Fabrication of Alternating Copolymers Based on Cyclopentadithiophene-Benzothiadiazole Dicarboxylic Imide with Reduced Optical Band Gap: Synthesis, Optical, Electrochemical, Thermal, and Structural Properties

 , ,
, , 
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Measurements
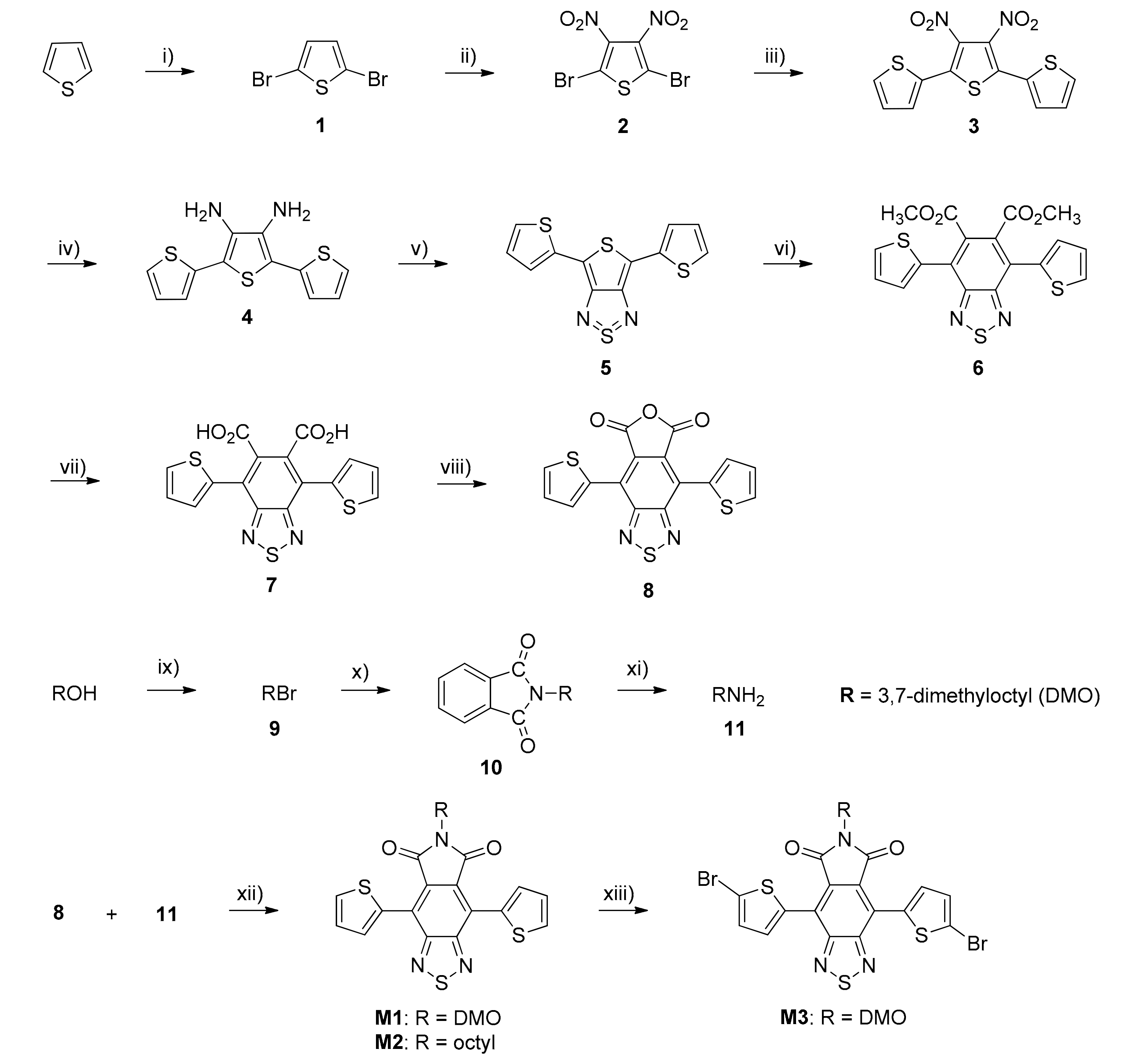
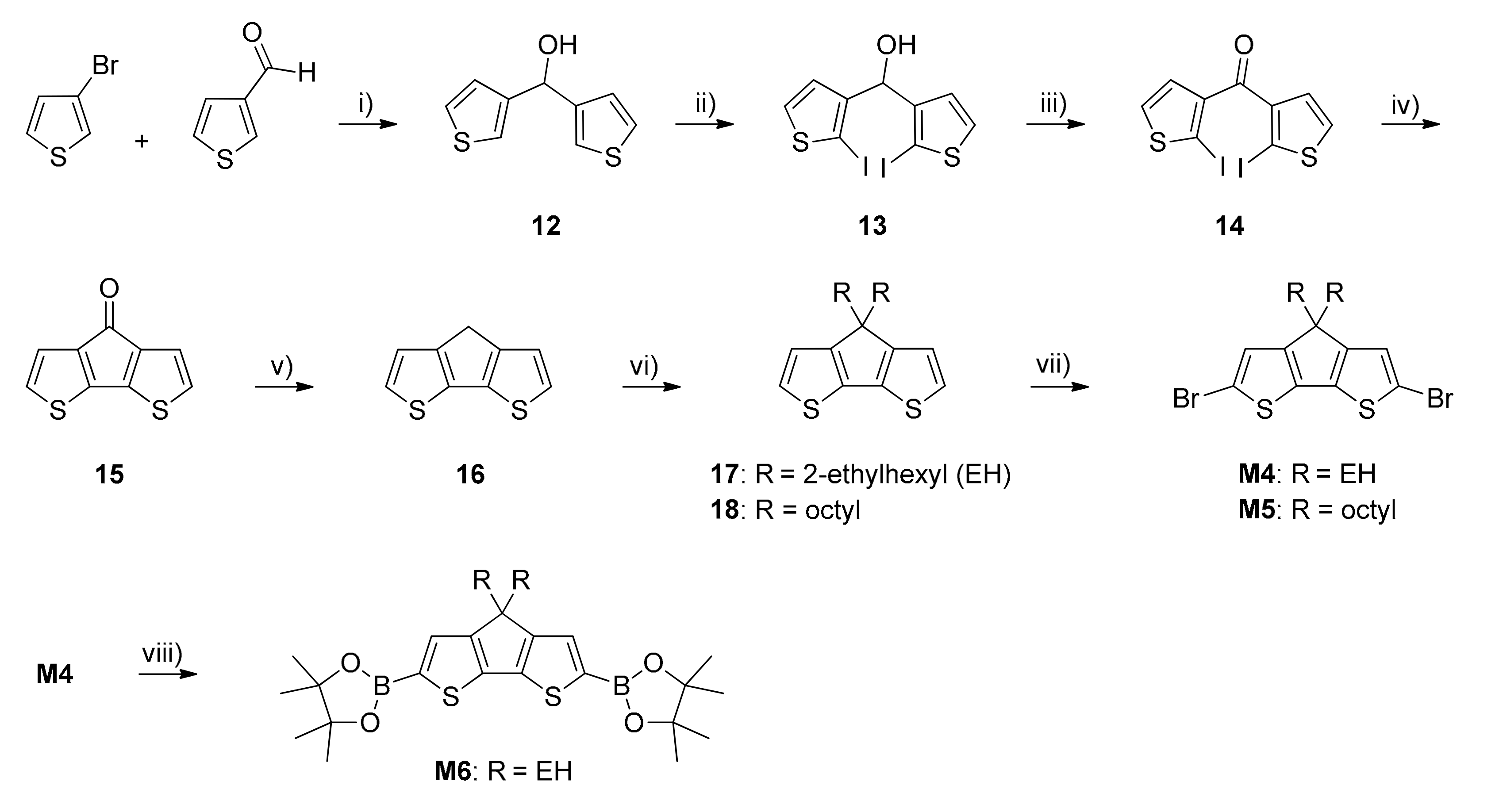
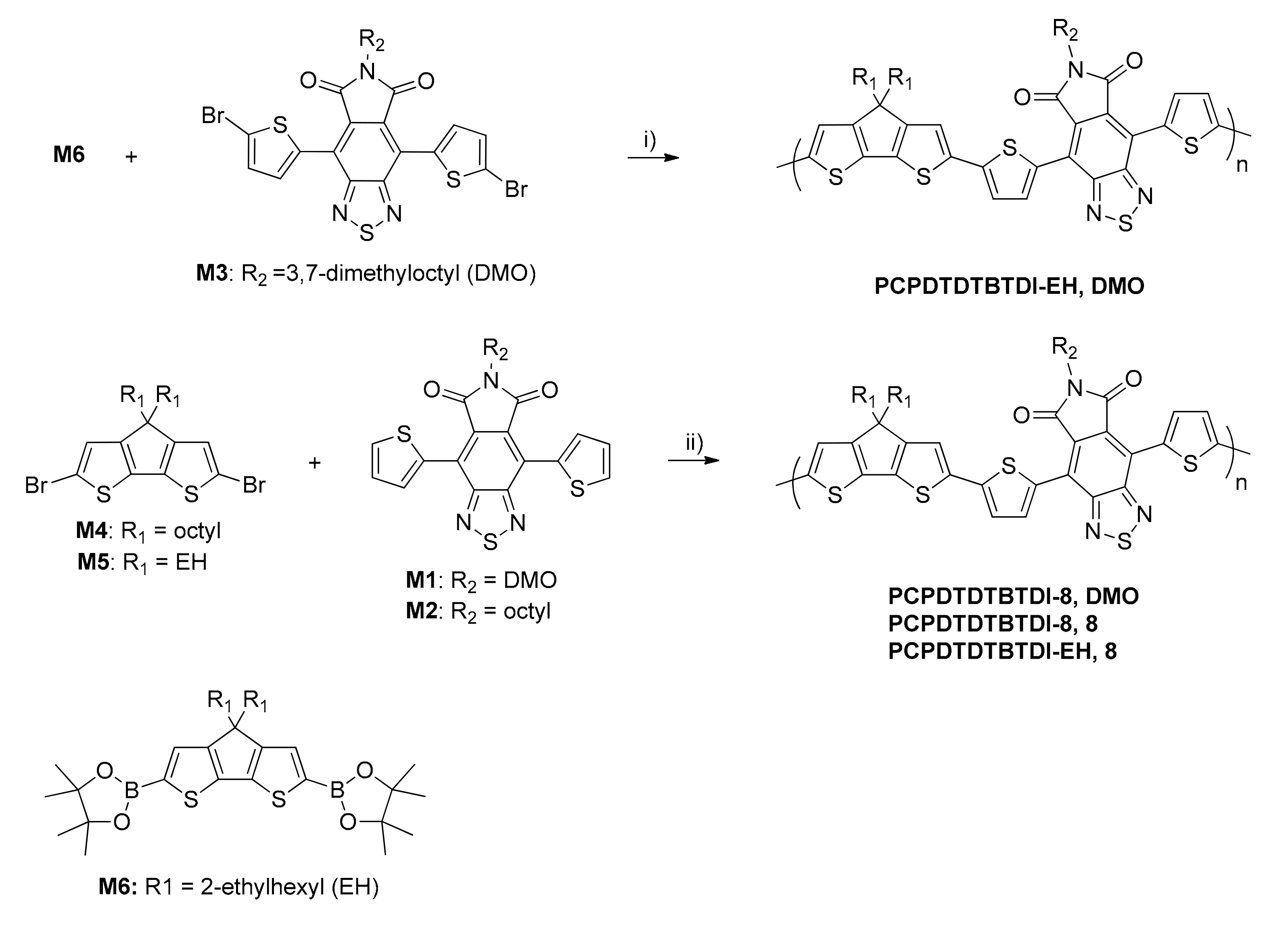
2.3. Monomers and Polymers Synthesis
3. Results and Discussion
3.1. Monomers and Polymers Synthesis
3.2. Polymers Synthesis
3.3. Molecular Weights and Yields
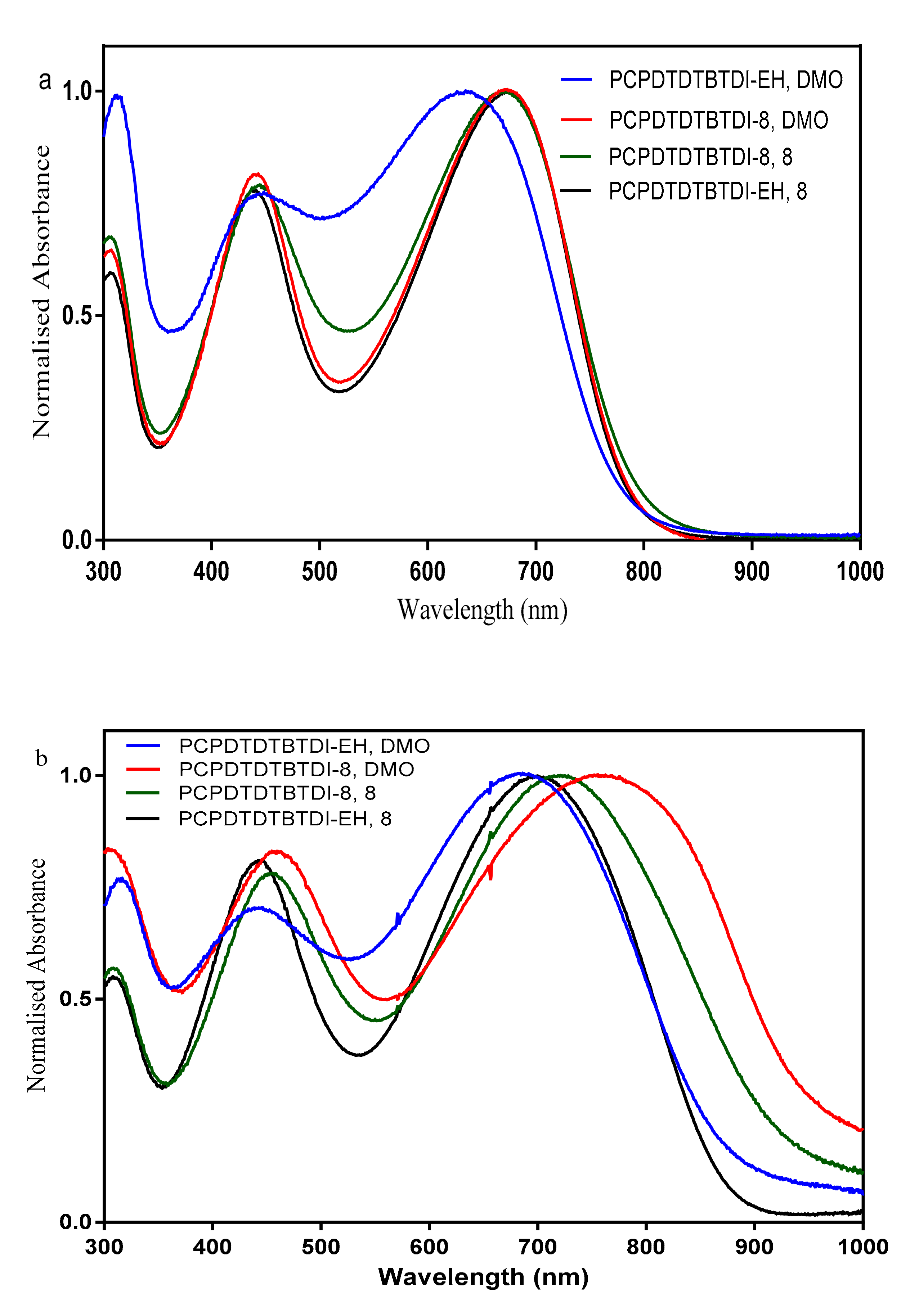
3.4. Optical Properties
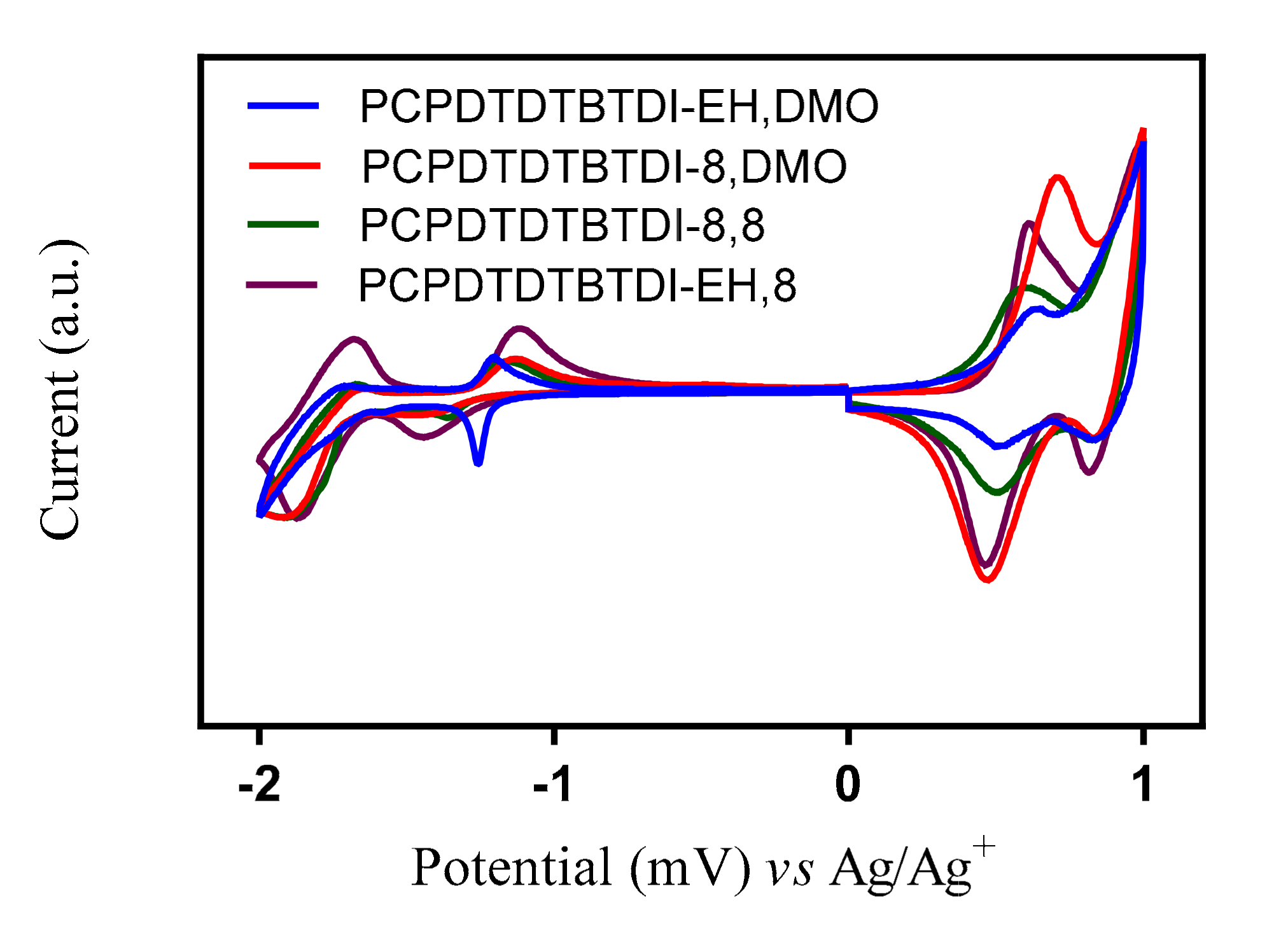
3.5. Electrochemical Properties
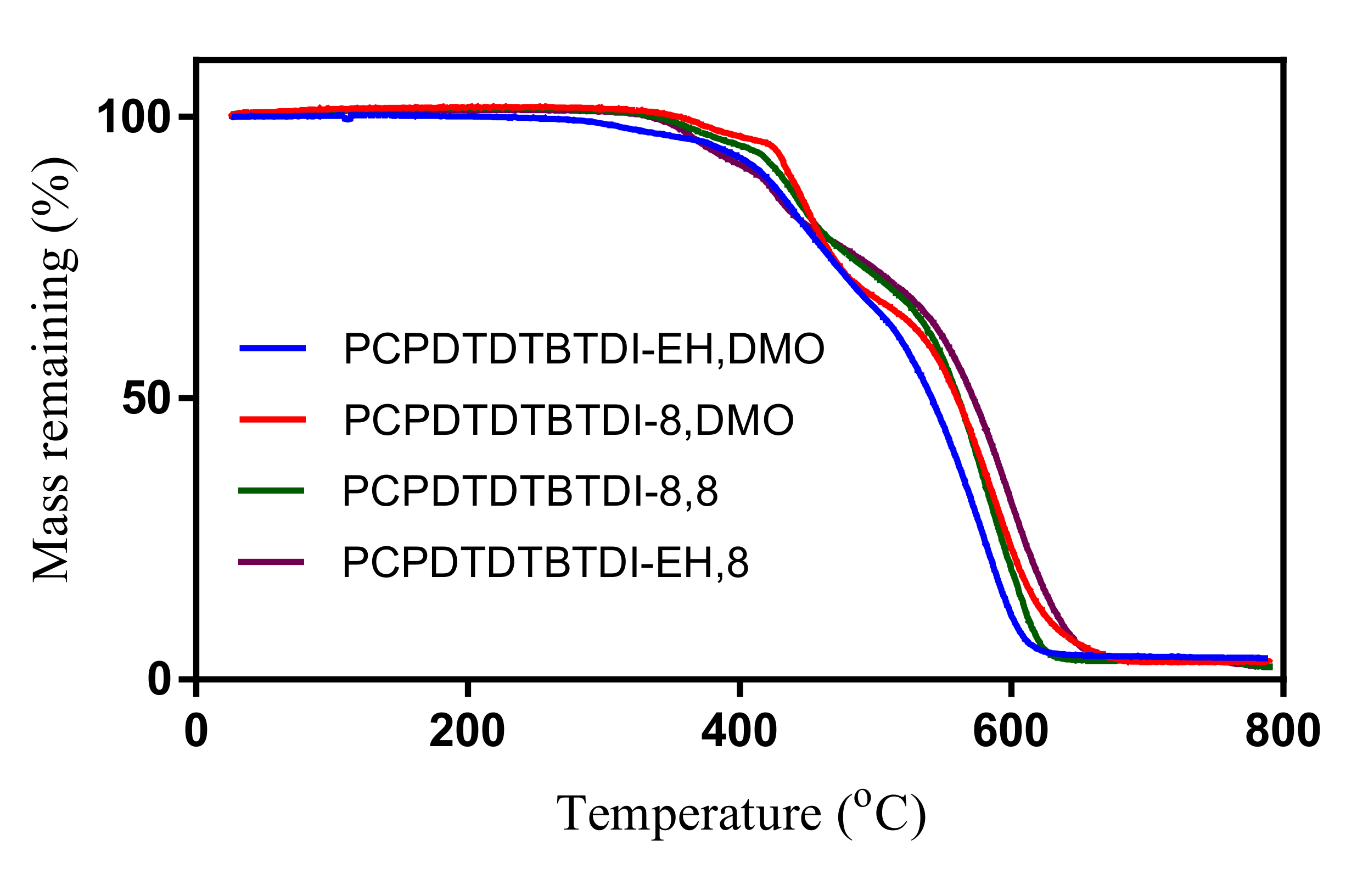
3.6. Thermal Properties
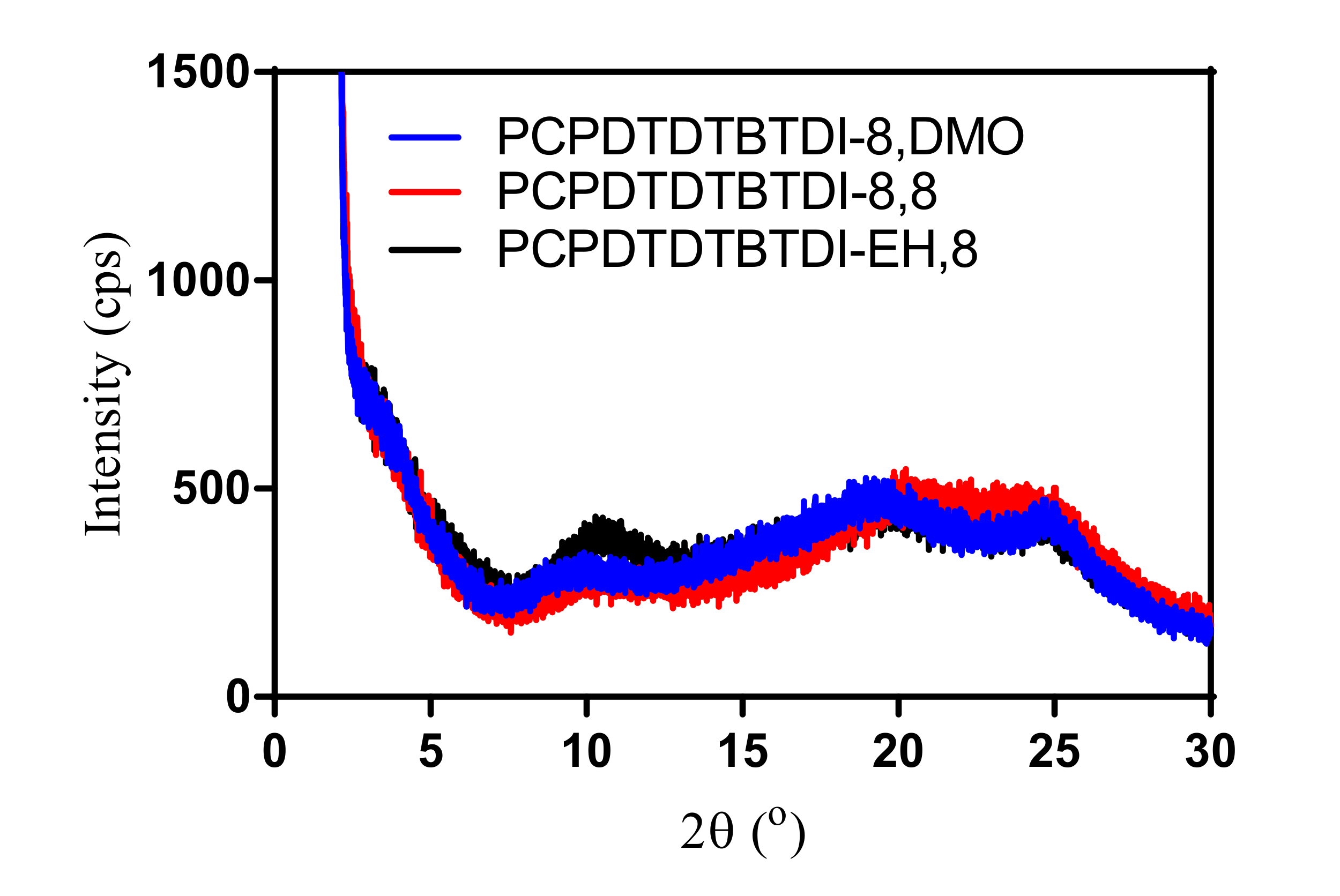
3.7. Powder X-ray Diffraction of the Polymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, Y.D.; Abate, S.Y.; Chung, H.C.; Liau, K.L.; Tao, Y.T.; Chow, T.J.; Sun, S.S. Donor-Acceptor-Donor-Type Cyclopenta[2,1-b;3,4-b′]dithiophene Derivatives as A New Class of Hole Transporting Materials for Highly Efficient and Stable Perovskite Solar Cells. ACS Appl. Energy Mater 2019, 2, 7070–7082. [Google Scholar] [CrossRef]
- Lin, Y.D.; Lee, K.M.; Ke, B.Y.; Chen, K.S.; Cheng, H.C.; Lin, W.J.; Chang, S.H.; Wu, C.G.; Kuo, M.C.; Chung, H.C.; et al. Rational Design of Cyclopenta[2,1-b;3,4-b′]dithiophene-bridged Hole Transporting Materials for Highly Efficient and Stable Perovskite Solar Cells. Energy Technol. 2019. [Google Scholar] [CrossRef]
- Caoa, J.; Liua, S.; Hua, W.; Xua, Y.; Zhoua, W.; Zenga, Y.; Yua, J.; Tanga, Z. Small-molecule acceptors based on 4H-cyclopenta[1,2-b:5,4-b′]dithiophene units with near-infrared absorption for nonfullerene polymer solar cells. Synth. Metals 2018, 240, 15–20. [Google Scholar] [CrossRef]
- Mühlbacher, D.; Scharber, M.; Morana, M.; Zhu, Z.; Waller, D.; Gaudiana, R.; Brabec, C. High photovoltaic performance of a low-bandgap polymer. Adv. Mater. 2006, 18, 2884–2889. [Google Scholar] [CrossRef]
- Zhu, Z.; Waller, D.; Gaudiana, R.; Morana, M.; Mühlbacher, D.; Scharber, M.; Brabec, C. Panchromatic conjugated polymers containing alternating donor/acceptor units for photovoltaic applications. Macromolecules 2007, 40, 1981–1986. [Google Scholar] [CrossRef]
- Soci, C.; Hwang, I.W.; Moses, D.; Zhu, Z.; Waller, D.; Gaudiana, R.; Brabec, C.J.; Heeger, A.J. Photoconductivity of a low-bandgap conjugated polymer. Adv. Funct. Mater. 2007, 17, 632–636. [Google Scholar] [CrossRef]
