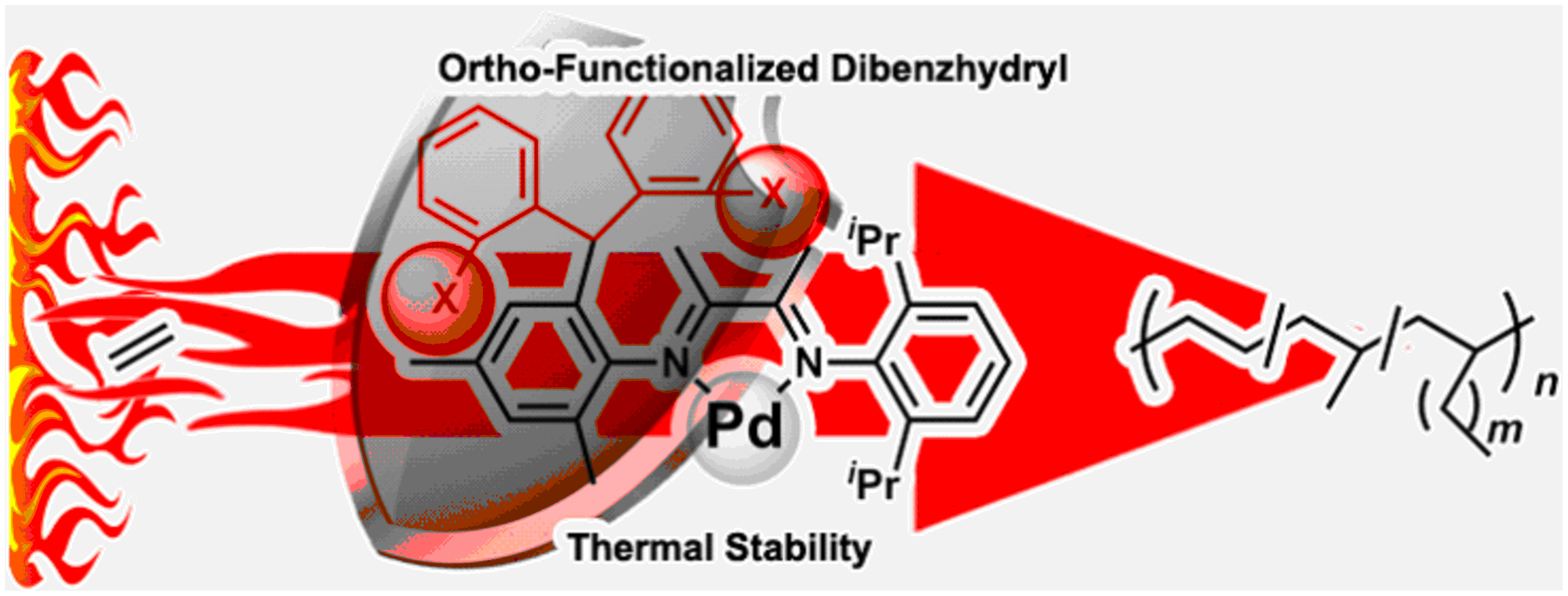
Ortho-Functionalized Dibenzhydryl Substituents in α-Diimine Pd Catalyzed Ethylene Polymerization and Copolymerization


1
Department of Polymer Science and Engineering, University of Science and Technology of China, Anhui, Hefei 230601, China
2
Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Institutes of Physical Science and Information Technology, Anhui University, Anhui, Hefei 230601, China
*
Authors to whom correspondence should be addressed.
Polymers 2020, 12(11), 2509; https://doi.org/10.3390/polym12112509
Submission received: 3 October 2020
/
Revised: 23 October 2020
/
Accepted: 24 October 2020
/
Published: 28 October 2020
(This article belongs to the Section Polymer Chemistry)
Abstract
:Sterically bulky diarylmethyl-based ligands have received increasing attention in the field of late-transition-metal catalyzed olefin polymerization. Ortho-substituents may have a significant impact on the performance of diarylmethyl-based α-diimine Pd catalysts. In this contribution, a series of α-diimine Pd catalysts bearing ortho-methoxyl/hydroxyl functionalized dibenzhydryl units were prepared, characterized, and investigated in ethylene polymerization and copolymerization with methyl acrylate (MA). The catalytic performances were improved by introducing more ortho-substituents. The catalysts exhibited good thermal stabilities at high temperatures, producing branched polyethylenes. The catalysts bearing hydroxyl groups possessing intramolecular H-bonding, resulted in slightly higher incorporation ratios of MA unit when compared with the catalysts bearing methoxyl groups.

1. Introduction
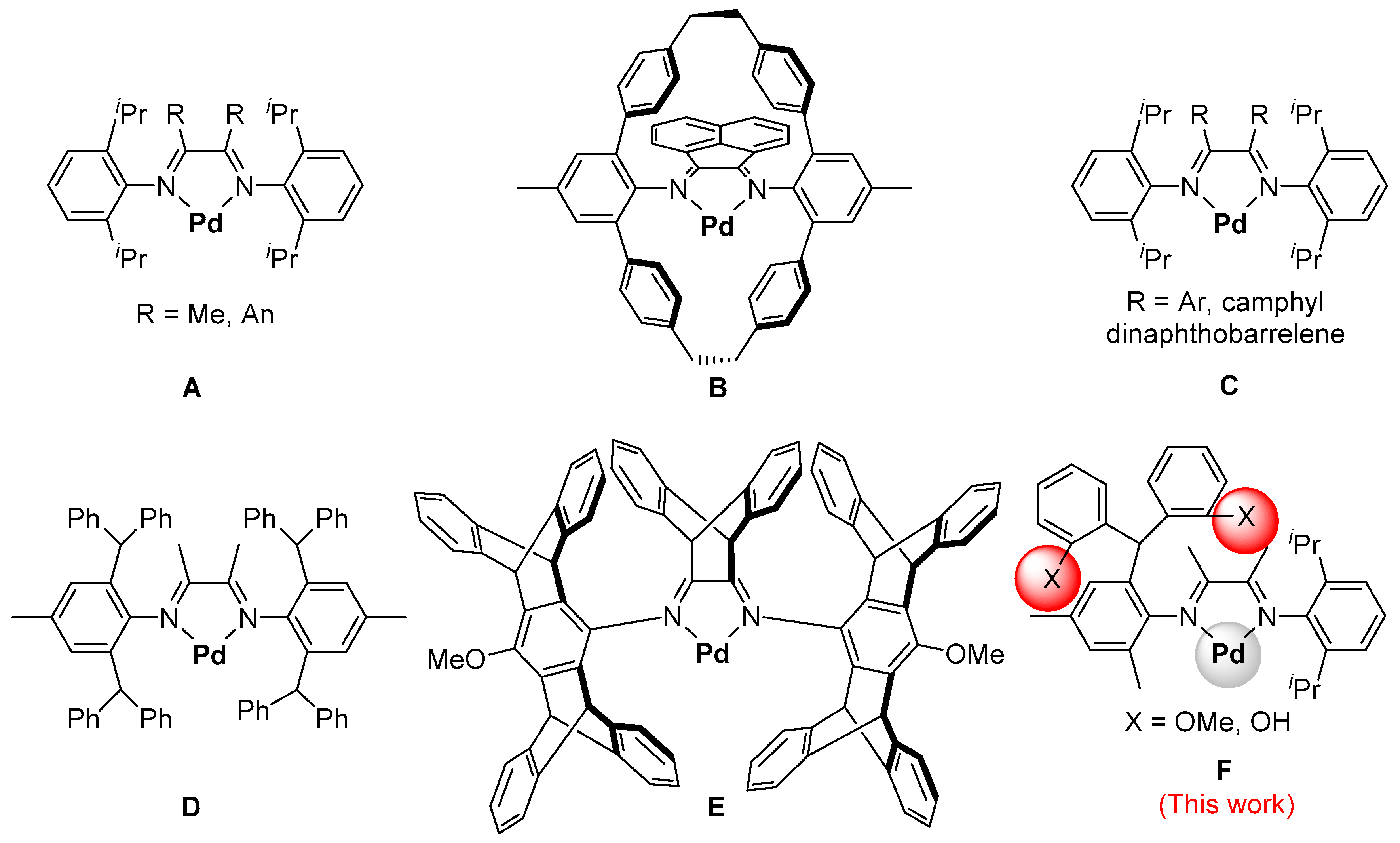
Polyolefins have been the world’s largest produced synthetic polymers [1]. Late-transition-metal catalysts can mediate olefin copolymerization with polar monomers [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21], providing a direct and potentially economic way to access functionalized polyolefins with some improved properties as well as some unique functions [22,23,24,25,26,27,28,29,30,31]. In the 1990s, Brookhart et al. reported the studies of α-diimine Pd catalysts for olefin polymerization and copolymerization with polar monomers (Chart 1A) [32,33,34]. These α-diimine Pd catalysts can produce polyolefins with branched even branch-on-branch structures while using ethylene as the only monomer via the “chain-walking” polymerization mechanism [35,36,37,38,39,40], providing a unique approach for modulating the polyolefin microstructures [41]. However, most α-diimine Pd catalysts exhibit poor thermal stability at high temperatures (>60 °C) due to decomposition reactions that are associated with C–H activation of ligand, associative chain transfer, and bisligation of M–H species, which has limited their practical applications [42,43,44,45,46,47,48,49,50]. It was demonstrated that the formation of a rigid conformation can keep the N-aryl bonds from rotation and, consequently, improve the thermal stabilities of α-diimine Pd catalysts. For instance, Guan et al. developed the α-diimine Pd catalysts containing sterically rigid cyclophane structures (Chart 1B) [51,52]. Wu and Gao et al. developed the steric bulk backbone strategies for preventing bond rotations in N-aryl moieties (Chart 1C) [53,54,55]. These catalysts exhibit much higher thermal stabilities at 80 °C when compared with the classical Brookhart’s Pd catalysts (generally have no activity above 60 °C). Recently, Chen et al. introduced the sterically bulky diarylmethyl moieties on the structures of the α-diimine Pd catalysts (Chart 1D). These catalysts bearing four diarylmethyl groups resulted in improved thermal stabilities of catalysts at high temperatures (80–100 °C), producing semi-crystalline products with low branching densities [46,47,48,49,50]. The polyethylene branching densities can be modulated by reducing the number of diarylmethyl groups [56,57]. The introduction of some para-substituents to the diarylmethyl moieties can result in the modulation of polyethylene branching densities via tuning polymerization temperature [58,59]. Jian et al. reported a series of diarylmethyl-based α-diimine Pd catalysts that contain bulky axial pentiptycenyl substituents and a bulky dibenzobarrelene backbone (Chart 1E), producing ultrahigh methyl branching polyethylenes at a wide range of temperature (0–130 °C) [60,61,62].
