Synthesis of Water Resistance and Moisture-Permeable Nanofiber Using Sodium Alginate–Functionalized Waterborne Polyurethane

 , , , and
, , , and
Abstract
:1. Introduction
2. Experimental
2.1. Materials
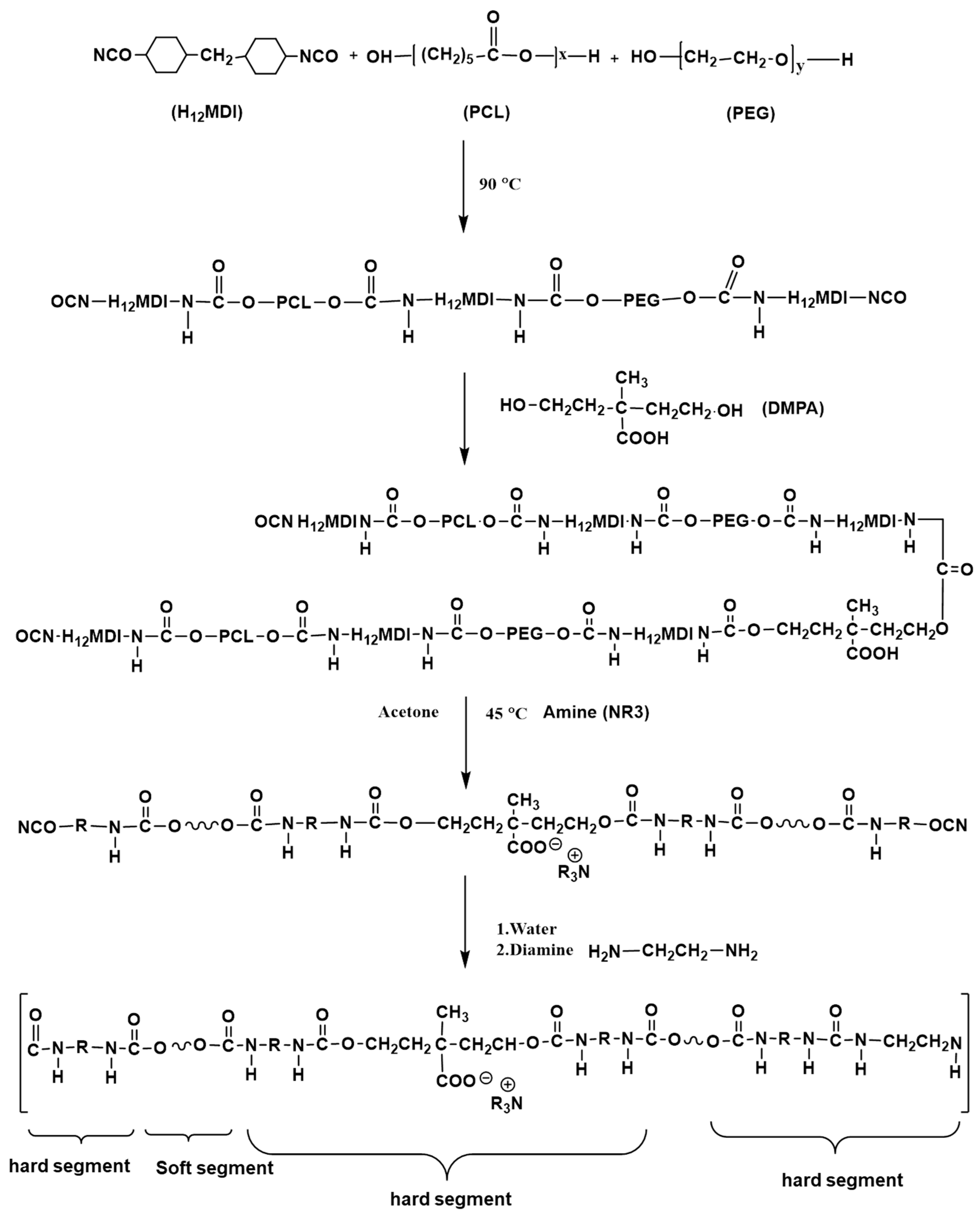
2.2. Synthesis of WPU
2.3. Preparing WPU/SA Blends and Crosslinked WPU/SA (WPU/CA) Blends
2.4. Measurements
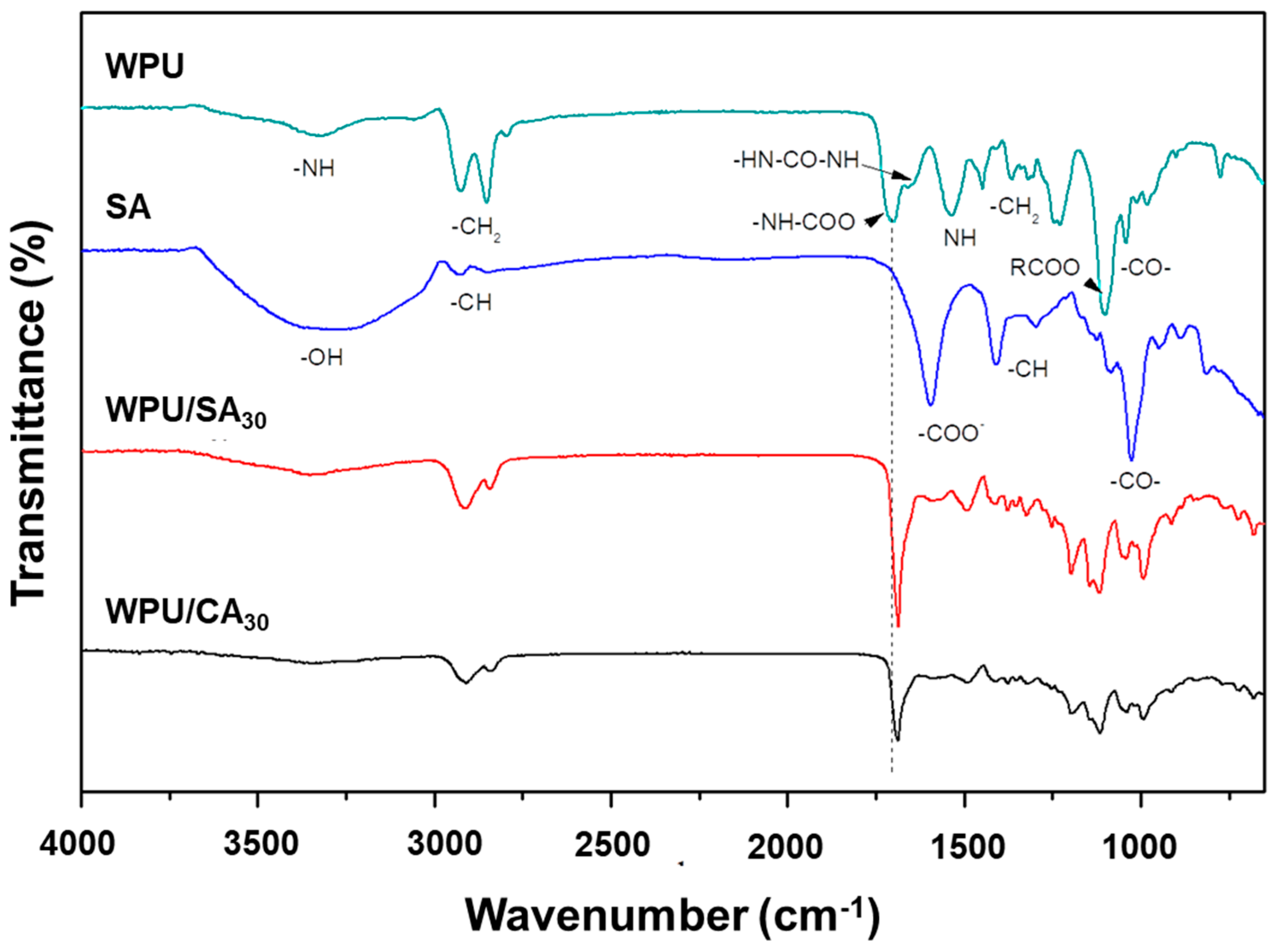
2.4.1. Fourier Transform Infrared Spectroscopy
2.4.2. Water Solubility and Degree of Crosslinking
2.4.3. Water Absorption
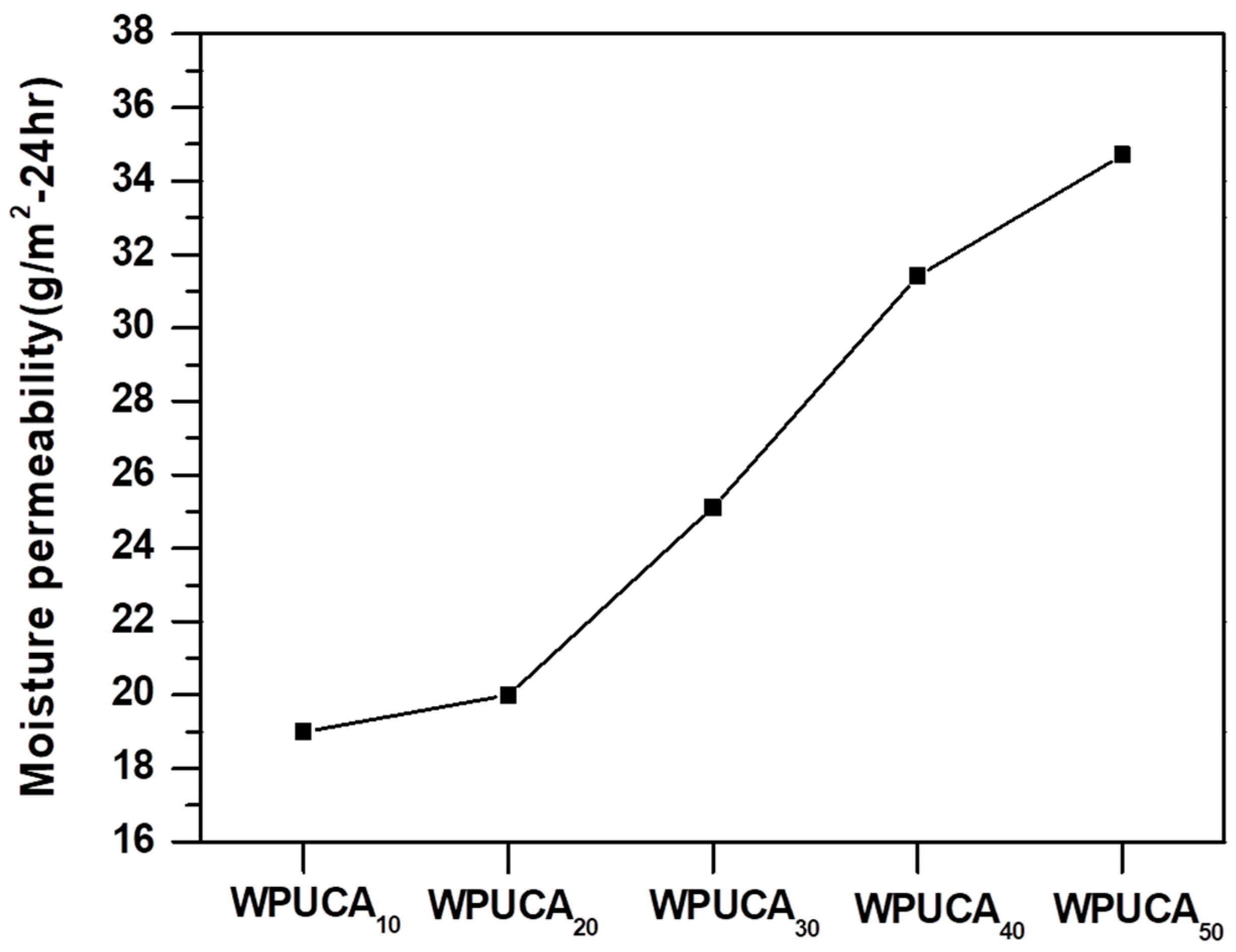
2.4.4. Moisture Permeability
2.4.5. Phase Transition Regions and Degree of Phase Separation
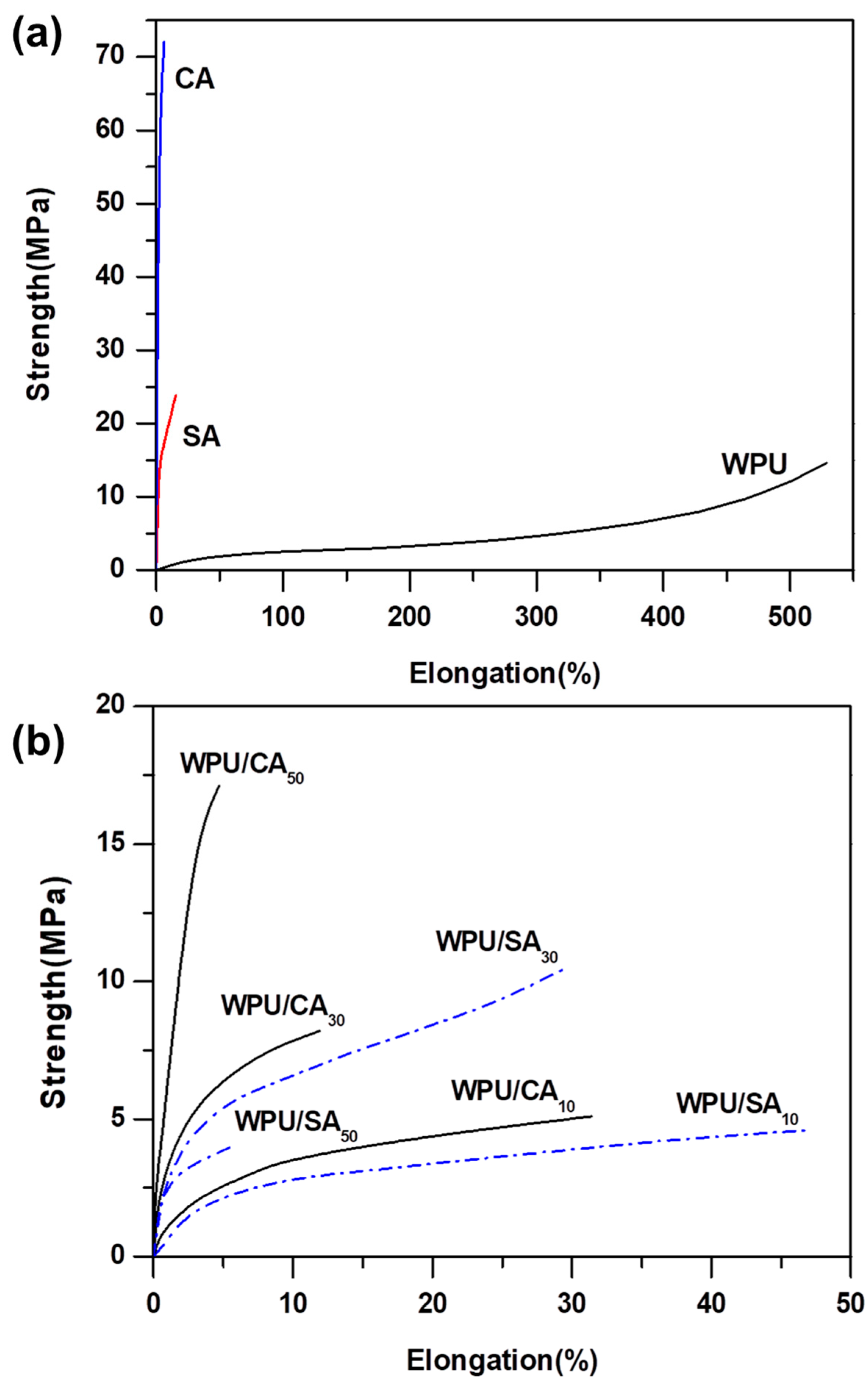
2.4.6. Tensile Strength
3. Results and Discussion
3.1. FTIR Spectrum Analysis of the WPU/SA and WPU/CA Blends Characteristics
3.2. Effect of Moisture on the WPU/CA Blends
3.2.1. Solubility of the WPU/CA Blends in Water
3.2.2. Water Absorption of the Crosslinked WPU/SA Blends
3.3. Moisture Permeability of the Crosslinked WPU/CA Blends
3.4. Phase Transition Regions of the WPU/SA Blends and WPU/CA Blends
3.5. Tensile Strength
3.6. SEM and EDS Analysis of Blends
3.7. Electrospinning of the WPU/SA Blends
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Król, P.; Król, B. Structures, properties and applications of the polyurethane ionomers. J. Mater. Sci. 2019, 55, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Prisacariu, C.; Scortanu, E.; Stoica, I.; Agapie, B.; Bărboiu, V. Morphological features and thermal and mechanical response in segmented polyurethane elastomers based on mixtures of isocyanates. Polym. J. 2011, 43, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Bankoti, K.; Dhara, S.; Datta, S.; Maity, P.P.; Goswami, P.; Datta, P.; Ghosh, S.K.; Mitra, A.; Dhara, S. Accelerated healing of full thickness dermal wounds by macroporous waterborne polyurethane-chitosan hydrogel scaffolds. Mater. Sci. Eng. C 2017, 81, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Christy, P.N.; Basha, S.K.; Kumari, V.S.; Bashir, A.; Maaza, M.; Kaviyarasu, K.; Arasu, M.V.; Al-Dhabi, N.A.; Ignacimuthu, S. Biopolymeric nanocomposite scaffolds for bone tissue engineering applications—A review. J. Drug Deliv. Sci. Technol. 2020, 55, 101452. [Google Scholar] [CrossRef]