- Peet, J.; Kim, J.Y.; Coates, N.E.; Ma, W.L.; Moses, D.; Heeger, A.J.; Bazan, G.C. Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat. Mater. 2007, 6, 497–500. [Google Scholar] [CrossRef]
- Lee, J.K.; Ma, W.L.; Brabec, C.J.; Yuen, J.; Moon, J.S.; Kim, J.Y.; Lee, K.; Bazan, G.C.; Heeger, A.J. Processing additives for improved efficiency from bulk heterojunction solar cells. J. Am. Chem. Soc. 2008, 130, 3619–3623. [Google Scholar] [CrossRef]
- Morana, M.; Wegscheider, M.; Bonanni, A.; Kopidakis, N.; Shaheen, S.; Scharber, M.; Zhu, Z.; Waller, D.; Gaudiana, R.; Brabec, C. Bipolar Charge Transport in PCPDTBT-PCBM Bulk-Heterojunctions for Photovoltaic Applications. Adv. Funct. Mater. 2008, 18, 1757–1766. [Google Scholar] [CrossRef]
- Zhang, M.; Tsao, H.N.; Pisula, W.; Yang, C.; Mishra, A.K.; Müllen, K. Field-effect transistors based on a benzothiadiazole-cyclopentadithiophene copolymer. J. Am. Chem. Soc. 2007, 129, 3472–3473. [Google Scholar] [CrossRef]
- Tsao, H.N.; Cho, D.M.; Park, I.; Hansen, M.R.; Mavrinskiy, A.; Yoon, D.Y.; Graf, R.; Pisula, W.; Spiess, H.W.; Muüllen, K. Ultrahigh mobility in polymer field-effect transistors by design. J. Am. Chem. Soc. 2011, 133, 2605–2612. [Google Scholar] [CrossRef]
- Bijleveld, J.C.; Shahid, M.; Gilot, J.; Wienk, M.M.; Janssen, R.A. Copolymers of Cyclopentadithiophene and Electron-Deficient Aromatic Units Designed for Photovoltaic Applications. Adv. Funct. Mater. 2009, 19, 3262–3270. [Google Scholar] [CrossRef]
- Moulé, A.J.; Tsami, A.; Bünnagel, T.W.; Forster, M.; Kronenberg, N.M.; Scharber, M.; Koppe, M.; Morana, M.; Brabec, C.J.; Meerholz, K. Two novel cyclopentadithiophene-based alternating copolymers as potential donor components for high-efficiency bulk-heterojunction-type solar cells. Chem. Mater. 2008, 20, 4045–4050. [Google Scholar] [CrossRef]
- Li, K.-C.; Huang, J.-H.; Hsu, Y.-C.; Huang, P.-J.; Chu, C.-W.; Lin, J.-T.S.; Wei, K.-H.; Lin, H.-C. Tunable novel cyclopentadithiophene-based copolymers containing various numbers of bithiazole and thienyl units for organic photovoltaic cell applications. Macromolecules 2009, 42, 3681–3693. [Google Scholar] [CrossRef]