Many efforts have been directed towards modifying structures of ligands by installing various substituents in order to improve the performances of α-diimine Pd catalysts. However, to the best of our knowledge, so far, no α-diimine Pd catalysts bearing ortho-functionalized dibenzhydryl substituents have been reported. In this contribution, a series of α-diimine Pd catalysts bearing ortho-methoxyl/hydroxyl functionalized dibenzhydryl substituents (Chart 1F) were designed, synthesized, characterized, and investigated in ethylene polymerization and copolymerization with methyl acrylate. On account of distance, the ortho-substituents may lead to a more significant improvement of the steric hindrance effect as well as larger impact on the performance of the catalysts compared with the corresponding meta- and para-substituents [63,64,65,66,67,68,69,70,71]. Consequently, this may lead to catalyst with enhanced thermal stabilities. Moreover, the ortho-functionalization provides an approach to install functional moieties to the structures of the catalysts. For instance, the ortho-hydroxyl functionalized dibenzhydryl substituents are Brønsted acidic moieties. The hydroxyl groups may form Brønsted acid-base interactions with some polar monomers and improve the performances of the catalysts in ethylene copolymerization. Chen et al. reported that the ligand–substrate effect can improve the catalytic performances of the phosphine-sulfonate palladium and nickel catalysts bearing Brønsted basic moieties in the copolymerization of ethylene with Brønsted acidic polar monomers [3,72]. Moreover, Jordan et al. reported that the α-diimine Pd catalyst bearing the Brønsted acidic secondary amide moiety resulted in a higher incorporation of polar monomers when compared with the analogue bearing the tertiary amide unit in the ethylene copolymerizations with methyl acrylate as well as acrylic acid [73], indicating the ligand–substrate effect based on the Brønsted acid-base interaction.
2. Experimental
2.1. Materials
All of the air sensitive experiments were performed under nitrogen atmosphere while using a Schlenk line and glove box. Deuterated solvents that were used for NMR were dried and distilled prior to use. Dichloromethane was pre-dried overnight with Na2SO4 and then stirred with CaH2 under dry nitrogen for 10–12 h. Subsequently, it was distilled under inert conditions. Toluene was dried with sodium metal while using benzophenone as an indicator and distilled under nitrogen.
2.2. Methods
(COD)PdMeCl (COD = 1,5-cyclooctadiene) [74] and sodium tetrakis(3,5-bis[trifluoromethyl]phenyl)borate (NaBArF) [75] were prepared according to previously reported procedures, respectively. Other solvents and chemicals were purchased from Energy Chemicals and J&K Chemical (Beijing, China) and used as received. 1H, 13C, 1H 1H COSY, and 1H 13C COSY NMR spectra were recorded by a Bruker Ascend Tm 400 spectrometer (Bruker, Rheinstetten, Germany). Chemical shifts were referenced to a tetramethylsilane signal (0 ppm). Elemental analyses were performed on a VarioELIII instrument (Shanghai, China). Mass spectra were obtained while using electrospray ionization (ESI) LCMS-2010A (Shimadzu, Japan). Matrix-assisted laser desorption ionization-time of flight mass spectroscopy (MALDI-TOFMS) was performed on a Bruker ultrafleXtreme (Bremen, Germany). X-ray diffraction data were collected at 298(2) K on a Bruker Smart CCD area detector (Bremen, Germany) with graphite-monochromated MoKα (λ = 0.71073 nm). Molecular weight and molecular weight distribution of the polymer were determined by gel permeation chromatography (GPC) with a PL320 that was equipped with two Agilent PLgel Olexis columns at 150 °C using o-dichlorobenzene as the solvent. The calibration was generated using a polystyrene standard and it was corrected for linear polyethylene by universal calibration using the Mark–Houwink parameters of Rudin: K = 1.75 × 10−2 cm3/g and R = 0.67 for polystyrene and K = 5.90 × 10−2 cm3/g and R = 0.69 for polyethylene.
2.3. Preparation of 1a and 1b
The compounds 1a and 1b were synthesized by following the literatures procedures [76].
2.4. General Procedure for the Synthesis of 2 and 3
A mixture of 2,4-dimethylaniline (1.21 g, 0.01 mol) and alcohols 1a (2.44 g, 0.01 mol) while alcohol 1b (2.14 g, 0.01 mol) and 12.8 g of LiClO4·3H2O in 20 mL of CH3NO2 (4M soln.) were heated separately at 80 °C for 10–12 h until the completion of reaction, as monitored by TLC. After the completion of reaction, the solvent was removed under vacuum and the crude product was purified by column chromatography with 1:8 EtOAc/ petroleum ether to afford aniline 2 as an off-white solid while aniline 3 as a light yellowish solid.
Preparation of 2. Off-white solid, Yield: 78%, 1H NMR (CDCl3, 400 MHz): δ 7.25–7.20 (m, 2H, Ar), 6.8920136.83 (m, 6H, Ar), 6.77 (d, J = 4 Hz, 1H, Ar), 6.33 (d, J = 4 Hz, 1H, Ar), 6.12 (s, 1H, CHPh2), 3.72 (s, 3H, Ar-OCH3), 3.07 (br s, 2H, NH2), 2.13 (s, 3H, Ar-CH3), 2.10 (s, 3H, Ar-CH3).13C NMR (101 MHz, CDCl3): δ 157.39, 139.51, 131.09, 130.07, 129.06, 128.61, 127.51, 127.37, 126.58, 122.31, 120.25, 110.78, 55.83 (Ar-OCH3), 37.91 (CHPh2), 20.75 (Ar-CH3), 17.79 (Ar-CH3). ESI-MS (m/z): 348.19 [M + H]+.
Preparation of 3. Light yellowish solid, Yield: 75%, 1H NMR (CDCl3, 400 MHz): δ 7.29–7.19 (m, 4H, Ar), 7.12–7.10 (m, 2H, Ar), 6.92–6.86 (m, 3H, Ar), 6.80 (d, J = 4 Hz, 1H, Ar), 6.35 (d, J = 4 Hz, 1H, Ar), 5.85 (s, 1H, CHPh2), 3.74 (s, 3H, Ar-OCH3), 3.15 (br s, 2H, NH2), 2.14 (s, 3H, Ar-CH3), 2.11 (s, 3H, Ar-CH3). 13C NMR (101 MHz, CDCl3): δ 157.14, 142.86, 139.63, 130.77, 130.32, 129.55, 129.29, 128.84, 128.24, 128.09, 127.84, 126.83, 126.23, 122.54, 120.50, 110.63, 55.71 (Ar-OCH3), 44.64 (CHPh2), 20.74 (Ar-CH3), 17.81 (Ar-CH3). ESI-MS (m/z): 318.18 [M + H]+.
2.5. General Procedure for the Synthesis of 4 and 5
The anilines 2 and 3 (1mol equiv.) were separately dissolved in appropriate amount of dry DCM under nitrogen and the mixtures were cooled to −40 °C. After this BBr3 (2.5 mol equiv.) was added dropwise and then the mixture was stirred overnight at room temperature. Finally, the reaction mixtures were quenched very cautiously with ice pieces and 5% NaHCO3 solution and extracted with DCM. The solvent was rotary evaporated to afford anilines 4 and 5 in good yield.
Preparation of 4. Light yellow solid, Yield: 95%, 1H NMR (CDCl3, 400 MHz): δ 7.18–7.14 (m, 2H, Ar), 6.91–6.82 (m, 7H, Ar), 6.51 (d, J = 4 Hz, 1H, Ar), 5.83 (s, 1H, CHPh2), 4.33 (br s, 4H, NH2 & OH), 2.13 (s, 3H, Ar-CH3), 2.09 (s, 3H, Ar-CH3). 13C NMR (101 MHz, CDCl3): δ 153.98, 138.92, 130.44, 129.95, 128.74, 128.45, 127.36, 127.30, 126.40, 124.04, 120.98, 116.56, 40.10 (CHPh2), 20.75 (Ar-CH3), 17.71 (Ar-CH3). ESI-MS (m/z): 320.16 [M + H]+.
Preparation of 5. Light yellow solid, Yield: 96%, 1H NMR (CDCl3, 400 MHz): δ 7.31–7.23 (m, 4H, Ar), 7.14–7.09 (m, 3H, Ar), 6.84–6.81 (m, 3H, Ar), 6.47 (s, 1H, Ar), 5.67 (s, 1H, CHPh2), 4.32 (br s, 2H, NH2, 1H, -OH), 2.12 (s, 3H, Ar-CH3), 2.09 (s, 3H, Ar-CH3). 13C NMR (101 MHz, CDCl3): δ 154.03, 141.43, 138.56, 130.32, 130.09, 129.49, 128.91, 128.68, 128.64, 128.25, 128.17, 128.03, 126.82, 124.04, 120.72, 116.46, 46.41 (CHPh2), 20.76 (Ar-CH3), 17.76 (Ar-CH3). ESI-MS (m/z): 304.16 [M + H]+.
2.6. Procedure for the Synthesis of 6
A mixture of 2,6-diisopropylaniline (1mol equiv.) and 2,3-buten-dione (2.5 mol equiv.) was heated at 80 °C in toluene for 24 h until the completion of reaction was checked by TLC. After the completion of reaction the solvent was rotary evaporated and the product was recrystallized in n-Hexane in order to afford yellowish solid in excellent yield. Yellow solid, Yield: 86%, 1H NMR (CDCl3, 400 MHz): δ 7.17 (d, J = 8 Hz, 2H, Ar), 7.10 (t, J = 8 Hz, 1H, Ar), 2.71 (m, 2H, CH(CH3)2), 2.07 (s, 3H, CH3-C = O), 1.32–1.17 (m, 15H, CH(CH3)2 & CH3-C = N). 13C NMR (101 MHz, CDCl3): δ 168.20 (C = O), 146.18 (C = N), 135.08, 123.75, 123.01, 29.73 (CH3-C = O), 28.51 (CH(CH3)2), 23.01 (CH(CH3)2), 22.72 (CH(CH3)2), 16.60 (CH3-CN).