- Dawlee, S.; Jayabalan, M. Development of segmented polyurethane elastomers with low iodine content exhibiting radiopacity and blood compatibility. Biomed. Mater. 2011, 6, 055002. [Google Scholar] [CrossRef] [PubMed]
- Mansur, S.; Othman, M.H.D.; Ismail, A.F.; Kadir, S.H.S.A.; Goh, P.S.; Hasbullah, H.; Ng, B.C.; Abdullah, M.S.; Kamal, F.; Abidin, M.N.Z.; et al. Synthesis and characterisation of composite sulphonated polyurethane/polyethersulphone membrane for blood purification application. Mater. Sci. Eng. C 2019, 99, 491–504. [Google Scholar] [CrossRef]
- Xue, L.; Greisler, H.P. Biomaterials in the development and future of vascular grafts. J. Vasc. Surg. 2003, 37, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Adipurnama, I.; Yang, M.-C.; Ciach, T.; Butruk-Raszeja, B. Surface modification and endothelialization of polyurethane for vascular tissue engineering applications: A review. Biomater. Sci. 2017, 5, 22–37. [Google Scholar] [CrossRef]
- Cherng, J.Y.; Hou, T.Y.; Shih, M.F.; Talsma, H.; Hennink, W.E. Polyurethane-based drug delivery systems. Int. J. Pharm. 2013, 450, 145–162. [Google Scholar] [CrossRef]
- Lowinger, M.B.; Barrett, S.E.; Zhang, F.; Williams, R.O. Sustained Release Drug Delivery Applications of Polyurethanes. Pharmaceutics 2018, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Klemm, D.; Cranston, E.D.; Fischer, D.; Gama, M.; Kedzior, S.A.; Kralisch, D.; Kramer, F.; Kondo, T.; Lindström, T.; Nietzsche, S.; et al. Nanocellulose as a natural source for groundbreaking applications in materials science: Today’s state. Mater. Today 2018, 21, 720–748. [Google Scholar] [CrossRef] [Green Version]
- Caracciolo, P.C.; Buffa, F.; Abraham, G.A. Effect of the hard segment chemistry and structure on the thermal and mechanical properties of novel biomedical segmented poly(esterurethanes). J. Mater. Sci. Mater. Electron. 2008, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-W.; Cheng, Y.; Lee, H.; Wang, C.; Chiu, C.-W.; Suen, M.-C. Synthesis and properties of castor oil-based polyurethane containing short fluorinated segment. J. Appl. Polym. Sci. 2020, 137, 49062. [Google Scholar] [CrossRef]
