer. Waste Manag. Res. 2019, 37, 135–141. [Google Scholar] [CrossRef]
- Li, J.; Ren, J.; Cao, Y.; Yuan, W. Synthesis of biodegradable penta-armed star-block copolymers via an asymmetric BIS-TRIS core by combination of ROP and RAFT: From star architectures to double responsive micelles. Polymer 2010, 51, 1301–1310. [Google Scholar] [CrossRef]
- Yu, L.; Dean, K.; Li, L. Polymer Blends and Composites from Renewable Resources. Prog. Polym. Sci. 2006, 36, 576–602. [Google Scholar] [CrossRef]
- Williams, C.K.; Hillmyer, M.A. Polymers from Renewable Resources: A Perspective for a Special Issue of Polymer Reviews. Polym. Rev. 2008, 48, 1–10. [Google Scholar] [CrossRef]
- Lim, L.T.; Auras, R.; Rubino, M. Processing Technologies for Poly (Lactic Acid). Prog. Polym. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S.; Hernandez, R. Mechanical, physical, and barrier properties of poly(lactide) films. J. Plast. Film. Sheeting 2003, 19, 123–135. [Google Scholar] [CrossRef]
- Auras, R.; Lim, L.; Selke, S.E. Poly (Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 457–467. [Google Scholar]
- Signori, F.; Coltelli, M.B.; Bronco, S. Thermal degradation of poly (lactic acid) (PLA) and poly (butylenes adipate-co-terephthalate) (PBAT) and their blends upon melt processing. Polym. Degrad. Stab. 2009, 94, 74–82. [Google Scholar] [CrossRef]
- Julien, J.M.; Benezet, J.C.; Lafranche, E.; Quantin, J.C.; Bergeret, A.; Lacrampe, M.F.; Krawczak, P. Development of poly (lactic acid) cellular materials: Physical and morphological characterizations. Polymer 2012, 53, 5885–5895. [Google Scholar] [CrossRef]
- Luong, N.D.; Moon, I.S.; Lee, D. Surface Modification of Poly (L-Lactide) Electrospun Fibers with Nanocrystal Hydroxyapatite for Engineered Scaffold Applications. Mat. Sci. Eng. C 2008, 28, 242–249. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramot, Y.; Haim-Zada, M.; Domb, A.J.; Nyska, A. Biocompatibility and safety of PLA and its copolymers. Adv. Drug Deliv. Rev. 2016, 107, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Tavanaie, M.A. Melt Recycling of Poly (lactic Acid) Plastic Wastes to Produce Biodegradable Fibers. Polym. Plast. Technol. Eng. 2014, 53, 742–751. [Google Scholar] [CrossRef]
- De Andrade, M.F.C.; Souza, P.M.; Cavalett, O. Life cycle assessment of poly (lactic acid) (PLA): Comparison between chemical recycling, mechanical recycling and composting. J. Polym. Environ. 2016, 24, 372–384. [Google Scholar] [CrossRef]
- Piemonte, V. Bioplastic Wastes: The Best Final Disposition for Energy Saving. J. Polym. Environ. 2011, 19, 988–994. [Google Scholar] [CrossRef]
- Kopinke, F.D.; Mackenzie, K. Mechanistic aspects of the thermal degradation of poly(lactic acid) and poly (b-hydroxybutyric acid). J. Anal. Appl. Pyrol. 1997, 40, 43–53. [Google Scholar] [CrossRef]
- Meng, Q.; Heuzey, M.C.; Carreau, P.J. Control of thermal degradation of polylactide/clay nanocomposites during melt processing by chain extension reaction. Polym. Degrad. Stab. 2012, 97, 2010–2020. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.S.; Maazouz, A. Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym. Degrad. Stab. 2012, 97, 1898–1914. [Google Scholar] [CrossRef]