2.7. Procedures for the Syntheses of the Ligands
Preparation of L1. Aniline 2 (1.2 mol equiv.) and compound 6 (1 mol equiv.) were heated in toluene using Dean-Stark apparatus in the presence of catalytic amount of p-toluenesulphonic acid. On completion of reaction, as checked by TLC, the solvent was rotatory evaporated and crude product was stirred in methanol overnight. The yellow precipitates were filtered and recrystallized in 2:1 DCM and methanol to afford pure product. Yellow solid, Yield: 76%, 1H NMR (CDCl3, 400 MHz): δ 7.20–7.11 (m, 4H, Ar), 7.05 (t, J = 8Hz, 1H, Ar), 6.90 (d, J = 4 Hz, 1H, Ar), 6.85–6.78 (m, 6H, Ar), 6.49 (d, J = 4 Hz, 1H, Ar), 6.05 (s, 1H, CHPh2), 3.65 (s, 3H, Ar-OCH3), 3.61 (s, 3H, Ar-OCH3), 2.64–2.52 (m, 2H, CH(CH3)2), 2.20 (s, 3H, CH3-C = N), 1.95 (s, 3H, Ar-CH3), 1.88 (s, 3H, Ar-CH3), 1.66 (s, 3H, CH3-C = N), 1.21 (d, J = 4Hz, 3H, CH(CH3)2), 1.11 (dd, J = 4 Hz, 8 Hz, 9H, CH(CH3)2). 13C NMR (101 MHz, CDCl3): δ 168.21 (C = N), 168.16 (C = N), 157.52, 157.04, 146.38, 145.41, 135.03, 134.81, 132.00, 131.69, 130.79, 130.10, 128.88, 127.15, 127.03, 123.88, 123.47, 123.00, 122.69, 120.18, 111.08, 110.50, 56.00 (Ar-OCH3), 55.42 (Ar-OCH3), 38.84 (CHPh2), 28.39 (CH(CH3)2), 28.16 (CH(CH3)2), 23.04 (CH(CH3)2), 22.97 (CH(CH3)2), 22.68 (CH(CH3)2), 22.61 (CH(CH3)2), 21.08 (Ar-CH3), 17.57(Ar-CH3), 16.00 (C = NCH3), 15.95 (C = NCH3). ESI-MS (m/z): 575.36 [M + H]+.
Preparation of L2. Aniline 3 (1.2 mol equiv.) and compound 6 (1 mol equiv.) were heated in toluene using Dean–Stark apparatus in the presence of catalytic amount of p-Toluenesulphonic acid. On completion of reaction, as checked by TLC, the solvent was rotatory evaporated and crude product was stirred in methanol overnight. The yellow precipitates were filtered and recrystallized in DCM to afford pure product. Yellow solid, Yield: 72%, 1H NMR (CDCl3, 400 MHz): δ 7.27–7.03 (m, 9H, Ar), 6.93–6.84 (m, 4H, Ar), 6.56–6.53 (m, 1H, Ar), 5.82, 5.69 (s, 1H, CHPh2), 3.69, 3.62 (s, 3H, Ar-OCH3), 2.62–2.49 (m, 2H, CH(CH3)2), 2.22 (s, 3H, CH3-C = N), 1.99, 1.96 (s, 3H, Ar-CH3), 1.95, 1.91 (s, 3H, Ar-CH3), 1.57, 1.55 (s, 3H, CH3-C = N), 1.22 (dd, J = 4Hz, 8 Hz, 3H, CH(CH3)2), 1.14–1.08 (m, 9H, CH(CH3)2). 13C NMR (101 MHz, CDCl3): δ 168.67 (C = N), 167.99 (C = N), 157.13, 156.93, 146.27, 145.44, 143.42, 143.28, 135.01, 134.71, 132.75, 132.22, 132.02, 131.83, 130.80, 130.50, 129.87, 129.57, 129.13, 128.14, 127.96, 127.89, 127.40, 127.32, 126.87, 125.88, 124.15, 124.00, 123.60, 123.56, 123.01, 122.94, 122.74, 120.33, 120.13, 110.84, 110.41, 55.82 (Ar-OCH3), 55.37(Ar-OCH3), 45.87 (CHPh2), 45.32 (CHPh2), 28.46 (CH(CH3)2), 28.16 (CH(CH3)2), 28.03 (CH(CH3)2), 23.34 (CH(CH3)2), 23.06 (CH(CH3)2), 22.96 (CH(CH3)2), 22.93(CH(CH3)2), 22.71(CH(CH3)2), 22.59 (CH(CH3)2), 21.08 (Ar-CH3), 17.88 (Ar-CH3), 17.55 (Ar-CH3), 16.49 (C = NCH3), 16.07(C = NCH3), 15.96 (C = NCH3), 15.79 (C = NCH3). ESI-MS (m/z): 545.36 [M + H]+.
Preparation of L3. Aniline 4 (1.2 mol equiv.) and compound 6 (1 mol equiv.) were heated in toluene and ethanol (1:1) keeping the temperature 105–110 °C in the presence of catalytic amount of p-toluenesulphonic acid. On completion of reaction as checked by TLC, the solvent was rotatory evaporated and crude product was stirred in small amount of DCM and excess of n-Hexane overnight. The yellow precipitates were filtered and recrystallized in ethanol in order to afford pure product. Yellow solid, Yield: 70%, 1H NMR (CDCl3, 400 MHz): δ 7.15–7.04 (m, 4H, Ar), 6.97–6.93 (m, 2H, Ar), 6.88–6.72 (m, 6H, Ar), 6.13 (br, s, 1H, -OH), 5.72 (s, 1H, CHPh2), 5.42 (br, s, 1H, -OH), 2.60–2.45 (m, 2H, CH(CH3)2), 2.22 (s, 3H, CH3-C = N), 1.98 (s, 3H, Ar-CH3), 1.87 (s, 3H, Ar-CH3), 1.54 (s, 3H, CH3-C = N), 1.19 (d, J = 4 Hz, 3H, CH(CH3)2) 1.09 (dd, J = 4Hz, 8Hz, 9H, CH(CH3)2). 13C NMR (101 MHz, CDCl3): δ 170.36 (C = N), 167.71(C = N), 153.87, 153.83, 145.93, 144.77, 134.99, 134.91, 133.39, 130.93, 130.38, 129.92, 129.85, 129.42, 128.24, 128.05, 128.00, 127.71, 127.49, 125.36, 123.80, 123.00, 122.92, 120.70, 116.28, 116.25, 40.67 (CHPh2), 28.54 (CH(CH3)2), 27.96 (CH(CH3)2), 23.39 (CH(CH3)2), 22.91 (CH(CH3)2), 22.84 (CH(CH3)2), 22.74 (CH(CH3)2), 21.04 (Ar-CH3), 17.62 (Ar-CH3), 16.32 (C = NCH3), 16.28 (C = NCH3). ESI-MS (m/z): 547.33 [M + H]+.
Preparation of L4. Aniline 5 (1.2 mol equiv.) and compound 6 (1 mol equiv.) were refluxed in toluene and ethanol (1:1) keeping the temperature 105–110 °C in the presence of catalytic amount of p-toluenesulphonic acid. On completion of reaction, as checked by TLC, the solvent was rotatory evaporated and the crude product was stirred in small amount of DCM and excess of n-hexane overnight. The yellow precipitates were filtered and recrystallized in DCM to afford pure product. Yellow solid, Yield: 68%, 1H NMR (CDCl3, 400 MHz): δ 7.23–7.13 (m, 3H, Ar), 7.09–6.97 (m, 6H, Ar), 6.92–6.68 (m, 5H, Ar), 5.57, 5.41 (s, 1H, CHPh2), 2.55–2.39 (m, 2H, CH(CH3)2), 2.18, 2.17 (s, 3H, CH3-C = N), 1.98, 1.92 (s, 3H, Ar-CH3), 1.89, 1.87 (s, 3H, Ar-CH3), 1.46, 1.40 (s, 3H, CH3-C = N), 1.14 (dd, J = 4 Hz, 8 Hz, 3H, CH(CH3)2), 1.08–1.02 (m, 9H, CH(CH3)2). 13C NMR (101 MHz, CDCl3): δ 170.33 (C = N), 167.88 (C = N), 167.51(C = N), 154.05, 153.98, 145.99, 145.11, 142.03, 141.66, 141.38, 139.59, 135.01, 134.93, 133.23, 132.81, 132.03, 131.18, 130.39, 130.30, 130.11, 129.79, 129.68, 129.44, 129.32, 128.93, 128.83, 128.72, 128.43, 128.38, 128.18, 127.92, 127.88, 127.85, 127.20, 126.86, 126.46, 125.07, 124.89, 123.83, 123.72, 123.45, 123.03, 122.98, 120.79, 120.68, 120.36, 116.43, 116.24, 46.86 (CHPh2), 46.17 (CHPh2), 28.53 (CH(CH3)2), 28.12 (CH(CH3)2), 27.92 (CH(CH3)2), 23.44 (CH(CH3)2), 23.34 (CH(CH3)2), 22.98 (CH(CH3)2), 22.90 (CH(CH3)2), 22.82 (CH(CH3)2), 22.75 (CH(CH3)2), 22.70 (CH(CH3)2), 21.06 (CH(CH3)2), 20.69 (Ar-CH3), 17.76(Ar-CH3), 17.62 (Ar-CH3), 17.58 (Ar-CH3), 16.66(C = NCH3), 16.37(C = NCH3), 16.29(C = NCH3), 15.95 (C = NCH3). ESI-MS (m/z): 531.33 [M + H]+.
2.8. General Procedure for the Synthesis of the Pd Catalysts
The ligands L1, L2, L3, L4 (1mol equiv.) and (COD)PdMeCl were stirred in a suitable amount of dry DCM in glove box for 3-4 days at room temperature. During stirring, the ligands were completely dissolved and the color of the solution was changed from yellow to red. At the end of the reaction, the desired compound was isolated while using column chromatography. The mixture was eluted on silica gel with first 1:1 hexane/CH2Cl2, and then pure CH2Cl2 as the mobile phase.
