Biodegradation of Crystalline Cellulose Nanofibers by Means of Enzyme Immobilized-Alginate Beads and Microparticles

 ,
,
Abstract
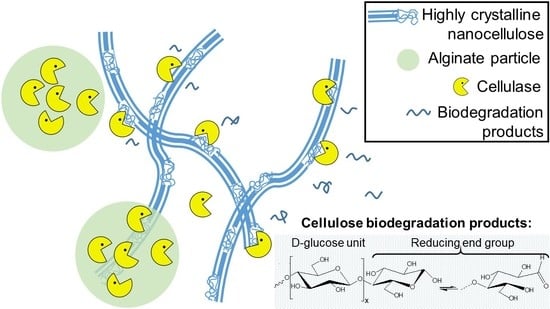
:
1. Introduction
2. Materials and Methods
2.1. Starting Materials—Preparation and Characterization
2.2. Preparation of Cellulase-Encapsulated Alginate Particles
2.3. Cellulose Enzymatic Degradation—Cellulase Activity Assay
2.4. Microscopical Observations
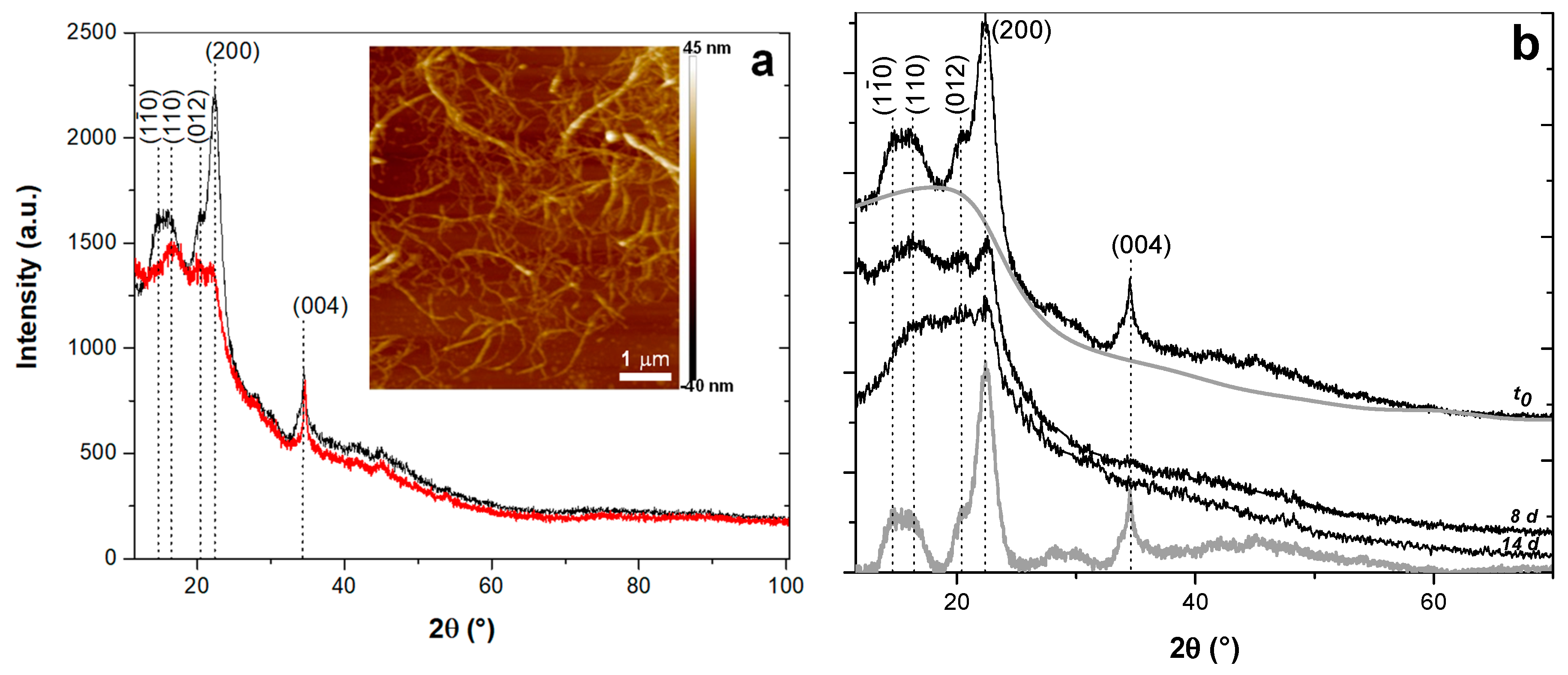
2.5. X-ray Diffraction
2.6. Thermogravimetric Analysis
3. Results and Discussion
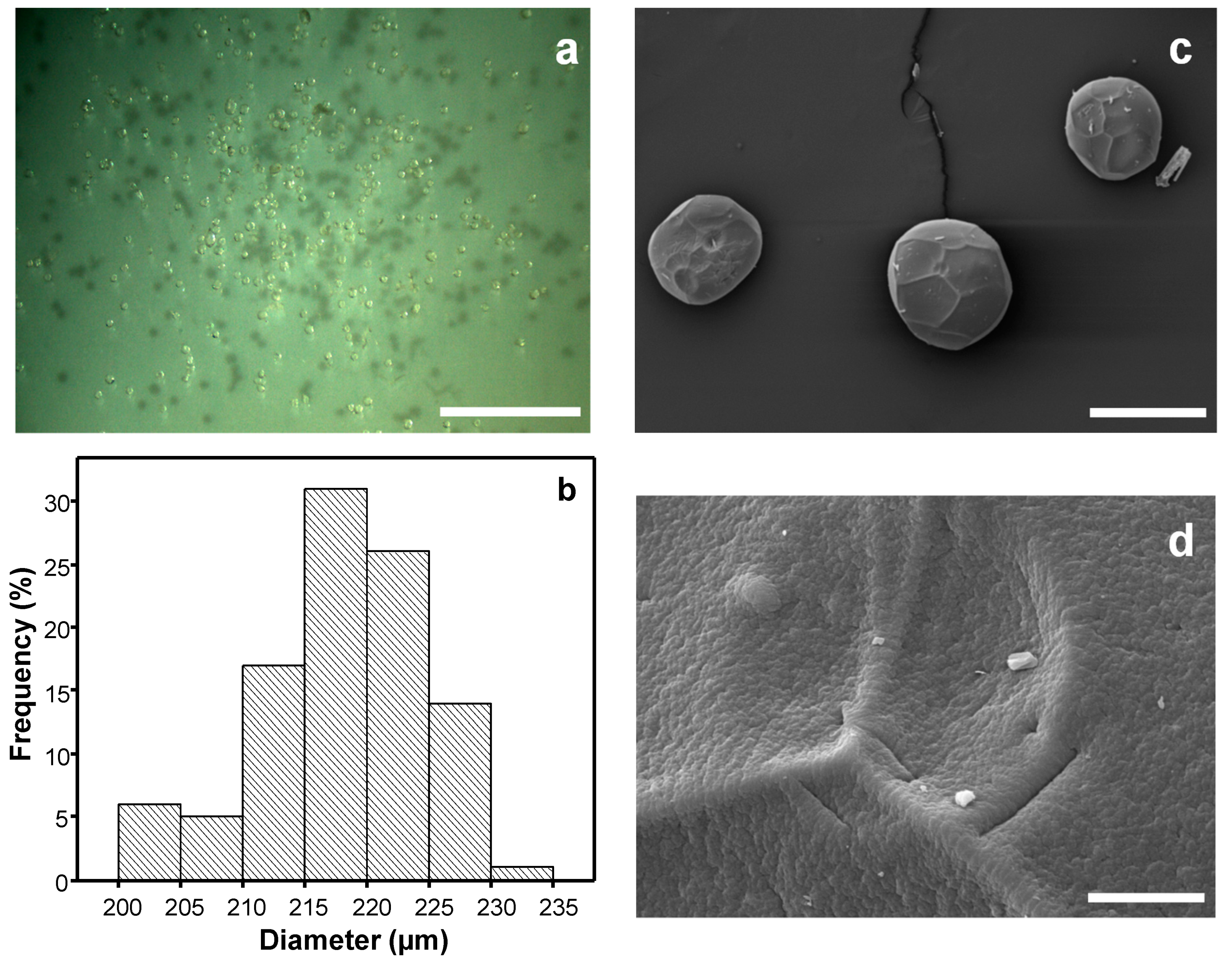
3.1. Particles Morphology and Dimensions
3.2. Activity of Encapsulated Cellulase towards Hydrolysis of Cellulose Substrate in Solution
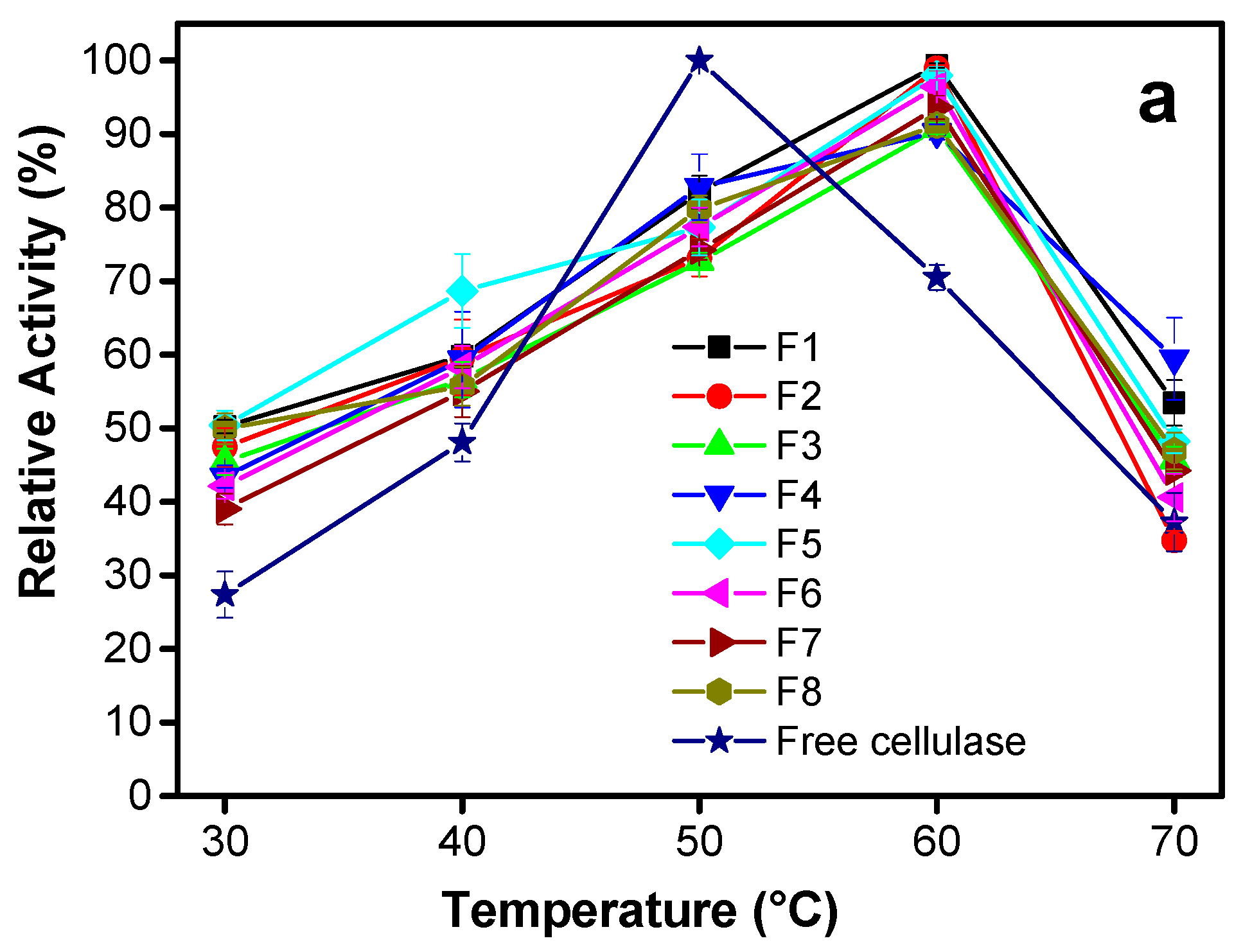
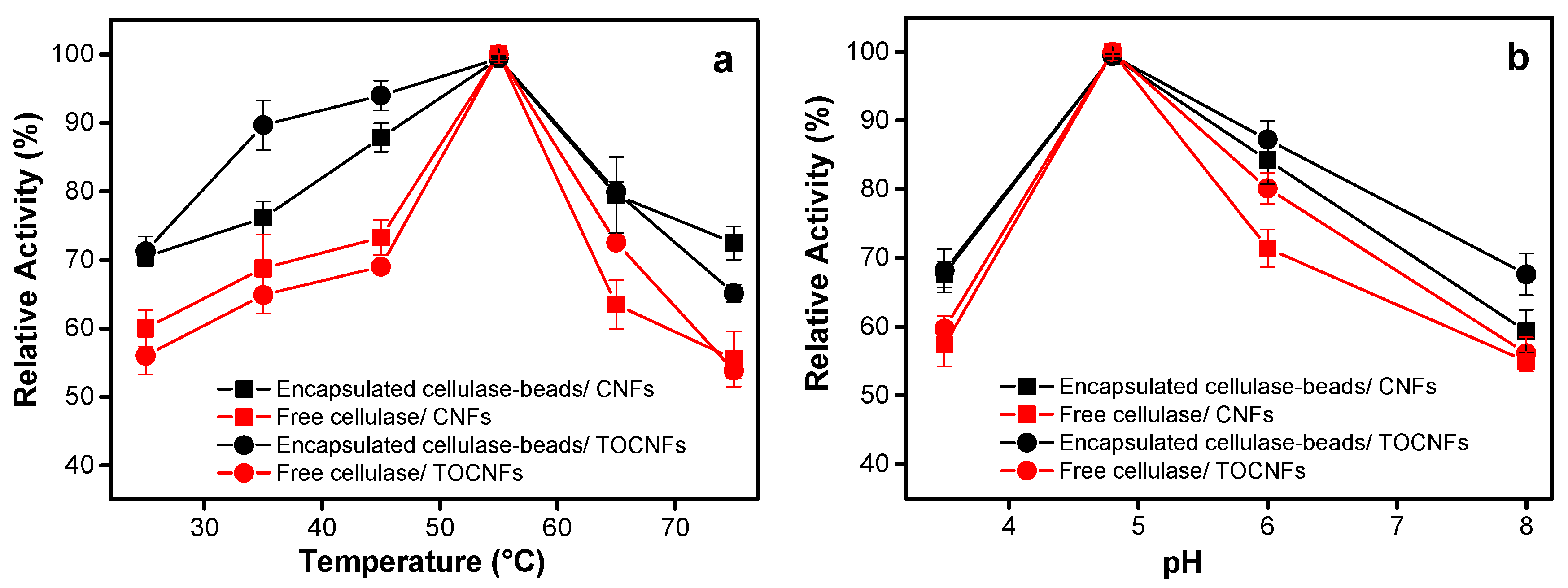
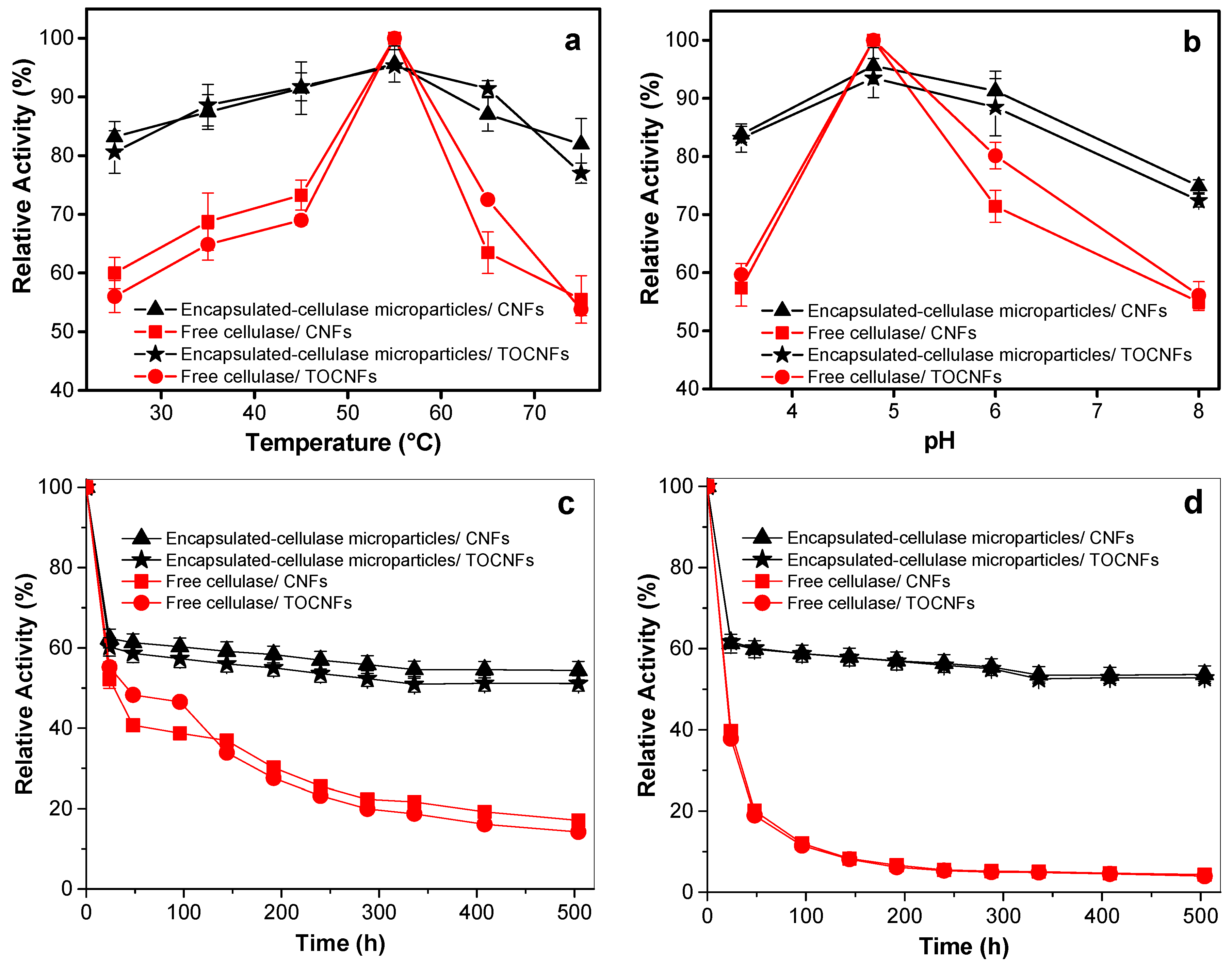
3.2.1. Effect of Temperature
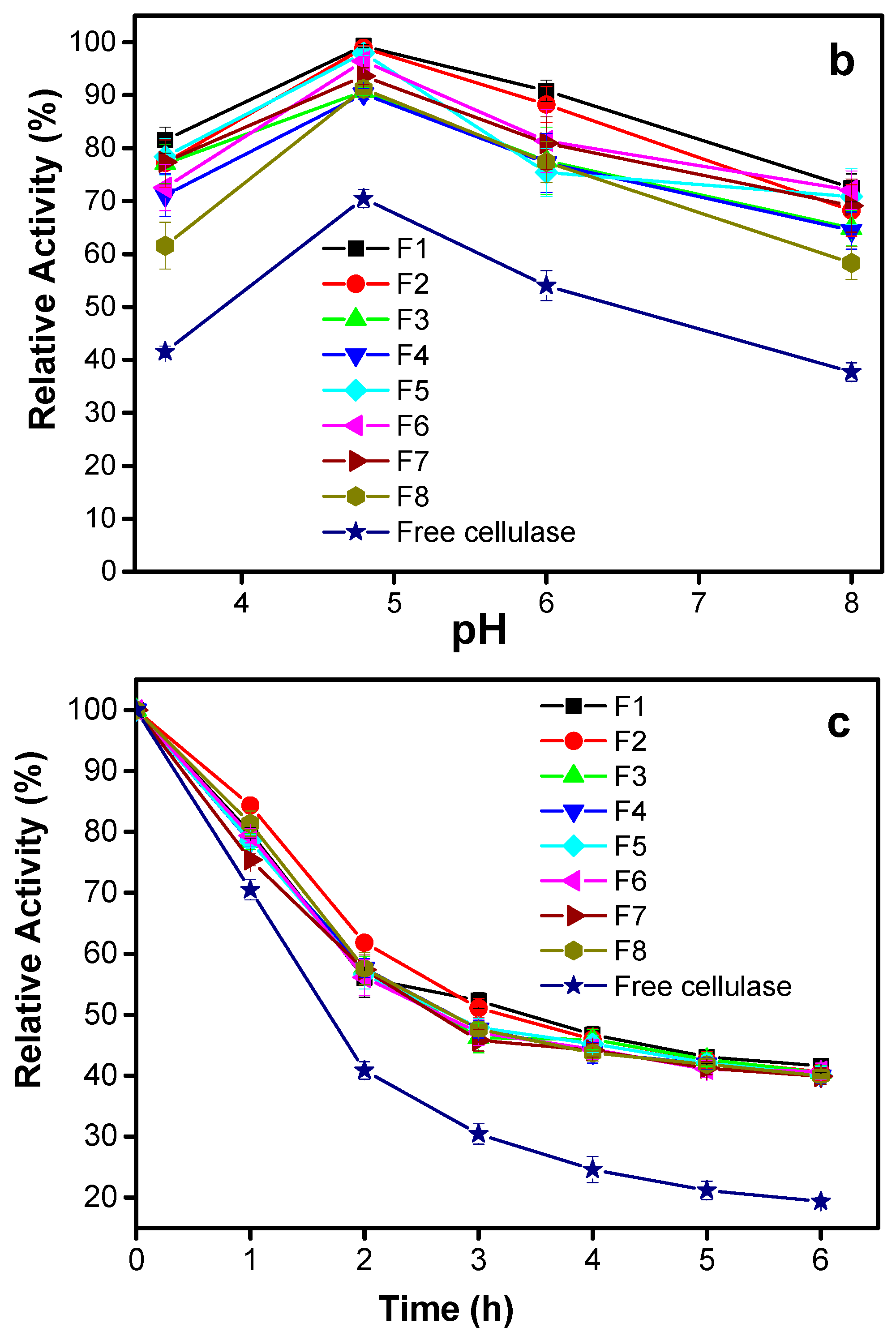
3.2.2. Effect of pH
3.2.3. Effect of Incubation Time
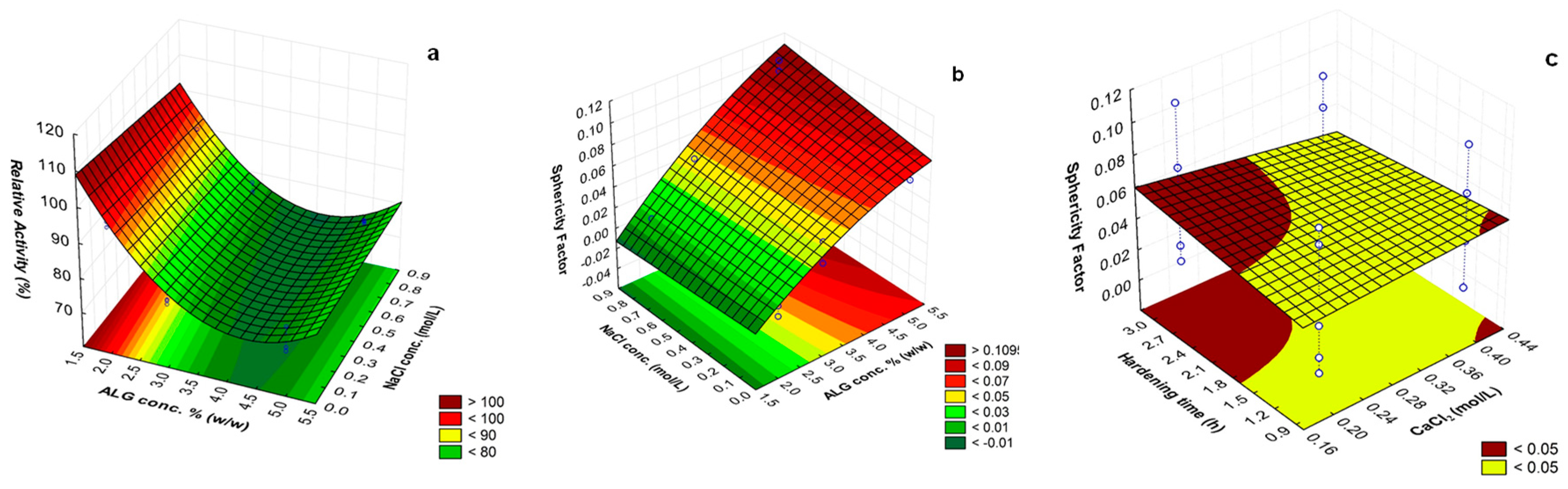
3.2.4. Optimization for High Catalytic Activity of the Encapsulated Cellulase
3.2.5. Multiple Response Optimization
3.3. Activity of Cellulase Encapsulated in Alginate Beads towards Degradation of Cellulose Nanofibers
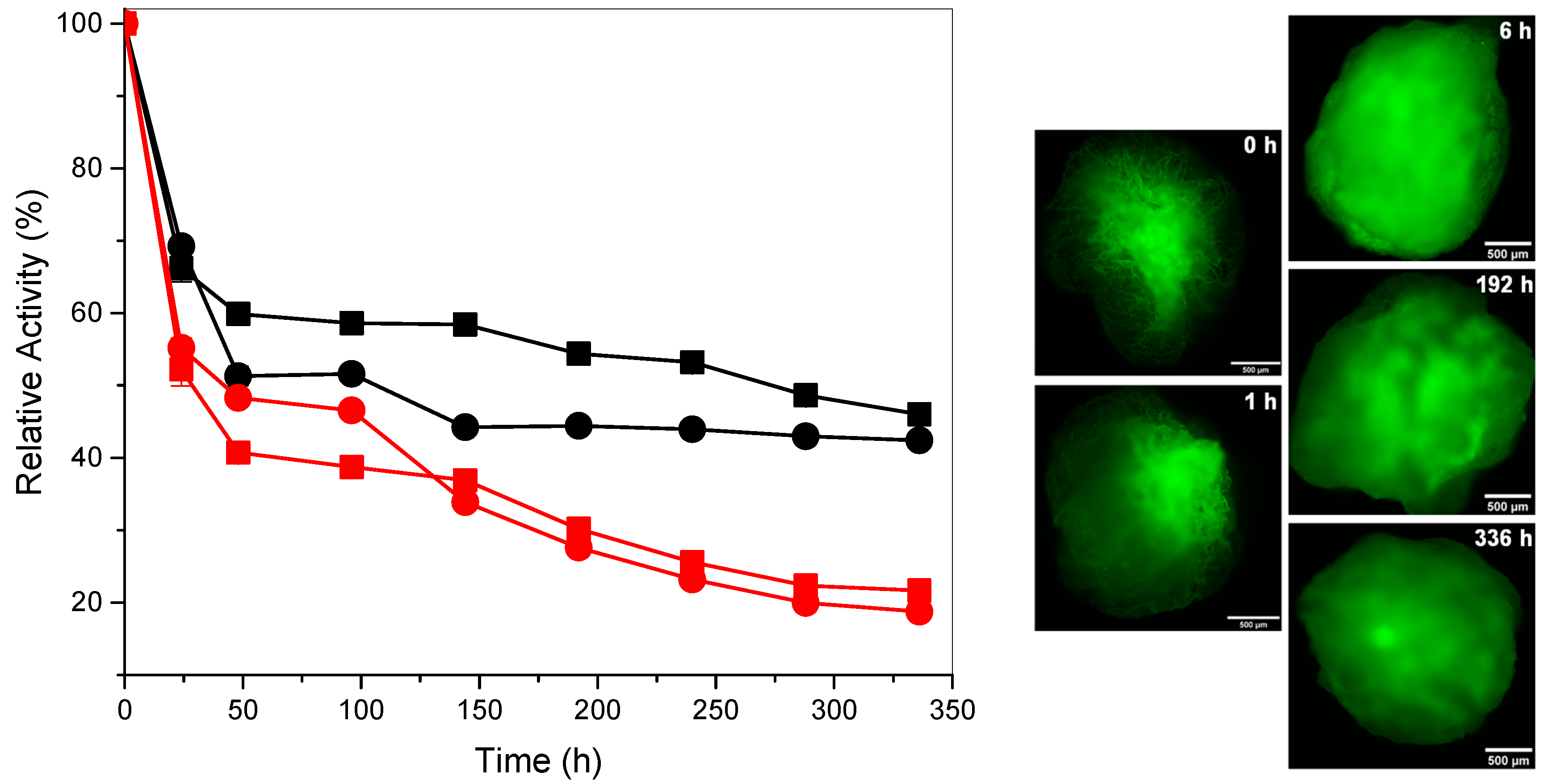
Storage Stability of Cellulase
3.4. Cellulase-Immobilized Alginate Microparticles
3.4.1. Microstructure, Dimensions and Properties
3.4.2. Catalytic Activity of Cellulase Encapsulated in Alginate Microparticles towards Cellulose Nanofibers Degradation
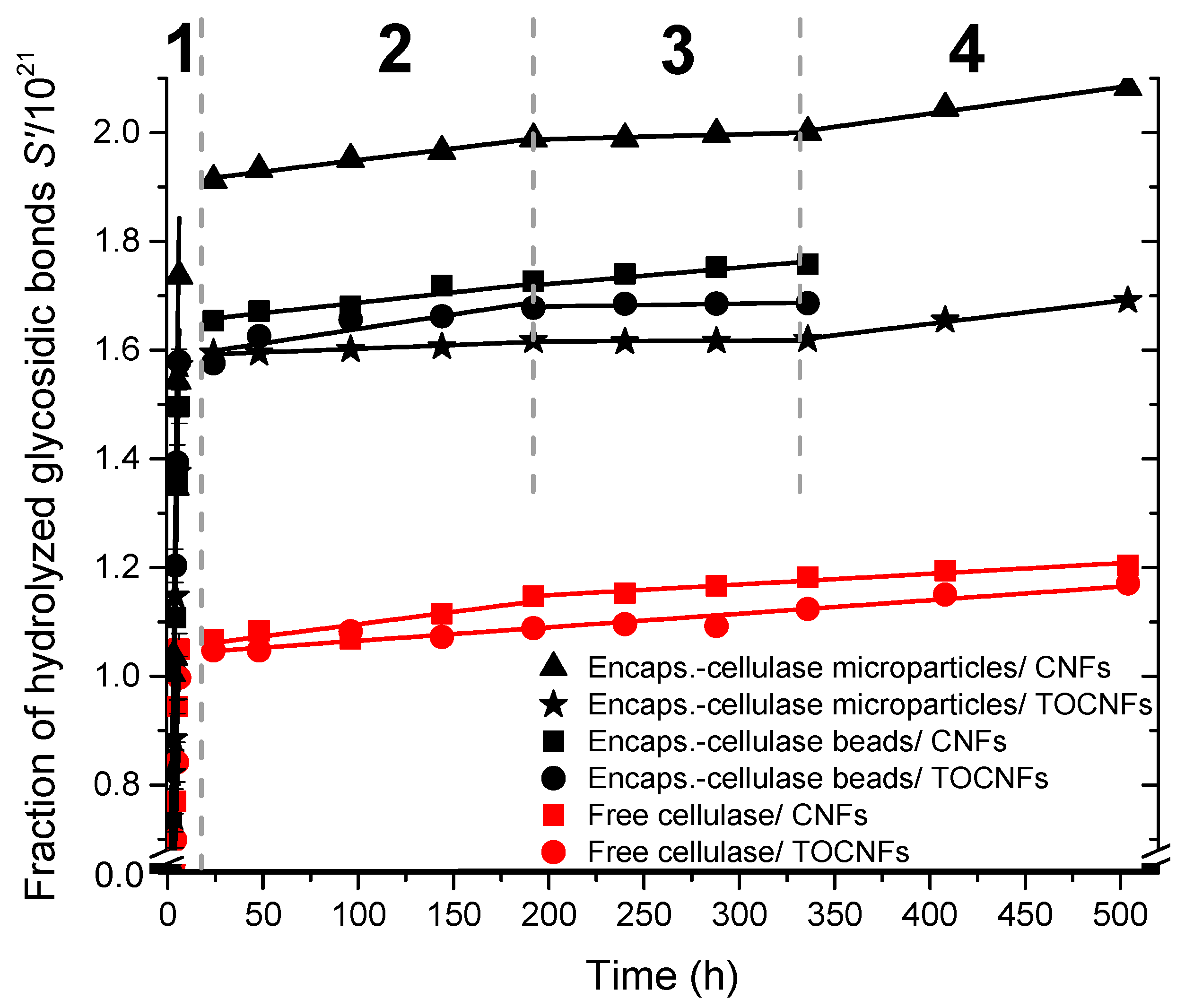
3.5. Kinetics Study of the Biodegradation of Cellulose Nanofibers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jorfi, M.; Foster, E.J. Recent advances in nanocellulose for biomedical applications. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Jozala, A.F.; De Lencastre-Novaes, L.C.; Lopes, A.M.; De Carvalho Santos-Ebinuma, V.; Mazzola, P.G.; Pessoa, A., Jr.; Grotto, D.; Gerenutti, M.; Chaud, M.V. Bacterial nanocellulose production and application: A 10-year overview. Appl. Microbiol. Biotechnol. 2016, 100, 2063–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio-Madrazo, A.; David, L.; Montembault, A.; Viguier, E.; Cachon, T. Hydrogel Composites Comprising Chitosan and Cellulose Nanofibers. International Patent Application No. WO 2019/175279 A1, 19 September 2019. [Google Scholar]
- Domingues, R.M.A.; Gomes, M.E.; Reis, R.L. The Potential of Cellulose Nanocrystals in Tissue Engineering Strategies. Biomacromolecules 2014, 15, 2327–2346. [Google Scholar] [CrossRef] [PubMed]
- De France, K.J.; Badv, M.; Dorogin, J.; Siebers, E.; Panchal, V.; Babi, M.; Moran-Mirabal, J.; Lawlor, M.; Cranston, E.D.; Hoare, T.; et al. Tissue Response and Biodistribution of Injectable Cellulose Nanocrystal Composite Hydrogels. ACS Biomater. Sci. Eng. 2019, 5, 2235–2246. [Google Scholar] [CrossRef]