- Li, J.-W.; Lee, H.-T.; Tsai, H.-A.; Suen, M.-C.; Chiu, C.-W. Synthesis and Properties of Novel Polyurethanes Containing Long-Segment Fluorinated Chain Extenders. Polymers 2018, 10, 1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zhang, P.; Fan, M.; Jiang, P.; Bao, Y.; Gao, X.; Xia, J. Dual bond synergy enhancement to mechanical and thermal properties of castor oil-based waterborne polyurethane composites. Polymers 2019, 182, 121832. [Google Scholar] [CrossRef]
- Trinca, R.B.; Abraham, G.A.; Felisberti, M.I. Electrospun nanofibrous scaffolds of segmented polyurethanes based on PEG, PLLA and PTMC blocks: Physico-chemical properties and morphology. Mater. Sci. Eng. C 2015, 56, 511–517. [Google Scholar] [CrossRef]
- Zhuc, T.; Yu, K.; Bhutto, M.A.; Guo, X.; Shen, W.; Wang, J.; Chen, W.; El-Hamshary, H.; Al-Deyab, S.S.; Mo, X.; et al. Synthesis of RGD-peptide modified poly(ester-urethane) urea electrospun nanofibers as a potential application for vascular tissue engineering. Chem. Eng. J. 2017, 315, 177–190. [Google Scholar] [CrossRef]
- Ambrogi, V.; Pietrella, D.; Donnadio, A.; Latterini, L.; Di Michele, A.; Luffarelli, I.; Ricci, M. Biocompatible alginate silica supported silver nanoparticles composite films for wound dressing with antibiofilm activity. Mater. Sci. Eng. C 2020, 112, 110863. [Google Scholar] [CrossRef]
- Summa, M.; Russo, D.; Penna, I.; Margaroli, N.; Bayer, I.S.; Bayer, I.S.; Athanassiou, A.; Bertorelli, R. A biocompatible sodium alginate/povidone iodine film enhances wound healing. Eur. J. Pharm. Biopharm. 2018, 122, 17–24. [Google Scholar] [CrossRef]
- Aadil, K.R.; Nathani, A.; Sharma, C.S.; Lenka, N.; Gupta, P. Fabrication of biocompatible alginate-poly(vinyl alcohol) nanofibers scaffolds for tissue engineering applications. Mater. Technol. 2018, 33, 507–512. [Google Scholar] [CrossRef]