- Li, Z.; Ding, J.; Song, N.; Lu, J.; Tao, Y. Development of a New s-Tetrazine-Based Copolymer for Efficient Solar Cells §. J. Am. Chem. Soc. 2010, 132, 13160–13161. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ding, J.; Song, N.; Du, X.; Zhou, J.; Lu, J.; Tao, Y. Alternating copolymers of dithienyl-s-tetrazine and cyclopentadithiophene for organic photovoltaic applications. Chem. Mater. 2011, 23, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Xin, H.; Kim, F.S.; Liyanage, A.D.; Jenekhe, S.A.; Watson, M.D. Thieno [3, 4-c] pyrrole-4, 6-dione-Based Donor− Acceptor Conjugated Polymers for Solar Cells. Macromolecules 2010, 44, 269–277. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, J.; Yip, H.-L.; Sun, Y.; Davies, J.A.; Chen, K.-S.; Acton, O.; Jen, A.K.-Y. Conjugated polymers based on C, Si and N-bridged dithiophene and thienopyrroledione units: Synthesis, field-effect transistors and bulk heterojunction polymer solar cells. J. Mater. Chem. 2011, 21, 3895–3902. [Google Scholar] [CrossRef]
- Li, Z.; Tsang, S.W.; Du, X.; Scoles, L.; Robertson, G.; Zhang, Y.; Toll, F.; Tao, Y.; Lu, J.; Ding, J. Alternating Copolymers of Cyclopenta [2, 1-b; 3, 4-b′] dithiophene and Thieno [3, 4-c] pyrrole-4, 6-dione for High-Performance Polymer Solar Cells. Adv. Funct. Mater. 2011, 21, 3331–3336. [Google Scholar] [CrossRef]
- Li, S.; Fan, Z. Encapsulation Methods of Sulfur Particles for Lithium-Sulfur Batteries: A Review. Energy Storage Mater. 2020, 34, 107–127. [Google Scholar] [CrossRef]
- Li, S.; Leng, D.; Li, W.; Qie, L.; Dong, Z.; Cheng, Z.; Fan, Z. Recent Progress in Developing Li2S Cathodes for Li-S Batteries. Energy Storage Mater. 2020, 27, 279–296. [Google Scholar] [CrossRef]
- Ponomarenko, S.; Muzafarov, A.; Borshchev, O.; Vodopyanov, E.; Demchenko, N.; Myakushev, V. Synthesis of bithiophenesilane dendrimer of the first generation. Russ. Chem. Bull. 2005, 54, 684–690. [Google Scholar] [CrossRef]
- Wen, L.; Rasmussen, S.C. Synthesis and structural characterization of 2, 5-dihalo-3, 4-dinitrothiophenes. J. Chem. Crystallogr. 2007, 37, 387–398. [Google Scholar] [CrossRef]
- Schwiderski, R.L.; Rasmussen, S.C. Synthesis and Characterization of Thieno [3, 4-b] pyrazine-Based Terthienyls: Tunable Precursors for Low Band Gap Conjugated Materials. J. Org. Chem. 2013, 78, 5453–5462. [Google Scholar] [CrossRef]
- Hailu, H.; Atsbeha, B.; Admassie, S.; Mammo, W.; Raju, V.; Chebude, Y. Variable denticity of a multidentate terthiophene derivative towards Ni (II) and Zn (II)–structural studies. Bull. Chem. Soc. Ethiop. 2011, 25, 221–231. [Google Scholar] [CrossRef]
- Delgado, M.R.; Hernandez, V.; Navarrete, J.L.; Tanaka, S.; Yamashita, Y. Combined spectroscopic and theoretical study of narrow band gap heterocyclic co-oligomers containing alternating aromatic donor and o-quinoid acceptor units. J. Phys. Chem. B 2004, 108, 2516–2526. [Google Scholar] [CrossRef]