- Torres-Rendon, J.G.; Femmer, T.; De Laporte, L.; Tigges, T.; Rahimi, K.; Gremse, F.; Zafarnia, S.; Lederle, W.; Ifuku, S.; Wessling, M.; et al. Bioactive Gyroid Scaffolds Formed by Sacrificial Templating of Nanocellulose and Nanochitin Hydrogels as Instructive Platforms for Biomimetic Tissue Engineering. Adv. Mater. 2015, 27, 2989–2995. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.-R.; Liisa Kanninen, L.; Kuisma, T.; Niklander, J.; Noon, L.A.; Burks, D.; Urtti, A.; Yliperttula, M. The Use of Nanofibrillar Cellulose Hydrogel as a Flexible Three-Dimensional Model to Culture Human Pluripotent Stem Cells. Stem Cells Dev. 2014, 23, 380–392. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Seabra, A.B.; Bernardes, J.S.; Fávaro, W.J.; Paula, A.J.; Durán, N. Cellulose nanocrystals as carriers in medicine and their toxicities: A review. Carbohydr. Polym. 2018, 181, 514–527. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; David, L.; Peniche-Covas, C.; Rochas, C.; Putaux, J.-L.; Trombotto, S.; Alcouffe, P.; Domard, A. Fine microstructure of processed chitosan nanofibril networks preserving directional packing and high molecular weight. Carbohydr. Polym. 2015, 131, 1–8. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; David, L.; Trombotto, S.; Lucas, J.-M.; Peniche-Covas, C.; Domard, A. Highly crystalline chitosan produced by multi-steps acid hydrolysis in the solid-state. Carbohydr. Polym. 2011, 83, 1730–1739. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; Fratzl, P.; David, L.; Urban, G.; Montembault, A.; Crepet, A.; Gorzelanny, C.; Mochales-Palau, C.; Heux, L.; Putaux, J.-L.; et al. Synthese und Charakterisierung von Biomaterialien (Polymere, Metalle, Keramiken, Komposite). P85: Hydrogel nanocomposite biomaterials for intervertebral disc tissue engineering. Preparation, characterization and application. Bionanomaterials 2015, 16, 236–255. [Google Scholar] [CrossRef]
- Doench, I.; Torres-Ramos, M.E.W.; Montembault, A.; Nunes de Oliveira, P.; Halimi, C.; Viguier, E.; Heux, L.; Siadous, R.; Thiré, R.M.S.M.; Osorio-Madrazo, A. Injectable and Gellable Chitosan Formulations Filled with Cellulose Nanofibers for Intervertebral Disc Tissue Engineering. Polymers 2018, 10, 1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, I.; Ahn Tran, T.; David, L.; Montembault, A.; Viguier, E.; Gorzelanny, C.; Sudre, G.; Cachon, T.; Louback-Mohamed, M.; Horbelt, N.; et al. Cellulose Nanofiber-Reinforced Chitosan Hydrogel Composites for Intervertebral Disc Tissue Repair. Biomimetics 2019, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- De France, K.J.; Yager, K.G.; Chan, K.J.W.; Corbett, B.; Cranston, E.D.; Hoare, T. Injectable Anisotropic Nanocomposite Hydrogels Direct in Situ Growth and Alignment of Myotubes. Nano Lett. 2017, 17, 6487–6495. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; Eder, M.; Rueggeberg, M.; Pandey, J.K.; Harrington, M.J.; Nishiyama, Y.; Putaux, J.-L.; Rochas, C.; Burgert, I. Reorientation of cellulose nanowhiskers in agarose hydrogels under tensile loading. Biomacromolecules 2012, 13, 850–856. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; David, L.; Trombotto, S.; Lucas, J.-M.; Peniche-Covas, C.; Domard, A. Kinetics study of the solid-state acid hydrolysis of chitosan: Evolution of the crystallinity and macromolecular structure. Biomacromolecules 2010, 11, 1376–1386. [Google Scholar] [CrossRef]
- Jonoobi, M.; Oladi, R.; Davoudpour, Y.; Oksman, K.; Dufresne, A.; Hamzeh, Y.; Davoodi, R. Different preparation methods and properties of nanostructured cellulose from various natural resources and residues: A review. Cellulose 2015, 22, 935–969. [Google Scholar] [CrossRef]