- Zhou, Q.; Kang, H.; Bielec, M.; Wu, X.; Cheng, Q.; Wei, W.; Dai, H. Influence of different divalent ions cross-linking sodium alginate-polyacrylamide hydrogels on antibacterial properties and wound healing. Carbohydr. Polym. 2018, 197, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Suzuki, Y.; Kitada, M.; Kataoka, K.; Wu, S.; Suzuki, K.; Endo, K.; Nishimura, Y.; Ide, C. Peripheral nerve regeneration through alginate gel: Analysis of early outgrowth and late increase in diameter of regenerating axons. Exp. Brain Res. 2002, 146, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Gombotz, W.R.; Wee, S.F. Protein release from alginate matrices. Adv. Drug Deliv. Rev. 2012, 64, 194–205. [Google Scholar] [CrossRef]
- Salimi, S.; Sotudeh-Gharebagh, R.; Zarghami, R.; Chan, S.Y.; Yuen, K.H. Production of Nanocellulose and Its Applications in Drug Delivery: A Critical Review. ACS Sustain. Chem. Eng. 2019, 7, 15800–15827. [Google Scholar] [CrossRef]
- Daemi, H.; Barikani, M.; Barmar, M. Compatible compositions based on aqueous polyurethane dispersions and sodium alginate. Carbohydr. Polym. 2013, 92, 490–496. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Yang, L.; Zhu, Q.; Ma, Q.; Wang, R.; Zhang, C.; Zhang, Z. Indoxacarb-Loaded Anionic Polyurethane Blend with Sodium Alginate Improves pH Sensitivity and Ecological Security for Potential Application in Agriculture. Polymers 2020, 12, 1135. [Google Scholar] [CrossRef]
- Corpuz, A.G.; Pal, P.; Banat, F.; Abu Haija, M. Enhanced removal of mixed metal ions from aqueous solutions using flotation by colloidal gas aphrons stabilized with sodium alginate. Sep. Purif. Technol. 2018, 202, 103–110. [Google Scholar] [CrossRef]
- Sharma, P.R.; Joshi, R.; Sharma, S.K.; Hsiao, B.S. A Simple Approach to Prepare Carboxycellulose Nanofibers from Untreated Biomass. Biomacromolecules 2017, 18, 2333–2342. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, Q.; Liu, J.; Sun, J.; Zhu, Q.; Chen, H. Processing nanocellulose to bulk materials: A review. Cellulose 2019, 26, 7585–7617. [Google Scholar] [CrossRef]