- Raffa, P.; Coltelli, M.B.; Savi, S.; Bianchi, S.; Castelvetro, V. Chain extension and branching of poly (ethylene terephthalate) (PET) with di- and multifunctional epoxy or isocyanate additives: An experimental and modelling study. React. Funct. Polym. 2012, 72, 50–60. [Google Scholar] [CrossRef]
- Li, B.H.; Yang, M.C. Improvement of thermal and mechanical properties of poly (L-lactic acid) with 4,4-methylene diphenyl diisocyanate. Polym. Adv. Technol. 2006, 17, 439–443. [Google Scholar] [CrossRef]
- Meng, X.; Shi, G.; Chen, W.; Wu, C.; Xin, Z.; Han, T.; Shi, Y. Structure effect of phosphite on the chain extension in PLA. Polym. Degrad. Stab. 2015, 120, 283–289. [Google Scholar] [CrossRef]
- Duarte, I.S.; Tavares, A.A.; Lima, P.S.; Andrade, D.L.; Carvalho, L.H.; Canedo, E.L.; Silva, S.M. Chain extension of virgin and recycled poly (ethylene terephthalate): Effect of processing conditions and reprocessing. Polym. Degrad. Stab. 2016, 124, 26–34. [Google Scholar] [CrossRef]
- Kim, Y.S.; Hwang, K.A.; Hyun, S.H.; Nam, K.H.; Lee, C.K.; Choi, K.C. Bisphenol A and Nonylphenol Have the Potential to Stimulate the Migration of Ovarian Cancer Cells by Inducing Epithelial–Mesenchymal Transition via an Estrogen Receptor Dependent Pathway. Chem. Res. Toxicol. 2015, 28, 662–671. [Google Scholar] [CrossRef]
- Awaja, F.; Daver, F.; Kosior, E.N. Recycled poly (ethylene terephthalate) chain extension by a reactive extrusion process. Polym. Eng. Sci. 2004, 44, 1579–1587. [Google Scholar] [CrossRef]
- Awaja, F.; Pavel, D. Recycling of PET. Eur. Polym. J. 2005, 41, 1453–1477. [Google Scholar] [CrossRef]
- Villalobos, M.; Awojulu, A.; Greeley, T.; Turco, G.; Deeter, G. Oligomeric chain extenders for economic reprocessing and recycling of condensation plastics. Energy 2006, 31, 3227–3234. [Google Scholar] [CrossRef]
- Karayannidis, G.P.; Psalida, E.A. Chain extension of recycled poly (ethylene terephthalate) with 2,2′-(1,4-phenylene)bis(2-oxazoline). J. Appl. Polym. Sci. 2000, 77, 2206–2211. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J.; Wood-Adams, P.M. Control of thermal degradation of polylactide (PLA)-clay nanocomposites using chain extenders. Polym. Degrad. Stab. 2012, 97, 554–655. [Google Scholar] [CrossRef]
- Snowdon, M.R.; Abdelwahab, M.; Mohanty, A.K.; Misra, M. Mechanical optimization of virgin and recycled poly (ethylene terephthalate) biocomposites with sustainable biocarbon through a factorial design. Results Mater. 2000, 5, 100060. [Google Scholar] [CrossRef]
- Pillin, I.; Montrelay, N.; Bourmaud, A.; Grohens, Y. Effect of thermo-mechanical cycles on the physico-chemical properties of poly (lactic acid). Polym. Degrad. Stab. 2008, 93, 321–328. [Google Scholar] [CrossRef]
- Mallet, B.; Lamnawar, K.; Maazouz, A. Improvement of blown film extrusion, of poly (lactic acid): Structure–processing–properties relationships. Polym. Eng. Sci. 2014, 54, 840–857. [Google Scholar] [CrossRef]