- Prakash Menon, M.; Selvakumar, R.; Suresh kumar, P.; Ramakrishna, S. Extraction and modification of cellulose nanofibers derived from biomass for environmental application. RSC Adv. 2017, 7, 42750–42773. [Google Scholar] [CrossRef] [Green Version]
- Osorio-Madrazo, A.; Laborie, M.P. Morphological and Thermal Investigations of Cellulosic Bionanocomposites. In Biopolymer Nanocomposites: Processing, Properties, and Applications; Dufresne, A., Thomas, S., Pothen, L.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 411–435. [Google Scholar] [CrossRef]
- Samyn, P.; Osorio-Madrazo, A. Native Crystalline Polysaccharide Nanofibers: Processing and Properties. In Handbook of Nanofibers; Barhoum, A., Bechelany, M., Makhlouf, A., Eds.; Springer: Cham, Switzerland, 2018; pp. 1–36. [Google Scholar] [CrossRef]
- Šturcová, A.; Davies, G.R.; Eichhorn, S.J. Elastic Modulus and Stress-Transfer Properties of Tunicate Cellulose Whiskers. Biomacromolecules 2005, 6, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, D.G.; Bruce, D.M. A method of calculating the mechanical properties of nanoscopic plant cell wall components from tissue properties. J. Mater. Sci. 2000, 35, 5861–5865. [Google Scholar] [CrossRef]
- Nishino, T.; Takano, K.; Nakamae, K. Elastic modulus of the crystalline regions of cellulose polymorphs. J. Polym. Sci. Part B Polym. Phys. 1995, 33, 1647–1651. [Google Scholar] [CrossRef]
- Mao, J.; Osorio-Madrazo, A.; Laborie, M.P. Novel preparation route for cellulose nanowhiskers. In Proceedings of the 245th ACS Annual Meeting (CELL: Division of Cellulose and Renewable Materials), New Orleans, LA, USA, 7–11 April 2013. [Google Scholar]
- Mao, J.; Osorio-Madrazo, A.; Laborie, M.-P. Preparation of cellulose I nanowhiskers with a mildly acidic aqueous ionic liquid: Reaction efficiency and whiskers attributes. Cellulose 2013, 20, 1829–1840. [Google Scholar] [CrossRef]
- Benítez, A.J.; Lossada, F.; Zhu, B.; Rudolph, T.; Walther, A. Understanding Toughness in Bioinspired Cellulose Nanofibril/Polymer Nanocomposites. Biomacromolecules 2016, 17, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Pääkkö, M.; Ankerfors, M.; Kosonen, H.; Nykänen, A.; Ahola, S.; Osterberg, M.; Ruokolainen, J.; Laine, J.; Larsson, P.T.; Ikkala, O.; et al. Enzymatic hydrolysis combined with mechanical shearing and high-pressure homogenization for nanoscale cellulose fibrils and strong gels. Biomacromolecules 2007, 8, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Abushammala, H.; Pontes, J.F.; Gomes, G.H.; Osorio-Madrazo, A.; Thiré, R.M.S.M.; Pereira, F.V.; Laborie, M.-P.G. Swelling, viscoelastic, and anatomical studies on ionic liquid-swollen Norway spruce as a screening tool toward ionosolv pulping. Holzforschung 2015, 69, 1059–1067. [Google Scholar] [CrossRef]
- Hubbell, J.A. Biomaterials in Tissue Engineering. Bio/Technology 1995, 13, 565–576. [Google Scholar] [CrossRef]
- Peppas, N.; Langer, R. New challenges in biomaterials. Science 1994, 263, 1715–1720. [Google Scholar] [CrossRef]
- Eyholzer, C.; Borges De Couraça, A.; Duc, F.; Bourban, P.E.; Tingaut, P.; Zimmermann, T.; Månson, J.A.E.; Oksman, K. Biocomposite Hydrogels with Carboxymethylated, Nanofibrillated Cellulose Powder for Replacement of the Nucleus Pulposus. Biomacromolecules 2011, 12, 1419–1427. [Google Scholar] [CrossRef]