- Wang, L.; Cai, D.; Zheng, Q.; Tang, C.; Chen, S.-C.; Yin, Z. Low Band Gap Polymers Incorporating a Dicarboxylic Imide-Derived Acceptor Moiety for Efficient Polymer Solar Cells. ACS Macro Lett. 2013, 2, 605–608. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Ashraf, R.S.; Treat, N.D.; Schroeder, B.C.; Donaghey, J.E.; White, A.J.; Stingelin, N.; McCulloch, I. 2, 1, 3-Benzothiadiazole-5, 6-Dicarboxylic Imide–A Versatile Building Block for Additive-and Annealing-Free Processing of Organic Solar Cells with Efficiencies Exceeding 8%. Adv. Mater. 2015, 27, 948–953. [Google Scholar] [CrossRef] [Green Version]
- Lan, L.; Chen, Z.; Li, Y.; Ying, L.; Huang, F.; Cao, Y. Donor–acceptor conjugated polymers based on cyclic imide substituted quinoxaline or dibenzo [a, c] phenazine for polymer solar cells. Polym. Chem. 2015, 6, 7558–7569. [Google Scholar] [CrossRef]
- Matsueda, Y.; Xu, S.; Negishi, E.-I. A novel highly enantio-and diastereoselective synthesis of vitamin E side-chain. Tetrahedron Lett. 2015, 56, 3346–3348. [Google Scholar] [CrossRef]
- Thomson, A.; O’Connor, S.; Knuckley, B.; Causey, C.P. Design, synthesis, and in vitro evaluation of an activity-based protein profiling (ABPP) probe targeting agmatine deiminases. Bioorg. Med. Chem. 2014, 22, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Yen, W.-C.; Yang, J.-S.; Chao, C.-Y.; Hung, Y.-C.; Lin, S.-T.; Chuang, C.-H.; Chen, C.-W.; Su, W.-F. Substituent Effect on the Optoelectronic Properties of Alternating Fluorene− Cyclopentadithiophene Copolymers. Macromolecules 2008, 41, 6664–6671. [Google Scholar] [CrossRef]
- Gibson, G.L.; McCormick, T.M.; Seferos, D.S. Atomistic band gap engineering in donor–acceptor polymers. J. Am. Chem. Soc. 2011, 134, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Hsieh, C.-H.; Dubosc, M.; Cheng, Y.-J.; Hsu, C.-S. Synthesis and characterization of bridged bithiophene-based conjugated polymers for photovoltaic applications: Acceptor strength and ternary blends. Macromolecules 2009, 43, 697–708. [Google Scholar] [CrossRef]
- Yen, W.C.; Pal, B.; Yang, J.S.; Hung, Y.C.; Lin, S.T.; Chao, C.Y.; Su, W.F. Synthesis and characterization of low bandgap copolymers based on indenofluorene and thiophene derivative. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 5044–5056. [Google Scholar] [CrossRef]
- Li, R.; Liu, J.; Cai, N.; Zhang, M.; Wang, P. Synchronously reduced surface states, charge recombination, and light absorption length for high-performance organic dye-sensitized solar cells. J. Phys. Chem. B 2010, 114, 4461–4464. [Google Scholar] [CrossRef]