- Jaszkiewicz, A.; Bledzki, A.K.; van der Meer, R.; Franciszczak, P.; Meljon, A. How does a chain-extended polylactide behave?: A comprehensive analysis of the material, structural and mechanical properties. Polym. Bull. 2014, 71, 1675–1690. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Nagarajan, V.; Misra, M.; Mohanty, A.K. Supertoughened Renewable PLA Reactive Multiphase Blends System: Phase Morphology and Performance. ACS Appl. Mater. Interfaces 2014, 6, 12436–12448. [Google Scholar] [CrossRef]
- Chen, C.W.; Liu, P.H.; Lin, F.J.; Cho, C.J.; Wang, L.Y.; Mao, H.I.; Chiu, Y.C.; Changm, S.H.; Rwei, S.P.; Kuo, C.C. Influence of Different Molecular Weights and Concentrations of Poly(glycidyl methacrylate) on Recycled Poly(ethylene terephthalate): A Thermal, Mechanical, and Rheological Study. J. Polym. Environ. 2020, 28, 2880–2892. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Barner-Kowollik, C. Handbook of RAFT Polymerization; Wiley-VCH: Weinheim, Germany, 2008; pp. 110–122. [Google Scholar]
- Benvenuta-Tapia, J.J.; Tenorio-López, J.A.; Martínez-Estrada, A.; Guerrero-Sánchez, C. Application of RAFT-synthesized reactive tri-block copolymers for the recycling of post-consumer R-PET by melt processing. Mat. Chem. Phys. 2019, 229, 474–481. [Google Scholar] [CrossRef]
- Benvenuta-Tapia, J.J.; Valdez, M.H.; Cortez, J.C.; García, V.M.D.; Barrios, H.L. Improving the rheological and mechanical properties of recycled PET modified by macromolecular chain extenders synthesized by controlled radical polymerization. J. Polym. Environ. 2018, 26, 4221–4232. [Google Scholar] [CrossRef]
- Rizzardo, E.; Solomon, D.H. On the Origins of Nitroxide Mediated Polymerization (NMP) and Reversible Addition–Fragmentation Chain Transfer (RAFT). Aust. J. Chem. 2012, 65, 945–969. [Google Scholar] [CrossRef] [Green Version]
- Marić, M. History of nitroxide mediated polymerization in Canada. Can. J. Chem. Eng. 2021, 99, 832–852. [Google Scholar] [CrossRef]
- Benoit, D.; Grimaldi, S.; Robin, S.; Finet, J.P.; Tordo, P.; Gnanou, Y. Kinetics and Mechanism of Controlled Free-Radical Polymerization of Styrene and n-Butyl Acrylate in the Presence of an Acyclic β-Phosphonylated Nitroxide. J. Am. Chem. Soc. 2000, 122, 5929–5939. [Google Scholar] [CrossRef]
- Grimaldi, S.; Finet, J.P.; Le Moigne, F.O.; Zeghdaoui, A.; Tordo, P.; Benoit, D.; Fontanille, M.; Gnanou, Y. Acyclic β-Phosphonylated Nitroxides: A New Series of Counter-Radicals for “Living”/Controlled Free Radical Polymerization. Macromolecules 2000, 33, 1141–1147. [Google Scholar] [CrossRef]
- Grassl, B.; Clisson, G.; Khoukh, A.; Billon, L. Nitroxide-mediated radical polymerization of acrylamide in water solution. Eur. Polym. J. 2008, 44, 50–58. [Google Scholar] [CrossRef]
- Vinas, J.; Chagneux, N.; Gigmes, D.; Trimaille, T.; Favier, A.; Bertin, D. SG1-based alkoxyamine bearing a N-succinimidyl ester: A versatile tool for advanced polymer synthesis. Polymer 2008, 49, 3639–3647. [Google Scholar] [CrossRef]
- Moayeri, A.; Lessard, B.; Maric, M. Nitroxide mediated controlled synthesis of glycidyl methacrylate-rich copolymers enabled by SG1-based alkoxyamines bearing succinimidyl ester groups. Polym. Chem. 2011, 2, 2084–2092. [Google Scholar] [CrossRef]
- Abdel-Azim, A.A.; AttA, A.M.; Farahat, M.S.; Boutros, W.Y. Determination of intrinsic viscosity of polymeric compounds through a single specific viscosity measurement. Polymer 1998, 39, 6827–6833. [Google Scholar] [CrossRef]