- Chen, W.; Yu, H.; Lee, S.; Wei, T.; Li, J.; Fan, Z. Nanocellulose: A promising nanomaterial for advanced electrochemical energy storage. Chem. Soc. Rev. 2018, 47, 2837–2872. [Google Scholar] [CrossRef]
- Thomas, B.; Raj, M.C.; B, A.K.; H, R.M.; Joy, J.; Moores, A.; Drisko, G.L.; Sanchez, C. Nanocellulose, a Versatile Green Platform: From Biosources to Materials and Their Applications. Chem. Rev. 2018, 118, 11575–11625. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Wang, C.; Xia, R.; Chen, P.; Miao, J.; Yang, B.; Qian, J.; Tang, Y. Preparation and performance of the modified high-strength/high-modulus polyvinyl alcohol fiber/polyurethane grouting materials. Constr. Build. Mater. 2018, 168, 482–489. [Google Scholar] [CrossRef]
- Karahaliloglu, Z. Electrospun PU-PEG and PU-PC hybrid scaffolds for vascular tissue engineering. Fibers Polym. 2017, 18, 2135–2145. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Y.; Fang, Z.; Yuan, W.; Khan, M. Co-electrospun blends of PU and PEG as potential biocompatible scaffolds for small-diameter vascular tissue engineering. Mater. Sci. Eng. C 2012, 32, 2306–2315. [Google Scholar] [CrossRef]
- Amjed, N.; Bhatti, I.A.; Zia, K.M.; Iqbal, J.; Jamil, Y. Synthesis and characterization of stable and biological active chitin-based polyurethane elastomers. Int. J. Biol. Macromol. 2020, 154, 1149–1157. [Google Scholar] [CrossRef]
- Cho, C.-J.; Chang, Y.-S.; Lin, Y.-Z.; Jiang, D.-H.; Chen, W.-H.; Lin, W.-Y.; Chen, C.-W.; Rwei, S.-P.; Kuo, C.-C. Green electrospun nanofiber membranes filter prepared from novel biomass thermoplastic copolyester: Morphologies and filtration properties. J. Taiwan Inst. Chem. Eng. 2020, 106, 206–214. [Google Scholar] [CrossRef]
- Jiang, D.-H.; Chiu, P.-C.; Cho, C.-J.; Veeramuthu, L.; Tung, S.-H.; Satoh, T.; Chiang, W.-H.; Cai, X.; Kuo, C.-C. Facile 3D Boron Nitride Integrated Electrospun Nanofibrous Membranes for Purging Organic Pollutants. Nanomaterials 2019, 9, 1383. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.-C.; Ku, H.-J.; Cho, C.-J.; Chen, W.-C.; Lee, W.-Y.; Chen, W.-C.; Rwei, S.-P.; Borsali, R.; Kuo, C.-C. An intrinsically stretchable and ultrasensitive nanofiber-based resistive pressure sensor for wearable electronics. J. Mater. Chem. C 2020, 8, 5361–5369. [Google Scholar] [CrossRef]