- Borges, A.C.; Eyholzer, C.; Duc, F.; Bourban, P.-E.; Tingaut, P.; Zimmermann, T.; Pioletti, D.P.; Månson, J.-A.E. Nanofibrillated cellulose composite hydrogel for the replacement of the nucleus pulposus. Acta Biomater. 2011, 7, 3412–3421. [Google Scholar] [CrossRef] [Green Version]
- Arola, S.; Tammelin, T.; Setälä, H.; Tullila, A.; Linder, M.B. Immobilization–Stabilization of Proteins on Nanofibrillated Cellulose Derivatives and Their Bioactive Film Formation. Biomacromolecules 2012, 13, 594–603. [Google Scholar] [CrossRef]
- Kolakovic, R.; Laaksonen, T.; Peltonen, L.; Laukkanen, A.; Hirvonen, J. Spray-dried nanofibrillar cellulose microparticles for sustained drug release. Int. J. Pharm. 2012, 430, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Dufresne, A. Nanocellulose in biomedicine: Current status and future prospect. Eur. Polym. J. 2014, 59, 302–325. [Google Scholar] [CrossRef] [Green Version]
- Torres-Rendon, J.G.; Köpf, M.; Gehlen, D.; Blaeser, A.; Fischer, H.; Laporte, L.D.; Walther, A. Cellulose Nanofibril Hydrogel Tubes as Sacrificial Templates for Freestanding Tubular Cell Constructs. Biomacromolecules 2016, 17, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Nieves, R.A.; Ehrman, C.I.; Adney, W.S.; Elander, R.T.; Himmel, M.E. Survey and analysis of commercial cellulase preparations suitable for biomass conversion to ethanol. World J. Microbiol. Biotechnol. 1997, 14, 301–304. [Google Scholar] [CrossRef]
- Berlin, A.; Maximenko, V.; Gilkes, N.; Saddler, J. Optimization of enzyme complexes for lignocellulose hydrolysis. Biotechnol. Bioeng. 2007, 97, 287–296. [Google Scholar] [CrossRef]
- Bhat, M.K.; Bhat, S. Cellulose degrading enzymes and their potential industrial applications. Biotechnol. Adv. 1997, 15, 583–620. [Google Scholar] [CrossRef]
- Hu, Y.; Catchmark, J.M. Integration of cellulases into bacterial cellulose: Toward bioabsorbable cellulose composites. J. Biomed. Mater. Res. Part B Appl. Biomater. 2011, 97, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbert, W.; Chanzy, H.; Husum, T.L.; Schülein, M.; Ernst, S. Fluorescent Cellulose Microfibrils as Substrate for the Detection of Cellulase Activity. Biomacromolecules 2003, 4, 481–487. [Google Scholar] [CrossRef]
- Mansfield, S.D.; Meder, R. Cellulose hydrolysis—The role of monocomponent cellulases in crystalline cellulose degradation. Cellulose 2003, 10, 159–169. [Google Scholar] [CrossRef]
- Pu, Y.; Ziemer, C.; Ragauskas, A.J. CP/MAS 13C NMR analysis of cellulase treated bleached softwood kraft pulp. Carbohydr. Res. 2006, 341, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Stipanovic, A.J.; Winter, W.T.; Wilson, D.B.; Kim, Y.-J. Effect of digestion by pure cellulases on crystallinity and average chain length for bacterial and microcrystalline celluloses. Cellulose 2007, 14, 283. [Google Scholar] [CrossRef]