Preparation of Pd1. Yellow solid, Yield: 88%, 1H NMR (CDCl3, 400 MHz): δ 7.26–7.18 (m, 5H, Ar), 7.06–6.96 (m, 3H, Ar), 6.91–6.82 (m, 5H, Ar), 6.19, 6.01 (s, 1H, CHPh2), 3.79, 3.75 (s, 3H, Ar-OCH3), 3.58, 3.54 (s, 3H, Ar-OCH3), 3.18–3.08, 3.05–2.94 (m, 2H, CH(CH3)2), 2.32, 2.27 (s, 3H, Ar-CH3), 2.23, 2.18 (s, 3H, Ar-CH3), 1.78, 1.72 (s, 3H, CH3-C = N), 1.57–1.20 (m, 9H, CH(CH3)2), 1.10–1.07 (m, 6H, CH(CH3)2 & CH3-C = N), 0.60, 0.45 (s, 3H, CH3Pd). 13C NMR (101 MHz, CDCl3): δ 174.82 (C = N), 174.01(C = N), 169.73 (C = N), 169.11 (C = N), 158.11, 157.75, 157.51, 157.34, 141.68, 141.63, 141.40, 141.24, 138.89, 138.51, 138.08, 137.81, 135.23, 134.68, 133.23, 132.51, 131.96, 131.78, 131.52, 131.07, 130.63, 130.37, 130.00, 129.18, 128.68, 128.09, 128.05, 127.93, 127.73, 127.57, 127.48, 127.40, 126.84, 123.93, 123.84, 123.25, 123.18, 121.10, 121.07, 120.35, 119.56, 113.51, 112.93, 111.00, 110.61, 56.96 (Ar-OCH3), 55.98 (Ar-OCH3), 55.18 (Ar-OCH3), 40.28 (CHPh2), 28.90 (CH(CH3)2), 28.56 (CH(CH3)2), 28.30 (CH(CH3)2), 27.91 (CH(CH3)2), 24.22 (CH(CH3)2), 24.16 (CH(CH3)2), 23.82 (CH(CH3)2), 23.68 (CH(CH3)2), 23.51(CH(CH3)2), 23.48 (CH(CH3)2), 23.19(CH(CH3)2), 21.33 (Ar-CH3), 21.19 (Ar-CH3), 20.60 (Ar-CH3), 19.66 (Ar-CH3), 19.02 (CH3-CN), 18.99 (CH3-CN), 18.89 (CH3-CN), 17.92 (CH3-CN), 2.65 (CH3Pd), 2.16 (CH3Pd). Key C-H HSQC: δ 6.19/40.28 (CHPh2), 3.79/56.97 (Ar-OCH3), 3.58/55.18 (Ar-OCH3), 3.12/28.71 (CH(CH3)2), 2.27/21.33 (Ar-CH3), 2.18/20.60 (Ar-CH3), 1.72/18.99 (CH3-CN), 1.10/17.99 (CH3-CN), 0.60/2.16 (CH3Pd). MALDI-TOF-MS (m/z): 766.57 [M + HCl]+. Anal. Calcd. for C40H49ClN2O2Pd: C, 65.66, H, 6.75, N, 3.83 Found: C, 65.62, H, 6.71, N, 3.79.
Preparation of Pd2. Yellowish solid, Yield: 89%, 1H NMR (CDCl3, 400 MHz): δ 7.31–7.09 (m, 10H, Ar), 7.00–6.84 (m, 4H, Ar), 6.21, 6.16, 6.02 (s, 1H, CHPh2), 3.91, 3.83, 3.79, 3.60, 3.59, 3.53 (s, 3H, Ar-OCH3), 3.28–3.21, 3.16–3.02, 2.98–2.90 (m, 2H, CH(CH3)2), 2.28, 2.26, (s, 3H, Ar-CH3), 2.23, 2.14 (s, 3H, Ar-CH3), 1.74, 1.73, 1.66 (s, 3H, CH3-C = N), 1.56 1.04 (m, 12H, CH(CH3)2), 0.97, 0.90, 0.88 (s, 3H, CH3-C = N), 0.65, 0.58, 0.54 (s, 3H, CH3Pd). 13C NMR (101 MHz, CDCl3): δ 175.55 (C = N), 175.28 (C = N), 174.35 (C = N), 174.27 (C = N), 170.63 (C = N), 169.49 (C = N), 169.19 (C = N), 158.46, 158.32, 157.09, 143.23, 142.55, 141.68, 141.54, 141.50, 141.47, 141.19, 138.76, 138.72, 138.56, 138.07, 137.98, 137.95, 137.77, 135.86, 135.57, 134.97, 134.76, 134.62, 134.44, 133.78, 132.79, 132.10, 131.98, 131.78, 130.96, 130.82, 130.75, 130.53, 130.34, 130.30, 129.83, 129.74, 129.48, 129.41, 129.39, 129.32, 129.25, 129.06, 128.64, 128.47, 128.45, 128.43, 128.11, 128.03, 127.94, 127.89, 127.74, 127.66, 127.63, 127.60, 127.20, 127.00, 126.92, 126.88, 126.62, 126.37, 126.32, 125.90, 123.99, 123.95, 123.86, 123.31, 123.30, 123.21, 121.16, 121.07, 119.57, 119.31, 114.04, 113.46, 110.87, 110.70, 100.90, 57.49 (Ar-OCH3), 56.90 (Ar-OCH3), 55.49 (Ar-OCH3), 55.04 (Ar-OCH3), 47.22 (CHPh2), 46.50 (CHPh2), 45.43 (CHPh2), 31.05 (CHPh2), 28.96 (CH(CH3)2), 28.94 (CH(CH3)2), 28.87 (CH(CH3)2), 28.62 (CH(CH3)2), 28.42 (CH(CH3)2), 28.33 (CH(CH3)2), 28.01 (CH(CH3)2), 27.66 (CH(CH3)2), 24.37 (CH(CH3)2), 24.26 (CH(CH3)2), 24.06 (CH(CH3)2), 23.96 (CH(CH3)2), 23.89 (CH(CH3)2), 23.85 (CH(CH3)2), 23.80(CH(CH3)2), 23.58(CH(CH3)2), 23.50 (CH(CH3)2), 23.48(CH(CH3)2), 23.35 (CH(CH3)2), 23.16(CH(CH3)2), 23.01(CH(CH3)2), 21.36 (Ar-CH3), 21.27(Ar-CH3), 21.24 (Ar-CH3), 21.15 (Ar-CH3), 20.62 (Ar-CH3), 20.47 (Ar-CH3), 19.50 (Ar-CH3), 19.29 (Ar-CH3), 19.13 (CH3-C = N), 19.00 (CH3-C = N), 18.62 (CH3-C = N), 18.32 (CH3-C = N), 18.21 (CH3-C = N), 17.78 (CH3-C = N), 17.64 (CH3-C = N), 17.56 (CH3-C = N), 3.00 (CH3Pd), 2.94(CH3Pd), 2.53 (CH3Pd), 2.19 (CH3Pd). MALDI-TOF-MS (m/z): 738.61 [M + 3H,Cl] +. Anal. Calcd. for C39H47ClN2OPd: C, 66.76, H, 6.75, N, 3.99 Found: C, 66.70, H, 6.69, N, 3.93.
Preparation of Pd3. Redish solid, Yield: 78%, 1H NMR (CDCl3, 400 MHz): δ 7.32–7.13 (m, 5H, Ar), 7.05–7.00 (m, 3H, Ar), 6.85–6.82 (m, 5H, Ar), 6.12 (br, s, 1H, -OH), 5.57 (s, 1H, CHPh2), 3.15–2.98 (m, 2H, CH(CH3)2), 2.27 (s, 3H, Ar-CH3), 2.09 (s, 3H, Ar-CH3), 1.71 (s, 3H, CH3-C = N), 1.39 (d, J = 8 Hz, 3H, CH(CH3)2), 1.30–1.24 (m, 9H, CH(CH3)2 & CH3-C = N). 1.05 (d, J = 8 Hz, 3H, CH(CH3)2), 0.57 (s, 3H, CH3Pd). 13C NMR (101 MHz, CDCl3): δ 174.98 (C = N), 171.35(C = N), 155.09, 141.29, 138.95, 138.56, 131.55, 130.72, 130.07, 128.80, 128.37, 127.89, 124.22, 123.86, 120.51, 120.21, 118.50, 29.71 (CHPh2), 28.57 (CH(CH3)2), 28.09 (CH(CH3)2), 24.23 (CH(CH3)2), 23.83(CH(CH3)2), 23.81(CH(CH3)2), 22.89 (CH(CH3)2), 21.30 (Ar-CH3), 20.52 (Ar-CH3), 19.06 (CH3-C = N), 17.59 (CH3-C = N), 4.38 (CH3Pd) MALDI-TOF-MS (m/z): 738.78 [M +HCl]+. Anal. Calcd. for C38H45ClN2O2Pd: C, 64.86, H, 6.45, N, 3.98 Found: C, 65.72, H, 6.38, N, 3.90.
Preparation of Pd4. Redish solid, Yield: 68%, 1H NMR (CDCl3, 400 MHz): δ 7.32–7.16 (m, 9H, Ar), 7.03–6.97 (m, 3H, Ar), 6.78–6.73 (m, 2H, Ar), 5.99 (s, 1H, CHPh2), 3.17–2.96 (m, 2H, CH(CH3)2), 2.26 (s, 3H, Ar-CH3), 2.08 (s, 3H, Ar-CH3), 1.68 (s, 3H, CH3-C = N), 1.46 (d, J = 4 Hz, 3H, CH(CH3)2), 1.33–1.26 (m, 6H, CH(CH3)2), 1.06 (d, J = 4 Hz, 3H, CH(CH3)2), 1.02 (s, 3H, CH3-C = N), 0.57 (s, 3H, CH3Pd). 13C NMR (101 MHz, CDCl3): δ 174.80 (C = N), 171.13 (C = N), 155.27, 142.87, 141.45, 141.32, 138.61, 138.45, 135.99, 132.18, 130.69, 130.20, 130.17, 128.54, 128.30, 127.87, 127.27, 126.84, 126.77, 126.60, 124.00, 119.22, 117.80, 48.03 (CHPh2), 28.51 (CH(CH3)2), 28.49 (CH(CH3)2), 24.17 (CH(CH3)2), 23.79 (CH(CH3)2), 23.59 (CH(CH3)2), 22.97 (CH(CH3)2), 21.28 (Ar-CH3), 20.46 (Ar-CH3), 18.58 (CH3-C = N), 17.52 (CH3-C = N), 4.20 (CH3Pd) Key C-H HSQC: δ 5.99/48.03 (CHPh2), 3.17–2.96/28.51–28.49 (m, 2H, CH(CH3)2), 2.26/21.28 (Ar-CH3), 2.08/20.46 (Ar-CH3), 1.68/18.58 (CH3-C = N), 1.02/17.52 (CH3-C = N), 0.57/4.20 (CH3Pd). MALDI-TOF-MS (m/z): 738.61 [M-CH3,Cl]+. Anal. Calcd. for C38H45ClN2OPd: C, 66.37, H, 6.60, N, 4.07 Found: C, 66.29, H, 6.59, N, 4.03.
2.9. General Procedure for Ethylene Polymerization
In a Glovebox, a 10 mL glass thick-walled pressure vessel was charged with sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (NaBAF, 1.2 mol eq.) in 2 mL toluene and a magnetic bar. The vessel was pressurized with ethylene and then allowed to equilibrate under constant pressure for a few minutes with stirring. Pre-catalysts (1 mol eq.) (Pd1), (Pd3), (Pd2), and (Pd4) were injected to initiate polymerization respectively and continuously stirred for the desired time under specific temperature. The polymerization was quenched via the addition of MeOH and the polymer was precipitated while using excess MeOH and dried in a vacuum oven to a constant weight. Branch density (BD) was analyzed by using 1HNMR spectrum: BD = 1000 × (2/3) × (ICH3)/(ICH2 and CH + ICH3). δ (ppm): CH3 (m, 0.77–0.95), CH2 and CH (m, ca. 1.0–1.45) [46].