- Veeramuthu, L.; Li, W.-L.; Liang, F.-C.; Cho, C.-J.; Kuo, C.-C.; Chen, W.-C.; Lin, J.-H.; Lee, W.-Y.; Wang, C.-T.; Lin, W.-Y.; et al. Smart garment energy generators fabricated using stretchable electrospun nanofibers. React. Funct. Polym. 2019, 142, 96–103. [Google Scholar] [CrossRef]
- Venkatesan, M.; Veeramuthu, L.; Liang, F.-C.; Chen, W.-C.; Cho, C.-J.; Chen, C.-W.; Chen, J.; Yan, Y.; Chang, S.-H.; Kuo, C.-C.; et al. Evolution of electrospun nanofibers fluorescent and colorimetric sensors for environmental toxicants, pH, temperature, and cancer cells–A review with insights on applications. Chem. Eng. J. 2020, 397, 125431. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Geng, L.-H.; Amiralian, N.; Martin, D.J.; Hsiao, B.S. Nanocellulose from Spinifex as an Effective Adsorbent to Remove Cadmium(II) from Water. ACS Sustain. Chem. Eng. 2018, 6, 3279–3290. [Google Scholar] [CrossRef]
- Sharma, P.R.; Sharma, S.K.; Lindström, T.; Hsiao, B.S. Nanocellulose-Enabled Membranes for Water Purification: Perspectives. Adv. Sustain. Syst. 2020, 4. [Google Scholar] [CrossRef]
- Cho, C.-J.; Lu, S.-T.; Kuo, C.-C.; Liang, F.-C.; Chen, B.-Y.; Chu, C.-C. Pyrene or rhodamine derivative–modified surfaces of electrospun nanofibrous chemosensors for colorimetric and fluorescent determination of Cu 2+, Hg 2+, and pH. React. Funct. Polym. 2016, 108, 137–147. [Google Scholar] [CrossRef]
- Liang, F.-C.; Kuo, C.-C.; Chen, B.-Y.; Cho, C.-J.; Hung, C.-C.; Chen, W.-C.; Borsali, R. RGB-Switchable Porous Electrospun Nanofiber Chemoprobe-Filter Prepared from Multifunctional Copolymers for Versatile Sensing of pH and Heavy Metals. ACS Appl. Mater. Interfaces 2017, 9, 16381–16396. [Google Scholar] [CrossRef]
- Chen, R.-D.; Huang, C.-F.; Hsu, S.-H. Composites of waterborne polyurethane and cellulose nanofibers for 3D printing and bioapplications. Carbohydr. Polym. 2019, 212, 75–88. [Google Scholar] [CrossRef]