- Coppo, P.; Cupertino, D.C.; Yeates, S.G.; Turner, M.L. Synthetic routes to solution-processable polycyclopentadithiophenes. Macromolecules 2003, 36, 2705–2711. [Google Scholar] [CrossRef]
- Zoombelt, A.P.; Mathijssen, S.G.; Turbiez, M.G.; Wienk, M.M.; Janssen, R.A. Small band gap polymers based on diketopyrrolopyrrole. J. Mater. Chem. 2010, 20, 2240–2246. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Wu, C.-Y. Synthesis, characterization, optical and electrochemical properties of cyclopentadithiophene and fluorene based conjugated polymers containing naphthalene bisimide. Dyes Pigment. 2014, 106, 81–86. [Google Scholar] [CrossRef]
- Yi, H.; Al-Faifi, S.; Iraqi, A.; Watters, D.C.; Kingsley, J.; Lidzey, D.G. Carbazole and thienyl benzo [1, 2, 5] thiadiazole based polymers with improved open circuit voltages and processability for application in solar cells. J. Mater. Chem. 2011, 21, 13649–13656. [Google Scholar] [CrossRef]
- Wakioka, M.; Kitano, Y.; Ozawa, F. A Highly Efficient Catalytic System for Polycondensation of 2, 7-Dibromo-9, 9-dioctylfluorene and 1, 2, 4, 5-Tetrafluorobenzene via Direct Arylation. Macromolecules 2013, 46, 370–374. [Google Scholar] [CrossRef]
- Kenning, D.D.; Mitchell, K.A.; Calhoun, T.R.; Funfar, M.R.; Sattler, D.J.; Rasmussen, S.C. Thieno [3, 4-b] pyrazines: Synthesis, structure, and reactivity. J. Org. Chem. 2002, 67, 9073–9076. [Google Scholar] [CrossRef] [PubMed]
- McNamara, L.E.; Liyanage, N.; Peddapuram, A.; Murphy, J.S.; Delcamp, J.H.; Hammer, N.I. Donor–Acceptor–Donor Thienopyrazines via Pd-Catalyzed C–H Activation as NIR Fluorescent Materials. J. Org. Chem. 2015, 81, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Zhao, Y.; Shao, S.; Tian, H.; Xie, Z.; Geng, Y.; Wang, F. Novel NIR-absorbing conjugated polymers for efficient polymer solar cells: Effect of alkyl chain length on device performance. J. Mater. Chem. 2009, 19, 2199–2206. [Google Scholar] [CrossRef]
- Brzezinski, J.Z.; Reynolds, J.R. A new, improved and convenient synthesis of 4H-cyclopenta [2, 1-b: 3, 4-b′]-dithiophen-4-one. Synthesis 2002, 2002, 1053–1056. [Google Scholar] [CrossRef]
- Brza, M.A.; Aziz, S.B.; Anuar, H.; Hazza, A.H.M. From green remediation to polymer hybrid fabrication with improved optical band gaps. Int. J. Mol. Sci. 2019, 20, 3910. [Google Scholar] [CrossRef] [Green Version]