2.10. General Procedure for Pd Catalyzed Ethylene Copolymerization
A 10 mL glass thick-walled pressure vessel was heated at 150 °C for 2–3 h under vacuum and then cooled to room temperature. The flask was pressurized with 3-atm ethylene and evacuated three times. 2 M solution of polar monomer (MA) and NaBAF (1.2 mol eq.) were introduced into the flask with an appropriate amount of toluene followed by the addition of Pd1–4 catalyst (30 μmol) in DCM under ethylene atmosphere. Ethylene pressure was maintained at 3-atm for 5 h by continuously feeding ethylene. Finally, the polymerization was terminated by the addition of methanol. Subsequently, the oily polymers were further washed with methanol and then dried in vacue at 50 °C. The incorporation of MA was calculated by 1H NMR spectrum. The branches ending with the functional group were added to the total branches. MA% = 4 IOMe/3(ICH3 + ICH2 + ICH) × 100%. OMe (s, ca. 3.61–3.76 ppm); CH2 and CH (m, ca. 1.0–1.45 ppm); CH3 (m, 0.77–0.95 ppm) [46].
3. Results and Discussion
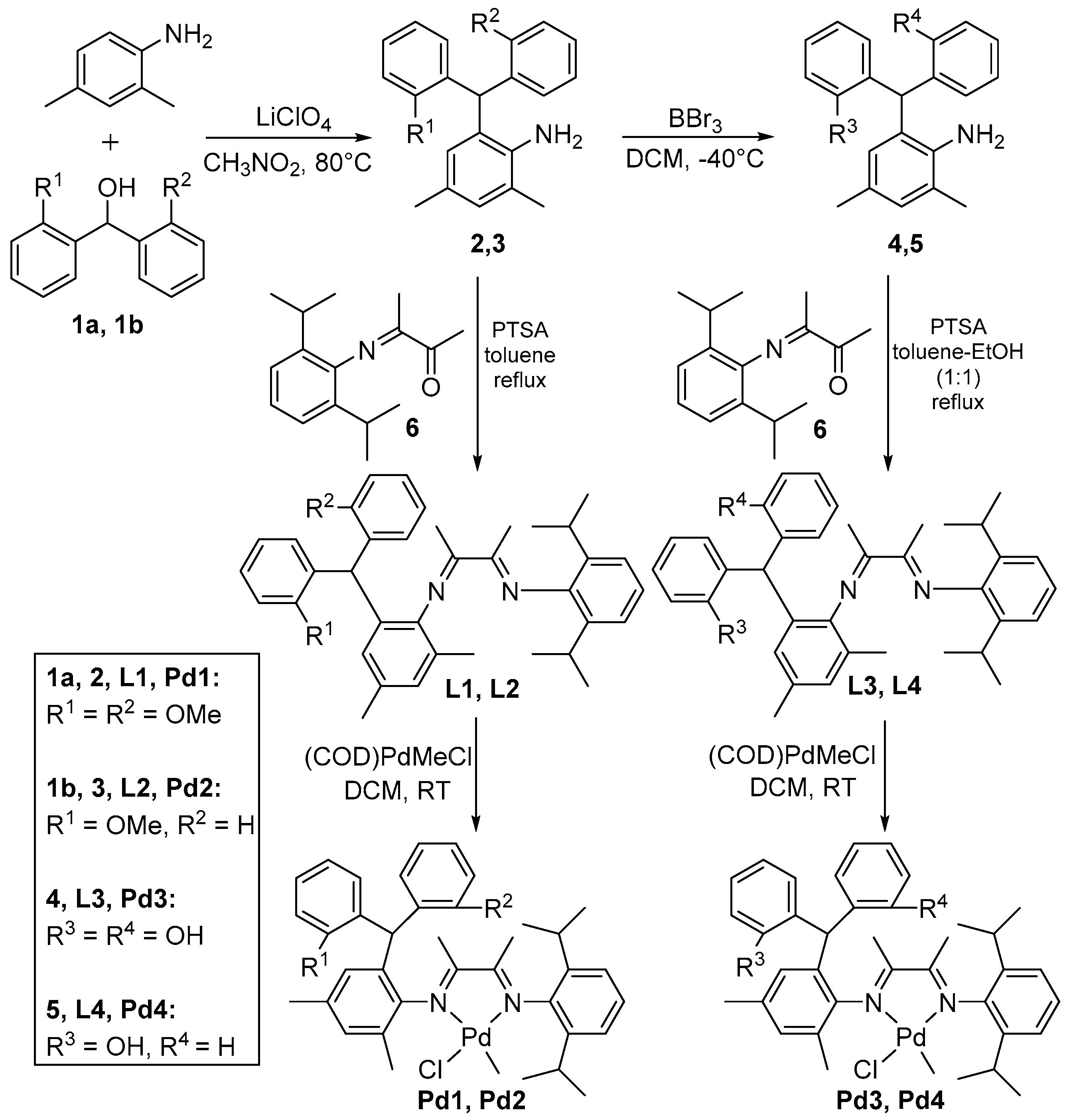
The anilines 2 and 3 were synthesized by the reactions of 2,4-dimethylaniline with 1,1-bis(2-methoxyphenyl)methanol or 1-(2-methoxyphenyl)-1-phenylmethanol in a solution of trihydrated lithiumperchlorate in nitromethane at 80 °C in 70–95% yields (Scheme 1) [76]. Demethylation was carried out by treating with boron tribromide at −40 °C in dichloromethane, forming the anilines 4 and 5 in 95% and 96% yields, respectively. The demethylation of anilines was confirmed by 1H NMR with the disappearance of methoxy signals and appearance of signals of hydroxyl proton at 4.3 ppm in CDCl3. Compound 6 was synthesized by heating 2,6-diisopropylaniline with 2,3-butandione in toluene at 80 °C. Ligands L1 and L2 were synthesized by refluxing 6 with the anilines 2 and 3 in toluene that was catalyzed by p-toluenesulfonic acid (PTSA). The ligand L3 and L4 were synthesized by heating the mixture of 6 with 4 or 5 in toluene and ethanol (1:1) at 105 °C in the presence of PTSA (Scheme 1). All of the ligands were characterized by 1H NMR, 13C NMR and ESI-MS. L1 and L3 were detected as a single isomer with single set of NMR peaks. L2 and L4 exhibit cis and trans isomers with two sets of peaks having different intensities. The more intense peaks may belong to trans-isomer due to its greater stability than cis-isomer at room temperature.
The palladium complexes were prepared by stirring the ligands with (COD)PdMeCl (COD = 1,5-cyclooctadiene) in dichloromethane (DCM) for 3–4 days at room temperature (Scheme 1) [46,47,48,49,50]. The formations of the metal complexes were confirmed by 1H NMR, 13C NMR, COSY, HSQC, and MALDI-TOF-MS. Pd1 was observed in two isomeric forms with respect to the orientation of methyl and chloride attached with palladium in unsymmetrical α-diimine system. Both of these isomers were detected in 1H NMR, 13C NMR, COSY, and HSQC spectra. For simplicity in 1H NMR spectrum assignment, two sets of peaks were treated for a single isomer, because many peaks were in overlapped form and also with different intensities indicating different proportion of isomers, but the differentiated peaks have been assigned with their corresponding values. Furthermore, the MALDI-TOF confirmed these are the isomers of same catalyst Pd1 with single mass value (Figure S41 from Supplementary Materials). Catalyst Pd2 has chiral center in mono-substituted dibenzhydryl unit, with racemic (RS) configuration. The orientation of methyl and chloride attached with palladium makes more isomers. Therefore, the 1H NMR and 13C NMR spectra of this catalyst were massive (Figures S24–S27 from Supplementary Materials). Although we separated some of these isomers using preparative TLC and compared the 1H NMR and 13C NMR spectra of these isomers, we were not able to separate them in complete proportions. The MALDI-TOF analysis confirmed that these are the isomers of same catalyst Pd2 with single mass value. On the other hand, catalysts Pd3 and Pd4 with hydroxyl substitution pattern were detected as single isomers in 1H NMR and 13CNMR spectra (Figures S28–S32 from Supplementary Materials).
Single crystals of catalysts Pd1 and Pd2 were obtained in DCM with the layering of n-hexane while complex Pd3 and Pd4 also in DCM, but with the slow diffusion of diethyl ether. The molecular structures were determined by X-ray diffraction (Figure 1), with observed bond lengths and bond angles typically for previously reported α-diimine palladium complexes [42,43,44,45,46,47,48,49,50]. The palladium center with square planner geometry has been observed in all of the complexes. The distance from metal center to oxygen of methoxy group was recorded 4.27 and 4.43 Å in Pd1 and Pd2, respectively. In Pd3 and Pd4, the distance between hydrogen of hydroxyl and chloride attached to palladium was found 2.49 and 2.33 Å, respectively, confirming the perfect intramolecular H-bonding. Furthermore, the N-Pd-N bite angle was comparable in all of catalysts, ranging from 77.4° to 77.9°.
By the activation of 1.2 equiv. NaBAF, the α-diimine Pd catalysts Pd1–Pd4 were found to be highly active in ethylene polymerization (Table 1, entries 1–12). Both catalytic activity and molecular weight increased significantly, along with the increasing number of ortho-substituents (Table 1, entries 1–3 versus 4–6, entries 7–9 versus 10–12), indicating that the ortho-substituents can lead to the significant improvement of steric-hindrance and improve the catalytic performance of the catalysts [42,43,44,45]. All of these catalysts exhibit high activities of up to 8.8 × 105 g PE (mol Pd h)−1 at 80 °C, and high polymer Mn (up to 3.93 × 104 g mol−1) in ethylene polymerization. The activities and molecular weights decreased less than an order of magnitude at 80 °C compared with at 40 °C. These results indicated that the sterically bulky ortho-functionalized dibenzhydryl groups can prevent the N-aryl bonds from rotation and, consequently, improve the thermal stabilities of the catalysts. The activities and molecular weights were comparable with the previously reported α-diimine Pd catalysts bearing four dibenzhydryl groups. However, the catalysts Pd1~Pd4 produced amorphous oily polyethylenes with high branching densities, while the α-diimine Pd catalysts bearing four dibenzhydryl groups produced semi-crystalline products with low branching densities [46,47,48,49,50]. The previously reported isopropyl-based α-diimine Pd catalysts bearing a dinaphthobarrelene-backbone or a nitrogen-containing second-coordination-sphere exhibited relatively low catalytic activities at 80 °C, and generated highly branched polyethylenes with low molecular weight and broad molecular weight distributions [55,77]. The catalysts (Pd3 and Pd4) bearing hydroxyl groups showed similar activities as well as molecular weights of polyethylenes at 40 °C when compared with the catalysts (Pd1 and Pd2) bearing methoxyl groups. They resulted in higher branching densities than those of their simulants bearing methoxyl groups, indicating that the methoxyl groups exhibit increased steric-hinerance effect when compared with the hydroxyl groups.