- Zhang, Y.-H.P.; Lynd, L.R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulase systems. Biotechnol. Bioeng. 2004, 88, 797–824. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.H.-Y.; Jang, J.; Wu, K.C.W. Cellulase immobilized mesoporous silica nanocatalysts for efficient cellulose-to-glucose conversion. Green Chem. 2011, 13, 2844–2850. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, S.; Singh, O.V. Bioconversion of lignocellulosic biomass: Biochemical and molecular perspectives. J. Ind. Microbiol. Biotechnol. 2008, 35, 377–391. [Google Scholar] [CrossRef]
- Tsai, C.-T.; Meyer, A.S. Enzymatic Cellulose Hydrolysis: Enzyme Reusability and Visualization of β-Glucosidase Immobilized in Calcium Alginate. Molecules 2014, 19, 19390–19406. [Google Scholar] [CrossRef] [Green Version]
- Mailin, M.; Hu, Z.; Bo, J. Nanobiocatalyst advancements and bioprocessing applications. J. R. Soc. Interface 2015, 12, 20140891. [Google Scholar] [CrossRef] [Green Version]
- Gross, R.A.; Kalra, B. Biodegradable Polymers for the Environment. Science 2002, 297, 803–807. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [Green Version]
- Toeri, J.; Osorio-Madrazo, A.; Laborie, M.-P. Preparation and Chemical/Microstructural Characterization of Azacrown Ether-Crosslinked Chitosan Films. Materials 2017, 10, 400. [Google Scholar] [CrossRef]
- Peniche, H.; Osorio, A.; Acosta, N.; De La Campa, A.; Peniche, C. Preparation and characterization of superparamagnetic chitosan microspheres: Application as a support for the immobilization of tyrosinase. J. Appl. Polym. Sci. 2005, 98, 651–657. [Google Scholar] [CrossRef]
- Bierhalz, A.C.K.; Da Silva, M.A.; Kieckbusch, T.G. Natamycin release from alginate/pectin films for food packaging applications. J. Food Eng. 2012, 110, 18–25. [Google Scholar] [CrossRef]
- Pérez-Madrigal, M.M.; Torras, J.; Casanovas, J.; Häring, M.; Alemán, C.; Díaz, D.D. Paradigm Shift for Preparing Versatile M2+-Free Gels from Unmodified Sodium Alginate. Biomacromolecules 2017, 18, 2967–2979. [Google Scholar] [CrossRef] [PubMed]
- Gombotz, W.R.; Wee, S. Protein release from alginate matrices. Adv. Drug Deliv. Rev. 1998, 31, 267–285. [Google Scholar] [CrossRef]
- Axpe, E.; Oyen, M.L. Applications of Alginate-Based Bioinks in 3D Bioprinting. Int. J. Mol. Sci. 2016, 17, 1976. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Filippov, M.P.; Kohn, R. Determination of composition of alginates by infrared spectroscopic method. Chem. Pap. 1974, 28, 817–819. [Google Scholar]
- Benítez, A.J.; Torres-Rendon, J.; Poutanen, M.; Walther, A. Humidity and Multiscale Structure Govern Mechanical Properties and Deformation Modes in Films of Native Cellulose Nanofibrils. Biomacromolecules 2013, 14, 4497–4506. [Google Scholar] [CrossRef]
- Røjel, N.; Kari, J.; Sørensen, T.H.; Borch, K.; Westh, P. pH profiles of cellulases depend on the substrate and architecture of the binding region. Biotechnol. Bioeng. 2020, 117, 382–391. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.-S. Preparation of Ca-alginate beads containing high oil content: Influence of process variables on encapsulation efficiency and bead properties. Carbohydr. Polym. 2011, 84, 1267–1275. [Google Scholar] [CrossRef]
- Chan, E.-S.; Lee, B.-B.; Ravindra, P.; Poncelet, D. Prediction models for shape and size of ca-alginate macrobeads produced through extrusion–dripping method. J. Colloid Interface Sci. 2009, 338, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Morales, E.; Rubilar, M.; Burgos-Díaz, C.; Acevedo, F.; Penning, M.; Shene, C. Alginate/Shellac beads developed by external gelation as a highly efficient model system for oil encapsulation with intestinal delivery. Food Hydrocoll. 2017, 70, 321–328. [Google Scholar] [CrossRef]