- Schindler, A.; Harper, D. Polylactic II: Viscosity-molecular weight relationships and unperturbed chain dimensions. J. Polym. Sci. Polym. Part A Chem. Ed. 1979, 17, 2593–2599. [Google Scholar] [CrossRef]
- Solomon, O.F.; Ciuta, I.Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]
- Hyon, S.H.; Jamshidi, K.; Ikada, Y. Synthesis of polylactides with different molecular weights. Biomaterials 1997, 16, 1503–1508. [Google Scholar] [CrossRef]
- American Society for Testing and Material. ASTM D1238–13: Standard Test Method for Melt Flow Rates of Thermoplastics by Extrusion Plastometer; ASTM International: West Conshohocken, PA, USA, 2013. [Google Scholar]
- Fisher, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reaction. Kolloid Z. Z. Polym. 1973, 251, 980–991. [Google Scholar] [CrossRef]
- Brar, A.S.; Kumar, A.A.; Goyal, A.K. Characterization and optimization of poly (glycidyl methacrylate-co-styrene) synthesized by atom transfer radical polymerization. Eur. Polym. J. 2008, 44, 4082–4091. [Google Scholar] [CrossRef]
- Darabi, A.; Shirin-Abadi, A.R.; Pinaud, J.; Jessop, P.G.; Cunningham, M.F. Nitroxide-mediated surfactant-free emulsion copolymerization of methyl methacrylate and styrene using poly (2-(diethyl)aminoethyl methacrylate-co-styrene) as a stimuli-responsive macroalkoxyamine. Polym. Chem. 2014, 5, 6163–6170. [Google Scholar] [CrossRef]
- Savelyeva, X.; Metafiot, A.; Li, L.; Bennett, I.; Maric, M. Stimuli-responsive 4-acryloylmorpholine/4-acryloylpiperidine copolymers via nitroxide mediated polymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2160–2170. [Google Scholar] [CrossRef]
- Benvenuta-Tapia, J.J.; Tenorio-López, J.A.; Vivaldo-Lima, E. Estimation of Reactivity Ratios in the RAFT Copolymerization of Styrene and Glycidyl Methacrylate. Macromol. React. Eng. 2018, 12, 1800003. [Google Scholar] [CrossRef]
- Inata, H.; Matsumura, S. Chain extenders for polyesters. I. Addition-type chain extenders reactive with carboxyl end groups of polyesters. J. Appl. Polym. Sci. 1985, 30, 3325–3337. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Japon, S.; Luciani, A.; Leterrier, Y.; Manson, J.A. Molecular characterization and rheological properties of modified poly (ethylene terephthalate) obtained by reactive extrusion. Polym. Eng. Sci. 2001, 41, 1299–1309. [Google Scholar] [CrossRef]
- Dhavalikar, R.; Yamaguchi, M.; Xanthos, M. Molecular and structural analysis of a triepoxide-modified poly (ethylene terephthalate) from rheological data. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 958–969. [Google Scholar] [CrossRef]
- Tuna, B.; Ozkoc, G. Effects of Diisocyanate and Polymeric Epoxidized Chain Extenders on the Properties of Recycled Poly (Lactic Acid). J. Polym. Environ. 2017, 25, 983–993. [Google Scholar] [CrossRef]
- Taubner, V.; Shishoo, R. Influence of processing parameters on the degradation of poly(L-lactide) during extrusion. J. Appl. Polym. Sci. 2001, 79, 2128–2135. [Google Scholar] [CrossRef]