All of these Pd catalysts exhibit high activities in ethylene copolymerization with methyl acrylate (MA) (Table 2, entries 1–4). The incorporation ratios of MA decreased with the number of ortho-substituents increasing (Table 2, entries 1 versus 2–4), which indicated that the steric-hindrance effect that was associated with the ortho-substituents prevents the polar monomer from copolymerization reaction. The catalysts (Pd3 and Pd4) bearing hydroxyl groups resulted in slightly higher incorporation ratios of MA compared with the catalysts (Pd1 and Pd2) bearing methoxyl groups (Table 2, entries 1 versus 3, and 2 versus 4). This could be due to the ligand-substrate effect based on the Brønsted acid-base interaction [73]. Nevertheless, it can also be due to the less steric-hinerance effect that is associated with the hydroxyl groups [57].
4. Conclusions
In summary, a series of α-diimine Pd catalysts bearing ortho-methoxyl/hydroxyl functionalized dibenzhydryl substituents have been designed, synthesized, characterized, and investigated in ethylene polymerization and copolymerization with methyl acrylate. The catalytic activity and molecular weight of polyethylene product increased significantly with the increasing number of ortho-substituents, which suggested that the ortho-substituents resulted in the significant improvement of steric-hindrance associated with the improved catalytic performance. These Pd catalysts displayed remarkable thermal stability in ethylene polymerization, maintaining high activity of up to 8.8 × 105 g PE (mol Pd h)−1 at 80 °C, and produced high molecular weight polyethylenes (Mn up to 3.93 × 104 g mol−1), suggesting that the sterically bulky ortho-functionalized dibenzhydryl groups can keep the N-aryl bonds from rotation at high temperatures and improve the thermal stabilities of the catalysts. The Pd catalysts exhibit high activities in ethylene copolymerization with MA. The catalysts bearing hydroxyl groups resulted in slightly higher incorporation ratios of MA unit when compared with the catalysts bearing methoxyl groups. It is envisaged that ortho-functionalized dibenzhydryl substituents will be applicable to other catalytic systems for olefin polymerizations.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4360/12/11/2509/s1. Figures S1–S32. NMR spectra of the compounds 2-6, ligands L1-L4 and catalysts Pd1-Pd4 in CDCl3; Figures S33–S44. MS spectra of the compounds 2-6, ligands L1-L4 and catalysts Pd1-Pd4; Figures S45–S60. NMR spectra of the (co)polymers; Figures S61–S76. GPC results of the (co)polymers. Crystal data structural refinement for Pd1-Pd4. CCDC 1941586, 1957450, 1941588 and 1959442 contain the supplementary crystallographic data for this paper. Copies of the data can be obtained free of charge by application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [Fax: (internat.) +44-1223/336-033; E-mail: [email protected]].
Author Contributions
Q.M. and W.P. performed experiments and data analyses. C.T. and F.W. wrote and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 22001004 and 21801002), Q. Muhammad is very thankful of CAS-TWAS President fellowship for financial support for Ph.D.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stgrzel, M.; Mihan, S.; Mulhaupt, R. From multisite polymerization catalysis to sustainable materials and all-polyolefin composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Chen, C.L. Nickel catalysts for the synthesis of ultra-high molecular weight polyethylene. Sci. Bull. 2020, 65, 1137–1138. [Google Scholar] [CrossRef]
- Chen, C.L. Designing transition metal catalysts for olefin polymerization and copolymerization: Beyond electronic and steric tuning. Nat. Rev. Chem. 2018, 2, 6–14. [Google Scholar] [CrossRef]
- Tan, C.; Chen, C.L. Emerging palladium and nickel catalysts for copolymerization of olefins with polar monomers. Angew. Chem. Int. Ed. 2019, 58, 7192–7200. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Chen, C.L. A continuing legend: The Brookhart-type α-diimine nickel and palladium catalysts. Polym. Chem. 2019, 10, 2354–2369. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J.M.; Long, B.K. Recent developments in redox-active olefin polymerization catalysts. Coord. Chem. Rev. 2018, 372, 141–152. [Google Scholar] [CrossRef]
- Ito, S. Palladium-catalyzed homo- and copolymerization of polar monomers: Synthesis of aliphatic and aromatic polymers. Bull. Chem. Soc. Jpn. 2018, 91, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Anselment, T.M.J.; Claverie, J.; Goodall, B.; Jordan, R.F.; Mecking, S.; Rieger, B.; Sen, A.; Van Leeuwen, P.W.N.M.; Nozaki, K. Ortho-phosphinobenzenesulfonate: A superb ligand for palladium-catalyzed coordination-insertion copolymerization of polar vinyl monomers. Acc. Chem. Res. 2013, 46, 1438–1449. [Google Scholar] [CrossRef]
- Carrow, B.P.; Nozaki, K. Transition-Metal-Catalyzed Functional Polyolefin Synthesis: Effecting Control through Chelating Ancillary Ligand Design and Mechanistic Insights. Macromolecules 2014, 47, 2541–2555. [Google Scholar] [CrossRef]
- Keyes, A.; Basbug, A.H.; Ordonez, E.; Ha, U.; Beezer, D.B.; Dau, H.; Liu, Y.S.; Tsogtgerel, E.; Jones, G.R.; Harth, E. Olefins and Vinyl Polar Monomers: Bridging the Gap for Next Generation Materials. Angew. Chem. Int. Ed. 2019, 58, 12370–12391. [Google Scholar] [CrossRef]
- Boucher-Jacobs, C.; Rabnawaz, M.; Katz, J.S.; Even, R.; Guironnet, D. Encapsulation of catalyst in block copolymer micelles for the polymerization of ethylene in aqueous medium. Nat. Commun. 2018, 9, 841. [Google Scholar] [PubMed]
- Liang, T.; Goudari, S.; Chen, C.L. A Simple and Versatile Nickel Platform for the Generation of Branched High Molecular Weight Polyolefins. Nat. Commun. 2020, 11, 372. [Google Scholar]
- Wang, G.H.; Peng, D.; Sun, Y.; Chen, C.L. Interplay of Supramolecular Chemistry and Photochemistry with Palladium-Catalyzed Ethylene Polymerization. CCS Chem. 2020, 2, 2025–2034. [Google Scholar]
- Chen, M.; Chen, C.L. Direct and Tandem Routes for the Copolymerization of Ethylene with Polar Functionalized Internal Olefins. Angew. Chem. Int. Ed. 2020, 59, 1206–1210. [Google Scholar]
- Wang, X.; Seidel, F.W.; Nozaki, K. Synthesis of Polyethylene with in-chain α,β-Unsaturated Ketone and Isolated Ketone Units by Pd-catalyzed Ring-opening Copolymerization of Cyclopropenones with Ethylene. Angew. Chem. Int. Ed. 2019, 58, 12955–12959. [Google Scholar]
- Xu, M.; Yu, F.; Li, P.; Xu, G.; Zhang, S.; Wang, F. Enhancing Chain Initiation Efficiency in the Cationic Allyl-Nickel Catalyzed (Co)Polymerization of Ethylene and Methyl Acrylate. Inorg. Chem. 2020, 59, 4475–4482. [Google Scholar]
- Yasuda, H.; Nakano, R.; Ito, S.; Nozaki, K. Palladium/IzQO-Catalyzed Coordination–Insertion Copolymerization of Ethylene and 1,1-Disubstituted Ethylenes Bearing a Polar Functional Group. J. Am. Chem. Soc. 2018, 140, 1876–1883. [Google Scholar]
- Wang, X.; Nozaki, K. Selective Chain-end Functionalization of Polar Polyethylenes: Orthogonal Reactivity of Carbene and Polar Vinyl Monomers in Their Copolymerization with Ethylene. J. Am. Chem. Soc. 2018, 140, 15635–15640. [Google Scholar] [CrossRef]
- Wang, C.; Luo, G.; Nishiura, M.; Song, G.; Yamamoto, A.; Luo, Y.; Hou, Z. Heteroatom-assisted olefin polymerization by rare-earth metal catalysts. Sci. Adv. 2017, 3, e1701011. [Google Scholar]
- Li, K.K.; Ye, J.H.; Wang, Z.; Mu, H.L.; Jian, Z.B. Indole-bridged bisphosphine-monoxide palladium catalysts for ethylene polymerization and copolymerization with polar monomers. Polym. Chem. 2020, 11, 2740–2748. [Google Scholar]
- Cui, L.; Jian, Z.B. A N-bridged strategy enables hemilabile phosphine–carbonyl palladium and nickel catalysts to mediate ethylene polymerization and copolymerization with polar vinyl monomers. Polym. Chem. 2020. [Google Scholar] [CrossRef]
- Chung, T.C. Functionalization of Polyolefins; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Dong, J.Y.; Hu, Y. Design and synthesis of structurally well-defined functional polyolefins via transition metal-mediated olefin polymerization chemistry. Coord. Chem. Rev. 2005, 250, 47–65. [Google Scholar] [CrossRef]
- Rünzi, T.; Mecking, S. Saturated Polar-Substituted Polyethylene Elastomers from Insertion Polymerization. Adv. Funct. Mater. 2014, 24, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Chen, C.L. Palladium-Catalyzed Direct Synthesis of Various Branched, Carboxylic Acid-Functionalized Polyolefins: Characterization, Derivatization, and Properties. Macromolecules 2018, 51, 6818–6824. [Google Scholar] [CrossRef]
- Na, Y.; Dai, S.; Chen, C.L. Direct synthesis of polar-functionalized Linear LowDensity Polyethylene (LLDPE) and Low-Density Polyethylene (LDPE). Macromolecules 2018, 51, 4040–4048. [Google Scholar] [CrossRef]
- Xiang, P.; Ye, Z.B. Hyperbranched Polyethylene Ionomers Containing Cationic Tetralkylammonium Ions Synthesized by Pd-Diimine-Catalyzed Direct Ethylene Copolymerization with Ionic Liquid Comonomers. Macromolecules 2015, 48, 6096–6107. [Google Scholar] [CrossRef]
- Zou, C.; Chen, C.L. Polar-Functionalized, Crosslinkable, Self-Healing and Photoresponsive Polyolefins. Angew. Chem. Int. Ed. 2020, 59, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.; Chen, C.L. Catechol Functionalized Polyolefins. Angew. Chem. Int. Ed. 2020, 59, 7953–7959. [Google Scholar] [CrossRef]
- Dai, S.; Li, S.; Xu, G.; Chen, C.L. Direct Synthesis of Polar Functionalized Polyethylene Thermoplastic Elastomer. Macromolecules 2020, 53, 2539–2546. [Google Scholar] [CrossRef]
- Gao, J.; Cai, W.; Hu, Y.; Chen, C.L. Improving the Flame Retardancy of Polyethylenes through Palladium-Catalyzed Incorporation of Polar Comonomers. Polym. Chem. 2019, 10, 1416–1422. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and alpha.-Olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Walsh, D.J.; Hyatt, M.G.; Miller, S.A.; Guironnet, D. Recent Trends in Catalytic Polymerizations. ACS Catal. 2019, 9, 11153–11188. [Google Scholar] [CrossRef]