- Hussein, A.M.; Dannoun, E.M.A.; Aziz, S.B.; Brza, M.A.; Abdulwahid, R.T.; Hussen, S.A.; Rostam, S.; Mustafa, D.M.T.; Muhammad, D.S. Steps Toward the Band Gap Identification in Polystyrene Based Solid Polymer Nanocomposites Integrated with Tin Titanate Nanoparticles. Polymers 2020, 12, 2320. [Google Scholar] [CrossRef]
- Brza, M.A.; Aziz, S.B.; Anuar, H.; Ali, F.; Elham, M.; Dannoun, A.; Saeed, S.R.; Mohammed, S.J.; Abdulwahid, R.T. Green coordination chemistry as a novel approach to fabricate polymer: Cd (II)-complex composites: Structural and optical properties. Opt. Mater. 2020, 100067. [Google Scholar] [CrossRef]
- Brza, M.A.; Aziz, S.B.; Anuar, H.; Ali, F.; Dannoun, E.M.A.; Mohammed, S.J.; Abdulwahid, R.T.; Zangana, S.A. Tea from the drinking to the synthesis of metal complexes and fabrication of PVA based polymer composites with controlled optical band gap. Sci. Rep. 2020, 10, 18108. [Google Scholar] [CrossRef]
- Murad, A.R.; Iraqi, A.; Aziz, S.B.; Abdullah, S.N.; Abdulwahid, R.T. Synthesis, Optical, Thermal and Structural Characteristics of Novel Thermocleavable Polymers Based on Phthalate Esters. Polymers 2020, 12, 2791. [Google Scholar] [CrossRef]
- Ameri, T.; Dennler, G.; Lungenschmied, C.; Brabec, C.J. Organic tandem solar cells: A review. Energy Environ. Sci. 2009, 2, 347–363. [Google Scholar] [CrossRef]
- Murad, A.R.; Iraqi, A.; Aziz, S.B.; Abdullah, S.N.; Abdulwahid, R.T.; Hussen, S.A. Optical, Electrochemical, Thermal, and Structural Properties of Synthesized Fluorene/Dibenzosilole-Benzothiadiazole Dicarboxylic Imide Alternating Organic Copolymers for Photovoltaic Applications. Coatings 2020, 10, 1147. [Google Scholar] [CrossRef]
- Murad, A.R.; Iraqi, A.; Aziz, S.B.; Hi, H.; Abdullah, S.N.; Brza, M.A.; Abdulwahid, R.T. Influence of Fluorine Substitution on the Optical, Thermal, Electrochemical and Structural Properties of Carbazole-Benzothiadiazole Dicarboxylic Imide Alternate Copolymers. Polymers 2020, 12, 2910. [Google Scholar] [CrossRef]
- Dannoun, E.M.A.; Aziz, S.B.; Brza, M.A.; Nofal, M.M.; Asnawi, A.S.F.M.; Yusof, Y.M.; Zangana, S.A.; Hamsan, M.H.; Kadir, M.F.Z.; Woo, H.J. The study of plasticized solid polymer blend electrolytes based on natural polymers and their application for energy storage EDLC devices. Polymers 2020, 12, 2531. [Google Scholar] [CrossRef]
- Nofal, M.M.; Aziz, S.B.; Hadi, J.M.; Abdulwahid, R.T.; Dannoun, E.M.A.; Marif, A.S.; Kadir, M.F.Z. Synthesis of Porous Proton Ion Conducting Solid Polymer Blend Electrolytes Based on PVA: CS Polymers: Structural, Morphological and Electrochemical Properties. Materials 2020, 13, 4890. [Google Scholar] [CrossRef] [PubMed]