- Kazan, A.; Heymuth, M.; Karabulut, D.; Akay, S.; Yildiz-Ozturk, E.; Onbas, R.; Muderrisoglu, C.; Sargin, S.; Heils, R.; Smirnova, I.; et al. Formulation of organic and inorganic hydrogel matrices for immobilization of β-glucosidase in microfluidic platform. Eng. Life Sci. 2017, 17, 714–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, G.E.A.; Abd El Aty, A.A.; Shehata, A.N.; Hassan, M.E.; Elnashar, M.M. Covalent immobilization of microbial naringinase using novel thermally stable biopolymer for hydrolysis of naringin. 3 Biotech 2016, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Davarcı, F.; Turan, D.; Ozcelik, B.; Poncelet, D. The influence of solution viscosities and surface tension on calcium-alginate microbead formation using dripping technique. Food Hydrocoll. 2017, 62, 119–127. [Google Scholar] [CrossRef]
- Constantin, M.; Fundueanu, G. Key Considerations in the Design of Polymeric Micro-and Nanoparticles for Drug Delivery Systems. In Intelligent Polymers for Nanomedicine and Biotechnologies, 1st ed.; Aflori, M., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2018; p. 244. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Senkal, B.F.; Arica, M.Y. Preparation of clay–poly (glycidyl methacrylate) composite support for immobilization of cellulase. Appl. Clay Sci. 2013, 85, 88–95. [Google Scholar] [CrossRef]
- Nawaz, M.A.; Rehman, H.U.; Bibi, Z.; Aman, A.; Ul Qader, S.A. Continuous degradation of maltose by enzyme entrapment technology using calcium alginate beads as a matrix. Biochem. Biophys. Rep. 2015, 4, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Şahin, F.; Demirel, G.; Tümtürk, H. A novel matrix for the immobilization of acetylcholinesterase. Int. J. Biol. Macromol. 2005, 37, 148–153. [Google Scholar] [CrossRef]
- Yahşi, A.; Şahin, F.; Demirel, G.; Tümtürk, H. Binary immobilization of tyrosinase by using alginate gel beads and poly(acrylamide-co-acrylic acid) hydrogels. Int. J. Biol. Macromol. 2005, 36, 253–258. [Google Scholar] [CrossRef]
- Jadhav, S.B.; Singhal, R.S. Pullulan-complexed α-amylase and glucosidase in alginate beads: Enhanced entrapment and stability. Carbohydr. Polym. 2014, 105, 49–56. [Google Scholar] [CrossRef]
- Tanford, C. Physical Chemistry of Macromolecules; Wiley: Hoboken, NJ, USA, 1961. [Google Scholar]
- Vårum, K.M.; Ottøy, M.H.; Smidsrød, O. Acid hydrolysis of chitosans. Carbohydr. Polym. 2001, 46, 89–98. [Google Scholar] [CrossRef]
- Montroll, E. Molecular Size Distributions and Depolymerization Reactions in Polydisperse Systems. J. Am. Chem. Soc. 1941, 63, 1215–1220. [Google Scholar] [CrossRef]
- Sharples, A. The hydrolysis of cellulose Part I. The fine structure of egyptian cotton. J. Polym. Sci. 1954, 13, 393–401. [Google Scholar] [CrossRef]
- Daruwalla, E.H.; Narsian, M.G. Detection and identification of acid-sensitive linkages in cellulose fiber substances. Tappi 1966, 49, 106–111. [Google Scholar]