- Chen, Z.; Brookhart, M. Exploring Ethylene/Polar Vinyl Monomer Copolymerizations Using Ni and Pd α-Diimine Catalysts. Acc. Chem. Res. 2018, 51, 1831–1839. [Google Scholar] [CrossRef]
- Zhong, L.; Du, C.; Liao, G.; Liao, H.; Zheng, H.; Wu, Q.; Gao, H. Effects of backbone substituent and intra-ligand hydrogen bonding interaction on ethylene polymerizations with α-diimine nickel catalysts. J. Catal. 2019, 375, 113–123. [Google Scholar] [CrossRef]
- Takeuchi, D.; Osakada, K. Controlled isomerization polymerization of olefins, cycloolefins, and dienes. Polymer 2016, 82, 392–405. [Google Scholar] [CrossRef]
- Brown, L.A.; Anderson, W.C.; Mitchell, N.E.; Gmernicki, K.R.; Long, B.K. High Temperature, Living polymerization of ethylene by a sterically-demanding nickel(II) α-diimine catalyst. Polymers 2018, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, X.; Behzadi, S.; Xu, M.; Yu, F.; Xu, G.; Wang, F. Living Chain-Walking (Co)Polymerization of Propylene and 1-Decene by Nickel α-Diimine Catalysts. Polymers 2020, 12, 1988. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Tian, S.S.; Li, R.P.; Li, W.M.; Chen, C.L. Ligand steric effects on naphthyl-α-diimine nickel catalyzed α-olefin polymerization. Chin. J. Polym. Sci. 2018, 36, 157–162. [Google Scholar] [CrossRef]
- Guo, L.H.; Zou, C.; Dai, S.Y.; Chen, C.L. Direct Synthesis of Branched Carboxylic Acid Functionalized Poly(1-octene) by α-Diimine Palladium Catalysts. Polymers 2017, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.H.; Dai, S.Y.; Sui, X.L.; Chen, C.L. Palladium and Nickel Catalyzed Chain Walking Olefin Polymerization and Copolymerization. ACS Catal. 2016, 6, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Tempel, D.J.; Johnson, L.K.; Huff, R.L.; White, P.S.; Brookhart, M. Mechanistic Studies of Pd (II) a-Diimine-Catalyzed Olefin Polymerizations. J. Am. Chem. Soc. 2000, 122, 6686–6700. [Google Scholar] [CrossRef]
- Liu, F.S.; Hu, H.B.; Xu, Y.; Guo, L.H.; Zai, S.B.; Song, K.M.; Gao, H.Y.; Zhang, L.; Zhu, F.M.; Wu, Q. Thermostable α-diimine Nickel(II) catalyst for ethylene polymerization: Effects of the substituted backbone structure on catalytic properties and branching structure of polyethylene. Macromolecules 2009, 42, 7789–7796. [Google Scholar] [CrossRef]
- Berkefeld, A.; Mecking, S. Deactivation pathways of neutral Ni (II) polymerization catalysts. J. Am. Chem. Soc. 2009, 131, 1565–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.P.; Chen, C.L. Influence of Backbone Substituents on the Ethylene (Co)polymerization Properties of α-diimine Pd(II) and Ni(II) Catalysts. Organometallics 2016, 35, 1794–1801. [Google Scholar] [CrossRef]
- Dai, S.Y.; Sui, X.L.; Chen, C.L. Highly Robust Pd(II) α–diimine Catalysts for Slow-Chain-Walking Polymerization of Ethylene and Copolymerization with Methyl Acrylate. Angew. Chem. Int. Ed. 2015, 54, 9948–9953. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.Y.; Chen, C.L. Direct Synthesis of Functionalized High-Molecular-Weight Polyethylene by Copolymerization of Ethylene with Polar Monomers. Angew. Chem. Int. Ed. 2016, 55, 13281–13285. [Google Scholar] [CrossRef]
- Dai, S.Y.; Chen, C.L. A Self-Supporting Strategy for Gas-Phase and Slurry-Phase Ethylene Polymerization using Late-Transition-Metal Catalysts. Angew. Chem. Int. Ed. 2020, 59, 14884–14890. [Google Scholar] [CrossRef]
- Muhammad, Q.; Tan, C.; Chen, C.L. Concerted steric and electronic effects on α-diimine nickel- and palladium-catalyzed ethylene polymerization and copolymerization. Sci. Bull. 2020, 65, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Pang, W.; Chen, C.L. A Phenol-Containing α-Diimine Ligand for Nickel- and Palladium-Catalyzed Ethylene Polymerization. Chin. J. Polym. Sci. 2019, 37, 974–980. [Google Scholar] [CrossRef]
- Camacho, D.H.; Salo, E.V.; Ziller, J.W.; Guan, Z. Cyclophane-based highly active late-transition-metal catalysts for ethylene polymerization. Angew. Chem. Int. Ed. 2004, 43, 1821–1825. [Google Scholar] [CrossRef]
- Popeney, C.S.; Rheingold, A.L.; Guan, Z. Nickel (II) and Palladium (II) Polymerization Catalysts Bearing a Fluorinated Cyclophane Ligand: Stabilization of the Reactive Intermediate. Organometallics 2009, 28, 4452–4463. [Google Scholar] [CrossRef]
- Guo, L.; Gao, H.; Guan, Q.; Hu, H.; Deng, J.; Liu, J.; Liu, F.; Wu, Q. Substituent effects of the backbone in α-diimine palladium catalysts on homo-and copolymerization of ethylene with methyl acrylate. Organometallics 2012, 31, 6054–6062. [Google Scholar] [CrossRef]
- Zhong, S.; Tan, Y.; Zhong, L.; Gao, J.; Jiang, H.L.; Gao, H.Y.; Wu, Q. Precision synthesis of ethylene and polar monomer copolymers by palladiumcatalyzed living coordination copolymerization. Macromolecules 2012, 50, 5661–5669. [Google Scholar] [CrossRef]
- Zhong, L.; Zheng, H.; Du, C.; Du, W.; Liao, G.; Cheung, C.S.; Gao, H. Thermally robust α-diimine nickel and palladium catalysts with constrained space for ethylene (co)polymerizations. J. Catal. 2020, 384, 208–217. [Google Scholar] [CrossRef]
- Sui, X.; Hong, C.; Pang, W.; Chen, C.L. Unsymmetrical α-diimine palladium catalysts and their properties in olefin (co)polymerization. Mater. Chem. Front. 2017, 1, 967–972. [Google Scholar] [CrossRef]
- Dai, S.; Zhou, S.X.; Zhang, W.; Chen, C.L. Systematic Investigations of Ligand Steric Effects on α-Diimine Palladium Catalyzed Olefin Polymerization and Copolymerization. Macromolecules 2016, 49, 2919–2926. [Google Scholar] [CrossRef]
- Sun, J.L.; Wang, F.Z.; Li, W.; Chen, M. Ligand steric effects on α-diimine nickel catalyzed ethylene and 1-hexene polymerization. RSC Adv. 2017, 7, 55051–55059. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Li, S.; Tan, C.; Kong, W.; Xu, G.; Zhang, S.; Liu, B.Y.; Dai, S. π–π interaction effect in insertion polymerization with α-Diimine palladium systems. J. Catal. 2019, 378, 184–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Mecking, S.; Jian, Z. Ultrahigh Branching of Main-Chain-Functionalized Polyethylenes by Inverted Insertion Selectivity. Angew. Chem. Int. Ed. 2019, 59, 14296–14302. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Q.; Zhang, Y.X.; Zhang, Y.; Jian, Z. Unsymmetrical Strategy Makes Significant Differences in α-Diimine Nickel and Palladium Catalyzed Ethylene (Co)Polymerizations. ChemCatChem 2020, 12, 2497–2505. [Google Scholar] [CrossRef]
- Liao, Y.D.; Zhang, Y.; Cui, L.; Mu, H.; Jian, Z. Pentiptycenyl Substituents in Insertion Polymerization with α-Diimine Nickel and Palladium Species. Organometallics 2019, 38, 2075–2083. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C.L. Rational Design of High-Performance Phosphine Sulfonate Nickel Catalysts for Ethylene Polymerization and Copolymerization with Polar Monomers. ACS Catal. 2017, 7, 1308–1312. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Chen, C.L. Position Makes the Difference: Electronic Effects in Nickel-Catalyzed Ethylene Polymerizations and Copolymerizations. Inorg. Chem. 2018, 57, 14913–14919. [Google Scholar] [CrossRef]
- Tan, C.; Qasim, M.; Pang, W.; Chen, C.L. Ligand-Metal Secondary Interaction in Phosphine-Sulfonate Palladium and Nickel Catalyzed Ethylene (Co)Polymerization. Polym. Chem. 2020, 11, 411–416. [Google Scholar] [CrossRef]
- Song, G.; Pang, W.; Li, W.; Chen, M.; Chen, C.L. PhosphineSulfonate-Based Nickel Catalysts: Ethylene Polymerization and Copolymerization with Polar-Functionalized Norbornenes. Polym. Chem. 2017, 8, 7400–7405. [Google Scholar] [CrossRef]
- Yang, B.; Xiong, S.; Chen, C.L. Manipulation of Polymer Branching Density in Phosphine-Sulfonate Palladium and Nickel Catalyzed Ethylene Polymerization. Polym. Chem. 2017, 8, 6272–6276. [Google Scholar] [CrossRef]
- Wu, Z.; Hong, C.; Du, H.; Pang, W.; Chen, C.L. Influence of Ligand Backbone Structure and Connectivity on the Properties of Phosphine-Sulfonate Pd(II)/Ni(II) Catalysts. Polymers 2017, 9, 168. [Google Scholar]
- Perrotin, P.; Mccahill, J.S.; Wu, G.; Scott, S.L. Linear, high molecular weight polyethylene from a discrete, mononuclear phosphinoarenesulfonate complex of nickel(II). Chem. Commun. 2011, 47, 6948–6950. [Google Scholar] [CrossRef]
- Dai, S.Y.; Sui, X.L.; Chen, C.L. Synthesis of High Molecular Weight Polyethylene Using Iminopyridyl Nickel Catalysts. Chem. Commun. 2016, 52, 9113–9116. [Google Scholar] [CrossRef]
- Wu, Z.X.; Chen, M.; Chen, C.L. Ethylene Polymerization and Copolymerization by Palladium and Nickel Catalysts Containing Naphthalene-Bridged Phosphine–Sulfonate Ligands. Organometallics 2016, 35, 1472–1479. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, C.L. Influence of Polyethylene Glycol Unit on Palladium and Nickel Catalyzed Ethylene Polymerization and Copolymerization. Angew. Chem. Int. Ed. 2017, 56, 14672–14676. [Google Scholar] [CrossRef]
- Feng, Z.; Solomon, J.B.; Jordan, R.F. Copolymerization of Ethylene with Acrylate Monomers by AmideFunctionalized α-Diimine Pd Catalysts. Organometallics 2017, 36, 1873–1879. [Google Scholar]
- Salo, E.V.; Guan, Z. Late-Transition-Metal Complexes with Bisazaferrocene Ligands for Ethylene Oligomerization. Organometallics 2003, 22, 5033–5046. [Google Scholar] [CrossRef]
- RajanBabu, T.V.; Smith, C.R.; Zhang, A.; Mans, D.; Xie, M. (R)-3-Methyl-3-phenyl-1-pentene via catalytic asymmetric hydrovinylation. Org. Synth. 2008, 85, 248–266. [Google Scholar]
- Kato, K.; Furukawa, K.; Mori, T.; Osuka, A. Porphyrin-Based Air-Stable Helical Radicals. Chem. Eur. J. 2018, 24, 572–575. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.B.; Luo, Y.; Chen, C.L. A Second-coordination-sphere Strategy to Modulate Nickel- and Palladium-catalyzed Olefin Polymerization and Copolymerization. Angew. Chem. Int. Ed. 2017, 56, 11604–11609. [Google Scholar] [CrossRef]
Chart 1.