- Liao, S.K.; Jang, S.C.; Lin, M.F. Phase Behavior and Mechanical Properties of Siloxane-Urethane Copolymer. J. Polym. Res. 2005, 12, 103–112. [Google Scholar] [CrossRef]
- Bistričić, L.; Baranović, G.; Leskovac, M.; Bajsić, E.G. Hydrogen bonding and mechanical properties of thin films of polyether-based polyurethane–silica nanocomposites. Eur. Polym. J. 2010, 46, 1975–1987. [Google Scholar] [CrossRef]
- Kwiatkowski, K.; Nachman, M. The Abrasive Wear Resistance of the Segmented Linear Polyurethane Elastomers Based on a Variety of Polyols as Soft Segments. Polymers 2017, 9, 705. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Filgueira, D.; Rodríguez, A.; Chinga-Carrasco, G. Nanocellulose-Based Inks—Effect of Alginate Content on the Water Absorption of 3D Printed Constructs. Bioengineering 2019, 6, 65. [Google Scholar] [CrossRef] [Green Version]
- Arahman, N.; Mulyati, S.; Fahrina, A.; Muchtar, S.; Yusuf, M.; Takagi, R.; Matsuyama, H.; Nordin, N.A.H.; Bilad, M.R. Improving Water Permeability of Hydrophilic PVDF Membrane Prepared via Blending with Organic and Inorganic Additives for Humic Acid Separation. Molecules 2019, 24, 4099. [Google Scholar] [CrossRef] [Green Version]
- Othman, S.H.; Edwal, S.A.M.; Risyon, N.P.; Basha, R.K.; Talib, R.A. Water sorption and water permeability properties of edible film made from potato peel waste. Food Sci. Technol. 2017, 37, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Bierhalz, A.C.K.; Moraes, A.M. Tuning the properties of alginate-chitosan membranes by varying the viscosity and the proportions of polymers. J. Appl. Polym. Sci. 2016, 133, 133. [Google Scholar] [CrossRef]
- Swamy, T.M.; Ramaraj, B.; Lee, J.H. Sodium alginate and its blends with starch: Thermal and morphological properties. J. Appl. Polym. Sci. 2008, 109, 4075–4081. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Water Solubility (wt%) | Exchange Degree of Sodium Alginate and Calcium Ions (Degree of Crosslinking) |
|---|---|---|
| SA 1 | dissolve | - |
| WPU/SA10 | 9.5 | - |
| WPU/SA20 | 19.8 | - |
| WPU/SA30 | 28.6 | - |
| WPU/SA40 | 39.4 | - |