- Sharples, A. Acid hydrolysis in alcoholysis. In Cellulose and Cellulose Derivatives; Bikales, N.M., Segal, L., Eds.; Wiley-Intersciences: Hoboken, NJ, USA, 1971; Volume 5, pp. 991–1006. [Google Scholar]
- Calvini, P. The Influence of Levelling-off Degree of Polymerisation on the Kinetics of Cellulose Degradation. Cellulose 2005, 12, 445–447. [Google Scholar] [CrossRef]
- Emsley, A.M.; Stevens, G.C. Kinetics and mechanisms of the low-temperature degradation of cellulose. Cellulose 1994, 1, 26–56. [Google Scholar] [CrossRef]
- Philipp, H.J.; Nelson, M.L.; Ziifle, H.M. Crystallinity of Cellulose Fibers as Determined by Acid Hydrolysis. Text. Res. J. 1947, 17, 585–596. [Google Scholar] [CrossRef]
- Feller, R.L.; Lee, S.B.; Bogaard, J. The kinetics of cellulose deterioration. In Historic Textile and Paper Materials: Conservation and Characterization; Needles, H.L., Zeronian, S.H., Eds.; American Chemical Society: Washington, DC, USA, 1986; pp. 329–346. [Google Scholar]
- Yachi, T.; Hayashi, J.; Takai, M.; Shimizu, Y. Supermolecular structure of cellulose: Stepwise decrease in LODP and particle size of cellulose hydrolyzed after chemical treatment. J. Appl. Polym. Sci. Appl. Polym. Symp. 1983, 1, 325–343. [Google Scholar]
- Calvini, P.; Gorassini, A. On the Rate of Paper Degradation: Lessons from the Past. Restaur. Int. J. Preserv. Libr. Arch. Mater. 2006, 27, 275–290. [Google Scholar] [CrossRef]
- Calvini, P.; Gorassini, A.; Merlani, A.L. On the kinetics of cellulose degradation: Looking beyond the pseudo zero order rate equation. Cellulose 2007, 15, 193. [Google Scholar] [CrossRef]
- Frisoni, G.; Baiardo, M.; Scandola, M.; Lednická, D.; Cnockaert, M.C.; Mergaert, J.; Swings, J. Natural Cellulose Fibers: Heterogeneous Acetylation Kinetics and Biodegradation Behavior. Biomacromolecules 2001, 2, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Habibi, Y.; Chanzy, H.; Vignon, M.R. TEMPO-mediated surface oxidation of cellulose whiskers. Cellulose 2006, 13, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, W.; Lin, X.; Xiao, H.; Miao, Q.; Huang, L.; Chen, L.; Wu, H. TEMPO-Oxidized Cellulose with High Degree of Oxidation. Polymers 2017, 9, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Responses | Particle Size | ||||||
|---|---|---|---|---|---|---|---|---|
| Formu-Lation | c(ALG) (% (w/v)) | c(CaCl2) (mol/L) | c(NaCl) (mol/L) | Hardening Time (h) | Sphericity Factor | Cellulase Rel. Act. (%) | Hydrated | Dried |
| (mm) | ||||||||
| F1 | 2 (−1) | 0.2 (−1) | 0 (−1) | 1 (−1) | 0.004 | 99.28 | 2.38 ± 0.20 | 1.37 ± 0.19 |
| F2 | 2 (−1) | 0.2 (−1) | 0 (−1) | 3 (1) | 0.016 | 98.91 | 2.37± 0.25 | 1.41 ± 0.16 |
| F3 | 2 (−1) | 0.2 (−1) | 0.8 (1) | 1 (−1) | 0.008 | 90.71 | 2.29 ± 0.20 | 1.33 ± 0.23 |
| F4 | 2 (−1) | 0.2 (−1) | 0.8 (1) | 3 (1) | 0.014 | 90.24 | 2.06 ± 0.21 | 1.17 ± 0.18 |
| F5 | 2 (−1) | 0.4 (1) | 0 (−1) | 1 (−1) | 0.011 | 97.94 | 2.29 ± 0.21 | 1.32 ± 0.22 |
| F6 | 2 (−1) | 0.4 (1) | 0 (−1) | 3 (1) | 0.003 | 96.43 | 2.00 ± 0.35 | 1.46 ± 0.25 |
| F7 | 2 (−1) | 0.4 (1) | 0.8 (1) | 1 (−1) | 0.009 | 93.60 | 2.19 ± 0.21 | 1.14 ± 0.16 |
| F8 | 2 (−1) | 0.4 (1) | 0.8 (1) | 3 (1) | 0.012 | 91.17 | 2.42 ± 0.21 | 1.54 ± 0.22 |
| F9 | 3 (0) | 0.2 (−1) | 0 (−1) | 1 (−1) | 0.039 | 79.86 | 2.29 ± 0.18 | 1.37 ± 0.16 |
| F10 | 3 (0) | 0.2 (−1) | 0 (−1) | 3 (1) | 0.054 | 84.32 | 2.39 ± 0.21 | 1.66 ± 0.20 |