Selected Brookhart-type α-diimine Pd catalysts (A) [32], (B) [51,52], (C) [53,54,55], (D) [46,47,48,49,50], (E) [60,61,62] and (F) Pd catalysts synthesized in this work.

Scheme 1.
Synthesis of the ligands and the Pd catalysts.

Figure 1.
Molecular structures of: (A) Pd1, (B) Pd2, (C) Pd3, and (D) Pd4. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) for Pd1 (CCDC. 1941586): Pd1-N1 = 2.158(5), Pd1-Cl1 = 2.3017(2), Pd1-N2 = 2.056(6), Pd1-C40 = 2.037(6); N1-Pd-N2 = 77.5(2), N1-Pd-C40 = 172.2(2), N2-Pd1-C40 = 95.6(2), N2-Pd1-Cl1 = 97.80(14), N1-Pd1-Cl1 = 175.32(16) C40-Pd1-Cl1 = 89.08(19); for Pd2 (CCDC. 1957450): Pd01-N003 = 2.052(4), Pd01-Cl02 = 2.297(1), Pd01-N004 = 2.149(4), Pd01-C007 = 2.044(4); N003-Pd-N004 = 77.44(14), N003-Pd-C007 = 95.27(17), N004-Pd01-C007 = 171.63(17), N004-Pd01-Cl02 = 97.82(11), N003-Pd01-Cl02 = 175.16(10), C007-Pd01-Cl02 = 89.53(14); for Pd3 (CCDC. 1941588): Pd1-N1 = 2.05(2), Pd1-Cl1 = 2.30(1), Pd1-N2 = 2.13(2), Pd1-C17 = 2.01(2); N1-Pd-N2 = 77.7(7), N1-Pd-C40 = 97.1(9), N2-Pd1-C17 = 174.6(10), N2-Pd1-Cl1 = 96.1(5), N1-Pd1-Cl1 = 172.1(6) C17-Pd1-Cl1 = 89.3(8); for Pd4 (CCDC. 1959442): Pd1-N1 = 2.018(6), Pd1-Cl1 = 2.2964(17), Pd1-N2 = 2.157(5), Pd1-C1 = 2.061(6); N1-Pd-N2 = 77.9(2), N1-Pd-C1 = 94.6(2), N2-Pd1-C1 = 172.5(3), N2-Pd1-Cl1 = 99.85(14), N1-Pd1-Cl1 = 177.03(19), C1-Pd1-Cl1 = 87.64(19).
Figure 1.
Molecular structures of: (A) Pd1, (B) Pd2, (C) Pd3, and (D) Pd4. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) for Pd1 (CCDC. 1941586): Pd1-N1 = 2.158(5), Pd1-Cl1 = 2.3017(2), Pd1-N2 = 2.056(6), Pd1-C40 = 2.037(6); N1-Pd-N2 = 77.5(2), N1-Pd-C40 = 172.2(2), N2-Pd1-C40 = 95.6(2), N2-Pd1-Cl1 = 97.80(14), N1-Pd1-Cl1 = 175.32(16) C40-Pd1-Cl1 = 89.08(19); for Pd2 (CCDC. 1957450): Pd01-N003 = 2.052(4), Pd01-Cl02 = 2.297(1), Pd01-N004 = 2.149(4), Pd01-C007 = 2.044(4); N003-Pd-N004 = 77.44(14), N003-Pd-C007 = 95.27(17), N004-Pd01-C007 = 171.63(17), N004-Pd01-Cl02 = 97.82(11), N003-Pd01-Cl02 = 175.16(10), C007-Pd01-Cl02 = 89.53(14); for Pd3 (CCDC. 1941588): Pd1-N1 = 2.05(2), Pd1-Cl1 = 2.30(1), Pd1-N2 = 2.13(2), Pd1-C17 = 2.01(2); N1-Pd-N2 = 77.7(7), N1-Pd-C40 = 97.1(9), N2-Pd1-C17 = 174.6(10), N2-Pd1-Cl1 = 96.1(5), N1-Pd1-Cl1 = 172.1(6) C17-Pd1-Cl1 = 89.3(8); for Pd4 (CCDC. 1959442): Pd1-N1 = 2.018(6), Pd1-Cl1 = 2.2964(17), Pd1-N2 = 2.157(5), Pd1-C1 = 2.061(6); N1-Pd-N2 = 77.9(2), N1-Pd-C1 = 94.6(2), N2-Pd1-C1 = 172.5(3), N2-Pd1-Cl1 = 99.85(14), N1-Pd1-Cl1 = 177.03(19), C1-Pd1-Cl1 = 87.64(19).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Pd catalyzed ethylene homo-polymerization. a
| Ent. | Cat. | Temp. (°C) | Yield (g) | Act. b | Mnc (×104) | Mw/Mnc | B d |
|---|---|---|---|---|---|---|---|
| 1 | Pd1 | 40 | 0.44 | 18 | 12.57 | 1.9 | 69 |
| 2 | Pd1 | 60 | 0.32 | 13 | 6.75 | 2.4 | 70 |
| 3 | Pd1 | 80 | 0.18 | 7.2 | 3.91 | 1.8 | 72 |
| 4 | Pd2 | 40 | 0.48 | 19 | 7.83 | 3.1 | 81 |
| 5 | Pd2 | 60 | 0.34 | 14 | 3.10 | 6.0 | 83 |
| 6 | Pd2 | 80 | 0.20 | 8.0 | 1.76 | 3.9 | 85 |
| 7 | Pd3 | 40 | 0.46 | 18 | 13.87 | 2.1 | 77 |
| 8 | Pd3 | 60 | 0.39 | 16 | 5.79 | 6.9 | 79 |
| 9 | Pd3 | 80 | 0.09 | 3.6 | 3.93 | 1.9 | 80 |
| 10 | Pd4 | 40 | 0.49 | 20 | 7.75 | 3.7 | 83 |
| 11 | Pd4 | 60 | 0.26 | 10 | 3.50 | 5.8 | 87 |
| 12 | Pd4 | 80 | 0.22 | 8.8 | 1.34 | 4.5 | 89 |
a General conditions: Pd catalyst (1 μmol), NaBAF (1.2 equiv), ethylene 0.8 MPa, CH2Cl2 0.1 mL, toluene 3 mL, 1 h. b Activity (Act.): 105 g PE (mol Pd h)−1. c Mn and Mw/Mn determined by GPC. d branches per 1000 carbons, determined by 1H NMR.
Table 2.
Pd catalyzed ethylene copolymerizations. a
| Ent. | Cat. | Yield (g) | Act. b | Xc c (%) | Mnd (×104) | Mw/Mnd | B e |
|---|---|---|---|---|---|---|---|
| 1 | Pd1 | 2.1 | 14 | 1.85 | 6.90 | 2.11 | 83 |
| 2 | Pd2 | 1.6 | 1.1 | 2.54 | 6.05 | 1.97 | 76 |
| 3 | Pd3 | 2.4 | 1.6 | 2.01 | 7.51 | 1.82 | 85 |
| 4 | Pd4 | 1.9 | 1.3 | 3.02 | 1.82 | 2.55 | 82 |
a General conditions: precatalyst (30 μmol), NaBAF (1.2 equiv), toluene (20 mL) and MA (2 M), 35 °C, ethylene 0.3 MPa, 5 h. b Activity (Act.): 104 g (mol Pd)−1 h−1. c Incorporation ratio (mol %) of co-monomer, determined by 1H NMR. d Mn and Mw/Mn determined by GPC. e Determined by 1H NMR.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Muhammad, Q.; Pang, W.; Wang, F.; Tan, C. Ortho-Functionalized Dibenzhydryl Substituents in α-Diimine Pd Catalyzed Ethylene Polymerization and Copolymerization. Polymers 2020, 12, 2509. https://doi.org/10.3390/polym12112509
AMA Style
Muhammad Q, Pang W, Wang F, Tan C. Ortho-Functionalized Dibenzhydryl Substituents in α-Diimine Pd Catalyzed Ethylene Polymerization and Copolymerization. Polymers. 2020; 12(11):2509. https://doi.org/10.3390/polym12112509
Chicago/Turabian StyleMuhammad, Qasim, Wenmin Pang, Fuzhou Wang, and Chen Tan. 2020. "Ortho-Functionalized Dibenzhydryl Substituents in α-Diimine Pd Catalyzed Ethylene Polymerization and Copolymerization" Polymers 12, no. 11: 2509. https://doi.org/10.3390/polym12112509
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.