| WPU/SA50 | - | |
| CA 1 | 0.5 | 99.5 |
| WPU/CA10 | 2.2 | 97.8 |
| WPU/CA20 | 3.4 | 96.6 |
| WPU/CA30 | 1.01 | 99.0 |
| WPU/CA40 | 1.68 | 98.3 |
| WPU/CA50 | 1.6 | 98.4 |
| Code | Tgs | Tgh | ΔB |
|---|---|---|---|
| WPU | −45 | 71 | 25 |
| SA | - | - | - |
| WPU/SA10 | −39 | 80 | 33 |
| WPU/SA20 | −33 | 82 | 37 |
| WPU/SA30 | −30 | 87 | 51 |
| WPU/SA40 | −27 | 90 | 42 |
| WPU/SA50 | −25 | 91 | 50 |
| CA | - | - | - |
| WPU/CA10 | −26 | 97 | 36 |
| WPU/CA20 | −21 | 100 | 42 |
| WPU/CA30 | −11 | - | 52 |
| WPU/CA40 | −15 | 103 | 44 |
| WPU/CA50 | −18 | 105 | 42 |
| Code | Initial Modulus (MPa) | Break Strength (MPa) | Elongation at Break (%) |
|---|---|---|---|
| WPU | 6.9 | 14.6 | 529 |
| SA | 797.2 | 23.8 | 7.8 |
| CA | 2516.2 | 72.4 | 4.9 |
| WPU/SA10 | 112.1 | 4.6 | 47.1 |
| WPU/SA20 | 154.0 | 5.9 | 21.9 |
| WPU/SA30 | 166.6 | 10.6 | 30.1 |
| WPU/SA40 | 196.0 | 5.0 | 16.3 |
| WPU/SA50 | 243.3 | 4.1 | 6.1 |
| WPU/CA10 | 98.6 | 5.1 | 31.4 |
| WPU/CA20 | 340.6 | 7.7 | 9.4 |
| WPU/CA30 | 403.1 | 8.2 | 11.9 |
| WPU/CA40 | 539.4 | 3.1 | 4.3 |
| WPU/CA50 | 1378.1 | 17.1 | 4.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, W.-C.; Chuang, F.-S.; Venkatesan, M.; Cho, C.-J.; Chen, P.-Y.; Tzeng, Y.-R.; Yu, Y.-Y.; Rwei, S.-P.; Kuo, C.-C. Synthesis of Water Resistance and Moisture-Permeable Nanofiber Using Sodium Alginate–Functionalized Waterborne Polyurethane. Polymers 2020, 12, 2882. https://doi.org/10.3390/polym12122882
Lu W-C, Chuang F-S, Venkatesan M, Cho C-J, Chen P-Y, Tzeng Y-R, Yu Y-Y, Rwei S-P, Kuo C-C. Synthesis of Water Resistance and Moisture-Permeable Nanofiber Using Sodium Alginate–Functionalized Waterborne Polyurethane. Polymers. 2020; 12(12):2882. https://doi.org/10.3390/polym12122882
Chicago/Turabian StyleLu, Wen-Chi, Fu-Sheng Chuang, Manikandan Venkatesan, Chia-Jung Cho, Po-Yun Chen, Yung-Ru Tzeng, Yang-Yen Yu, Syang-Peng Rwei, and Chi-Ching Kuo. 2020. "Synthesis of Water Resistance and Moisture-Permeable Nanofiber Using Sodium Alginate–Functionalized Waterborne Polyurethane" Polymers 12, no. 12: 2882. https://doi.org/10.3390/polym12122882