| F11 | 3 (0) | 0.2 (−1) | 0.8 (1) | 1 (−1) | 0.031 | 78.41 | 2.44 ± 0.20 | 1.56 ± 0.21 |
| F12 | 3 (0) | 0.2 (−1) | 0.8 (1) | 3 (1) | 0.045 | 78.41 | 2.48 ± 0.19 | 1.43 ± 0.14 |
| F13 | 3 (0) | 0.4 (1) | 0 (−1) | 1 (−1) | 0.041 | 78.50 | 2.21 ± 0.16 | 1.47 ± 0.17 |
| F14 | 3 (0) | 0.4 (1) | 0 (−1) | 3 (1) | 0.031 | 79.42 | 2.23 ± 0.14 | 1.57 ± 0.18 |
| F15 | 3 (0) | 0.4 (1) | 0.8 (1) | 1 (−1) | 0.035 | 75.44 | 2.53 ± 0.23 | 1.42 ± 0.16 |
| F16 | 3 (0) | 0.4 (1) | 0.8 (1) | 3 (1) | 0.041 | 81.36 | 2.07 ± 0.18 | 1.70 ± 0.17 |
| F17 | 5 (1) | 0.2 (−1) | 0 (-1) | 1 (−1) | 0.077 | 80.18 | 2.29 ± 0.22 | 1.55 ± 0.19 |
| F18 | 5 (1) | 0.2 (−1) | 0 (−1) | 3 (1) | 0.065 | 76.86 | 2.13 ± 0.57 | 1.73 ± 0.17 |
| F19 | 5 (1) | 0.2 (−1) | 0.8 (1) | 1 (−1) | 0.087 | 77.06 | 2.44 ± 0.14 | 1.44 ± 0.13 |
| F20 | 5 (1) | 0.2 (−1) | 0.8 (1) | 3 (1) | 0.111 | 77.21 | 2.29 ± 0.18 | 1.43 ± 0.19 |
| F21 | 5 (1) | 0.4 (1) | 0 (−1) | 1 (−1) | 0.068 | 74.18 | 2.22 ± 0.17 | 1.09 ± 0.14 |
| F22 | 5 (1) | 0.4 (1) | 0 (−1) | 3 (1) | 0.069 | 73.11 | 2.20 ± 0.17 | 1.19 ± 0.26 |
| F23 | 5 (1) | 0.4 (1) | 0.8 (1) | 1 (−1) | 0.099 | 70.19 | 2.10 ± 0.13 | 1.19 ± 0.25 |
| F24 | 5 (1) | 0.4 (1) | 0.8 (1) | 3 (1) | 0.086 | 76.41 | 2.25 ± 0.13 | 1.24 ± 0.26 |
| Levels | (−1): 2% (w/v); (0): 3% (w/v); (1): 5% (w/v) | (−1): 0.2 mol/L; (1): 0.4 mol/L | (−1): 0; (1): 0.8 mol/L | (−1): 1 h; (1): 3h | ||||
| Pseudo-Rate Constants/1014 (s−1) | Immobilized-Cellulase Microparticles | Immobilized-Cellulase Beads | Free Cellulase | |||
|---|---|---|---|---|---|---|
| CNFs | TOCNFs | CNFs | TOCNFs | CNFs | TOCNFs | |
| at 55 °C, pH 4.8: | ||||||
| k′1 | 760 ± 80 | 690 ± 60 | 660 ± 20 | 650 ± 30 | 350 ± 30 | 260 ± 30 |
| k′2 | 1.20 ± 0.10 | 0.38 ± 0.10 | 1.10 ± 0.20 | 1.50 ± 0.40 | 1.30 ± 0.30 | 0.69 ± 0.06 |
| k′3 | 0.25 ± 0.06 | 0.042 ± 0.01 | 0.85 ± 0.10 | 0.15 ± 0.06 | 0.54 ± 0.05 | - |
| k′4 | 1.40 ± 0.2 | 1.2 ± 0.40 | n.d. | n.d. | - | - |
| at 37 °C, pH 6.5: | ||||||
| k′1 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| k′2 | 2.60 ± 0.70 | 2.30 ± 0.60 | n.d. | n.d. | 1.20 ± 0.30 | 1.10 ± 0.30 |
| k′3 | 1.60 ± 0.40 | 1.30 ± 0.50 | n.d. | n.d. | - | - |
| k′4 | 3.10 ± 0.05 | 2.70 ±0.30 | n.d. | n.d. | - | - |
| Time of Enzymatic Degradation (h) | CrI (%) |
|---|---|
| 0 | 23 |
| 192 | 9.2 |
| 336 | 7.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamdem Tamo, A.; Doench, I.; Morales Helguera, A.; Hoenders, D.; Walther, A.; Madrazo, A.O. Biodegradation of Crystalline Cellulose Nanofibers by Means of Enzyme Immobilized-Alginate Beads and Microparticles. Polymers 2020, 12, 1522. https://doi.org/10.3390/polym12071522
Kamdem Tamo A, Doench I, Morales Helguera A, Hoenders D, Walther A, Madrazo AO. Biodegradation of Crystalline Cellulose Nanofibers by Means of Enzyme Immobilized-Alginate Beads and Microparticles. Polymers. 2020; 12(7):1522. https://doi.org/10.3390/polym12071522
Chicago/Turabian StyleKamdem Tamo, Arnaud, Ingo Doench, Aliuska Morales Helguera, Daniel Hoenders, Andreas Walther, and Anayancy Osorio Madrazo. 2020. "Biodegradation of Crystalline Cellulose Nanofibers by Means of Enzyme Immobilized-Alginate Beads and Microparticles" Polymers 12, no. 7: 1522. https://doi.org/10.3390/polym12071522