- Hamsan, H.M.; Aziz, S.B.; Kadir, M.F.Z.; Brza, M.A.; Karim, W.O. The study of EDLC device fabricated from plasticized magnesium ion conducting chitosan based polymer electrolyte. Polym. Test. 2020, 90, 106714. [Google Scholar] [CrossRef]
- Marf, A.S.; Aziz, S.B.; Abdullah, R.M. Plasticized H+ ion-conducting PVA: CS-based polymer blend electrolytes for energy storage EDLC application. J. Mater. Sci. Mater. Electron. 2020, 31, 18554–18568. [Google Scholar] [CrossRef]
- Aziz, S.B.; Brevik, I.; Hamsan, M.H.; Brza, M.A.; Nofal, M.M.; Abdullah, A.M.; Kadir, M.F.Z. Compatible solid polymer electrolyte based on methyl cellulose for energy storage application: Structural, electrical, and electrochemical properties. Polymers 2020, 12, 2257. [Google Scholar] [CrossRef]
- Aziz, S.B.; Brza, M.A.; Nofal, M.M.; Abdulwahid, R.T.; Hussen, S.A.; Hussein, A.M.; Karim, W.O. A comprehensive review on optical properties of polymer electrolytes and composites. Materials 2020, 13, 3675. [Google Scholar] [CrossRef]
- Brza, M.A.; Aziz, S.B.; Anuar, H.; Ali, F.; Hamsan, M.H.; Kadir, M.F.Z.; Abdulwahid, R.T. Metal framework as a novel approach for the fabrication of electric double layer capacitor device with high energy density using plasticized Poly(vinyl alcohol): Ammonium thiocyanate based polymer electrolyte. Arab. J. Chem. 2020, 13, 7247–7263. [Google Scholar] [CrossRef]
- Brza, M.A.; Aziz, S.B.; Anuar, H.; Dannoun, E.M.A.; Ali, F.; Abdulwahid, R.T.; Zangana, S.A.; Kadir, M.F.Z. The study of EDLC device with high electrochemical performance fabricated from proton ion conducting PVA-based polymer composite electrolytes plasticized with glycerol. Polymers 2020, 12, 1896. [Google Scholar] [CrossRef] [PubMed]
- Asnawi, A.S.F.M.; Aziz, S.B.; Nofal, M.M.; Yusof, Y.M.; Iver, B.; Hamsan, M.H.; Brza, M.A.; Abdulwahid, R.T.; Kadir, M.F.Z. Metal complex as a novel approach to enhance the amorphous phase and improve the EDLC performance of plasticized proton conducting chitosan-based polymer electrolyte. Membranes 2020, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, D.S.; Brza, M.A.; Nofal, M.M.; Aziz, S.B.; Hussen, S.A.; Abdulwahid, R.T. Optical Dielectric Loss as a Novel Approach to Specify the Types of Electron Transition: XRD and UV-vis as a Non-Destructive Techniques for Structural and Optical Characterization of PEO Based Nanocomposites. Materials 2020, 13, 2979. [Google Scholar] [CrossRef]