- Beltrán, F.R.; Lorenzo, V.; Acosta, J. Effect of simulated mechanical recycling processes on the structure and properties of poly (lactic acid). J. Environ. Manag. 2018, 216, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Cosate de Andrade, M.F.; Fonseca, G.; Morales, A.R.; Innocentini, L.H. Mechanical recycling simulation of polylactide using a chain extender. Adv. Polym. Technol. 2018, 37, 2053–2060. [Google Scholar] [CrossRef]
- Hung, C.Y.; Wang, C.; Chen, C.Y. Enhanced the thermal stability and crystallinity of polylactic acid (PLA) by incorporated reactive PS-b-PMMA-b-PGMA and PS-bPGMA block copolymers as chain extenders. Polymer 2013, 54, 1860–1866. [Google Scholar] [CrossRef]
- Carvalho, J.L.; Cormier, S.L.; Lin, N.; Dalnokiveress, K. Crystal growth rate in a blend of long and short polymer chains. Macromolecules 2012, 45, 1688–1691. [Google Scholar] [CrossRef]
- Feng, L.; Bian, X.; Li, G.; Chen, Z.; Chen, X. Compatibility, mechanical properties and stability of blends of polylactide and polyurethane based on poly (ethylene glycol)-b-polylactide copolymers by chain extension with diisocyanate. Polym. Degrad. Stab. 2016, 125, 148–155. [Google Scholar] [CrossRef]
- Han, C.D. Rheology and Processing of Polymeric Materials; Oxford University Press: New York, NY, USA, 2007; Volume 1. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expt. ID | [GMA]o mol L−1 | [S]o mol L−1 | [NHS-SG1] mol L−1 | fGMA a | Mn g mol−1 | Mw g mol−1 | Ð |
|---|---|---|---|---|---|---|---|
| P(S-co-GMA)-1 | 2.490 | 5.810 | 0.0320 | 35 | 27,000 | 32,900 | 1.22 |
| P(S-co-GMA)-2 | 3.276 | 4.915 | 0.3130 | 45 | 27,600 | 34,800 | 1.26 |
| P(S-co-GMA)-3 | 4.043 | 4.043 | 0.0326 | 55 | 28,400 | 36,600 | 1.29 |
| Expt. ID | FGMA a mol% | Mn g mol−1 | Mw g mol−1 | Ð |
|---|---|---|---|---|
| P(S-co-GMA)-b-PS-1 | 28 | 35,500 | 46,100 | 1.30 |
| P(S-co-GMA)-b-PS-2 | 35 | 36,300 | 47,900 | 1.32 |
| P(S-co-GMA)-b-PS-3 | 46 | 37,200 | 50,600 | 1.36 |
| Sample | Tg (°C) | Tcc (°C) | Tm (°C) | Xc (%) |
|---|---|---|---|---|
| PLA | 60 | 113 | 153 | 19 |
| rPLA | 60 | 111 | 153 | 21 |
| bPLA−1 | 59 | 108 | 153 | 24 |
| bPLA−2 | 59 | 105 | 152 | 27 |
| bPLA−3 | 59 | 98 | 151 | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benvenuta-Tapia, J.J.; Champagne, P.; Tenorio-López, J.A.; Vivaldo-Lima, E.; Guerrero-Santos, R. Improving Recycled Poly(lactic Acid) Biopolymer Properties by Chain Extension Using Block Copolymers Synthesized by Nitroxide-Mediated Polymerization (NMP). Polymers 2021, 13, 2791. https://doi.org/10.3390/polym13162791
Benvenuta-Tapia JJ, Champagne P, Tenorio-López JA, Vivaldo-Lima E, Guerrero-Santos R. Improving Recycled Poly(lactic Acid) Biopolymer Properties by Chain Extension Using Block Copolymers Synthesized by Nitroxide-Mediated Polymerization (NMP). Polymers. 2021; 13(16):2791. https://doi.org/10.3390/polym13162791
Chicago/Turabian StyleBenvenuta-Tapia, Juan José, Pascale Champagne, José Alfredo Tenorio-López, Eduardo Vivaldo-Lima, and Ramiro Guerrero-Santos. 2021. "Improving Recycled Poly(lactic Acid) Biopolymer Properties by Chain Extension Using Block Copolymers Synthesized by Nitroxide-Mediated Polymerization (NMP)" Polymers 13, no. 16: 2791. https://doi.org/10.3390/polym13162791