- Chen, H.Y.; Hou, J.; Hayden, A.E.; Yang, H.; Houk, K.; Yang, Y. Silicon Atom Substitution Enhances Interchain Packing in a Thiophene-Based Polymer System. Adv. Mater. 2010, 22, 371–375. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | % Yield | Time (h) | Toluene Fraction | ||
|---|---|---|---|---|---|
| Mn (g mol−1) | Mw (g mol−1) | PDI | |||
| PCPDTDTBTDI-EH, DMO | 8 | 48 | 5200 | 10,100 | 1.9 |
| PCPDTDTBTDI-8, DMO | 95 | 17 | 7800 | 18,100 | 2.3 |
| PCPDTDTBTDI-8, DMO * | 86 | 72 | 10,000 | 30,900 | 3.0 |
| PCPDTDTBTDI-8, 8 | 72 | 51 | 4900 | 20,800 | 4.2 |
| PCPDTDTBTDI-8, 8 | 76 | 96 | 9100 | 18,300 | 2.0 |
| PCPDTDTBTDI-EH, 8 | 72 | 96 | 15,900 | 29,700 | 1.8 |
| Polymer | ε (M−1 cm−1) | Solution | Film | ||
|---|---|---|---|---|---|
| λmax (nm) | λmax (nm) | λonset (nm) | Eg (eV) | ||
| PCPDTDTBTDI-EH, DMO | 28,200 | 635 | 686 | 865 | 1.43 |
| PCPDTDTBTDI-8, DMO | 37,500 | 673 | 759 | 936 | 1.32 |
| PCPDTDTBTDI-8, 8 | 27,500 | 672 | 724 | 922 | 1.34 |
| PCPDTDTBTDI-EH, 8 | 28,400 | 675 | 704 | 873 | 1.42 |
| Polymer | Td | Eox 0 | HOMO | Ered 0 | LUMO | Eg (elec) |
|---|---|---|---|---|---|---|
| (°C) | (V) | (eV) | (V) | (eV) | (eV) | |
| PCPDTDTBTDI-EH, DMO | 378 | 0.44 | −5.15 | −1.19 | −3.52 | 1.63 |
| PCPDTDTBTDI-8 | 419 | 0.49 | −5.20 | −1.24 | −3.47 | 1.73 |
| PCPDTDTBTDI-8, 8 | 402 | 0.39 | −5.10 | −1.27 | −3.44 | 1.66 |
| PCPDTDTBTDI-EH, 8 | 377 | 0.51 | −5.22 | −1.17 | −3.54 | 1.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murad, A.R.; Iraqi, A.; Aziz, S.B.; Abdullah, S.N.; Brza, M.A.; Saeed, S.R.; Abdulwahid, R.T. Fabrication of Alternating Copolymers Based on Cyclopentadithiophene-Benzothiadiazole Dicarboxylic Imide with Reduced Optical Band Gap: Synthesis, Optical, Electrochemical, Thermal, and Structural Properties. Polymers 2021, 13, 63. https://doi.org/10.3390/polym13010063
Murad AR, Iraqi A, Aziz SB, Abdullah SN, Brza MA, Saeed SR, Abdulwahid RT. Fabrication of Alternating Copolymers Based on Cyclopentadithiophene-Benzothiadiazole Dicarboxylic Imide with Reduced Optical Band Gap: Synthesis, Optical, Electrochemical, Thermal, and Structural Properties. Polymers. 2021; 13(1):63. https://doi.org/10.3390/polym13010063
Chicago/Turabian StyleMurad, Ary R., Ahmed Iraqi, Shujahadeen B. Aziz, Sozan N. Abdullah, Mohamad A. Brza, Salah R. Saeed, and Rebar T. Abdulwahid. 2021. "Fabrication of Alternating Copolymers Based on Cyclopentadithiophene-Benzothiadiazole Dicarboxylic Imide with Reduced Optical Band Gap: Synthesis, Optical, Electrochemical, Thermal, and Structural Properties" Polymers 13, no. 1: 63. https://doi.org/10.3390/polym13010063